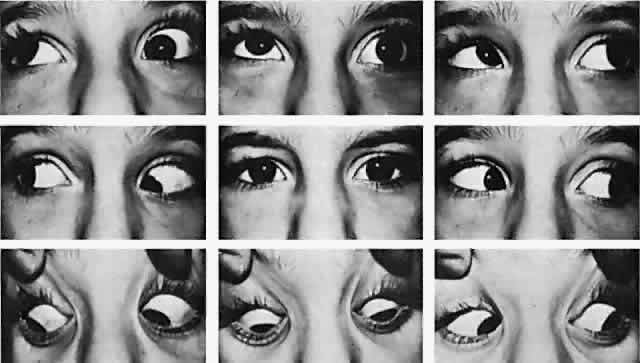
Clinical presentation of Duane retraction syndrome is varied. Invariably, in addition to retraction of the adducted globe, the patient exhibits a defect in horizontal motility. In addition, a vertical motility defect frequently occurs in adduction. Various contributors to the literature have categorized Duane syndrome4–10 into subtypes, but this may have its limitations, given that one type may merge with another. Furthermore, the various subclassifications are confusing because some are based on clinical findings4–6 whereas others are based on electromyographic findings.7–12 Huber,5 using electromyography, classified Duane retraction syndrome into three types:
[*] Type I: Absence of abduction, normal or restricted adduction with associated retraction
of the globe, and widening of the palpebral fissure on attempted
abduction. Electromyography shows absence of electrical activity
in the lateral rectus muscle on abduction but paradoxical electrical activity
on adduction.
[*] Type II: Exotropia with restricted adduction and abduction and retraction of the
globe on adduction. Electromyography reveals electrical activity with
contraction of the lateral rectus muscle on both abduction and adduction.
[*] Type III: Severe restriction of both abduction and adduction, with either minimal
esotropia or exotropia or near orthophoria and retraction on adduction
and widening of the palpebral fissure on abduction. The electromyogram
demonstrates cocontraction of the horizontal rectus muscles on both
adduction and abduction.
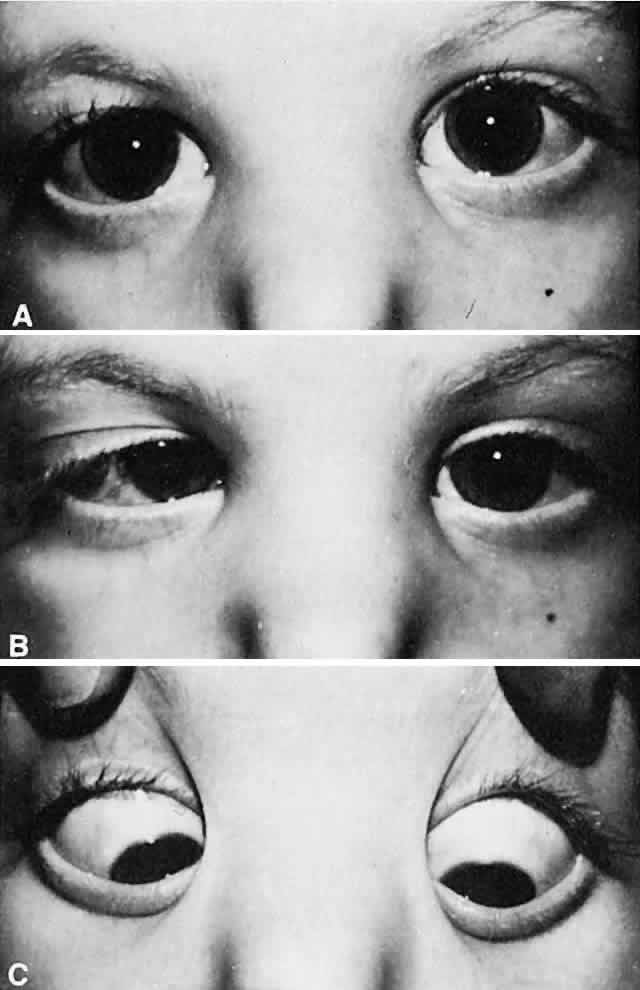
The most characteristic clinical presentation of Duane syndrome is an absence of abduction of an eye with some degree of restricted adduction and retraction when an attempt is made to adduct (Fig. 1). The retraction is variable: it is conspicuous in some but minimal in others. Additionally, either an upshooting or downshooting, or both, of the adducted eye frequently occurs, particularly as the adducting eye begins to move in the oblique position of up and in or down and in. This overshoot simulates overaction of the inferior and superioroblique muscles. Occasionally, the upshoot or down-shoot is so marked that the cornea is driven completely out of the palpebral fissure, hiding behind the upper or lower lid. Some patients manifest only the upshoot and a few only the downshoot, but most patients have various degrees of both vertical abnormalities. Magnetic resonance imaging (MRI) has been used in the cine mode13 to assess muscle contractility. In a patient with Duane type II the restriction of movement caused by the cocontraction of the horizontal rectus muscles was evident with the cine motion picture created by the MRI scanning unit.
|

Duane retraction syndrome may be bilateral,14–20 and although it most commonly involves the left eye, it may involve only the right eye. Duane retraction syndrome occurs more frequently in female than in male patients. Several excellent indepen-dent statistical studies have confirmed these facts.Kirkham15 determined that 1% of the people with strabismus have Duane retraction syndrome. In his study of 100 patients, 65 were female. The left eye was involved in 60% of the patients, the right eye was involved in 22%, and there was bilateral involvement in 18%. In reporting on 186 patientswith Duane syndrome, Pfaffenbach and partners16 found that 57% of the patients were female; the left eye was involved in 60% of the patients, the right eye in 21%, and both eyes in 19%. Isenberg and Urist18 reported on 101 patients and found 57% female; 84% were unilateral and the left eye wasinvolved in 66% of the unilateral cases, and 16% ofthe bilateral cases. In a review of 97 patients,O'Malley and associates19 found 62% female; 82% were unilateral, of which 67% involved the left eye, and 18% were bilateral. Raab20 reviewed 70 patients, of whom 64% were female; 90% were unilateral, with the left eye involved in 67%, and 10% were bilateral.
In Duane retraction syndrome, there is frequently a concurrent Klippel-Feil anomaly, which occurred in 4% of the patients from Kirkham's series15 and in 3% of the patients from Pfaffenbach and coworkers' series.16 Congenital labyrinthine deafness is alsooften associated with this syndrome; 11% ofKirkham's patients and 7.5% of the patients inPfaffenbach and coworkers' series manifested this association. Ro and associates21 found evidence of hearing impairment in 15.9% when testing a series of 44 patients with Duane syndrome with audiograms and auditory brainstem responses. Duane retraction syndrome, the Klippel-Feil anomaly, and congenital labyrinthine deafness constitute the syndrome of Wildervanck22 Kirkham15 has suggested that Duane syndrome, perceptual deafness, and the Klippel-Feil anomaly are inherited as a single gene trait, with incomplete penetrance and incomplete expressivity. The clinical presentation in the heterozygote is a gene with pleiotropic effects inherited in an irregularly dominant manner. The abnormal gene may be partly gender limited in such a way that male patients are more resistant, and female patients more susceptible to the action of the gene.
Several articles23–27 have reported localizations of genes for patients with Duane syndrome, including 8q insertion and deletion,23 localization to chromosome 2q31,24,25 chromosome loci at 4q, 8q, and 22q,26 and 22q11 deletion syndrome.27
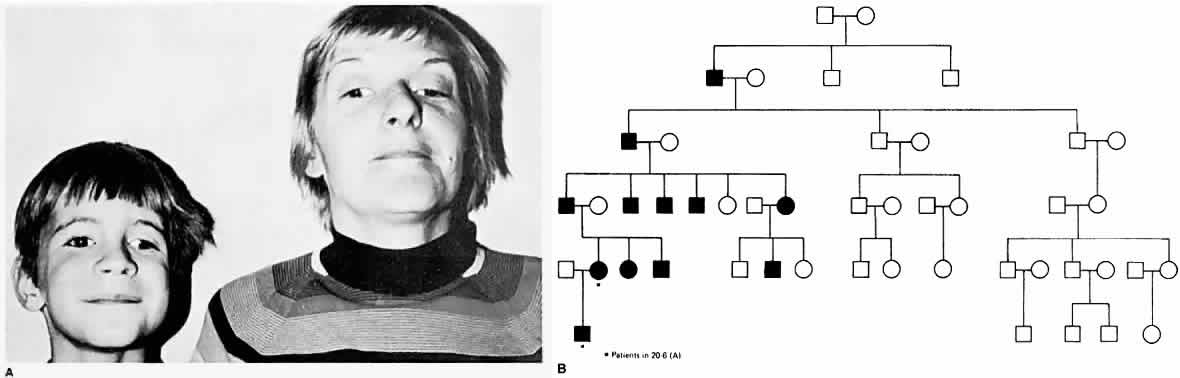
The epibulbar dermoids and preauricular skin tags of Goldenhar syndrome occur more frequently in patients with Duane syndrome than in people without Duane syndrome. Most cases of Duane syndrome seem to be sporadic; however, the syndrome has been reported by some and observed by one of the authors of this chapter (Parks) in three instances as a dominant inherited defect and has been reported by others, either as a unilateral28 or bilateral29 inherited syndrome. Chung and coworkers30 found 25 affected family members, distributed in an autosomal dominant pattern, in studying 110 members of 114 living relatives in an extended family with Duane syndrome. Twenty-four patients had bilateral Duane retraction syndrome, with a broad spectrum of severity. Amblyopia was present in 48% and strabismus in 76%. Other associated findings included cranial nerve (CN) IV palsy, partial CN III palsy, nystagmus, seizures, and deafness. Strabismus and amblyopia are far more common in bilateral than in unilateral Duane syndrome. Within families, a great deal of phenotypic variability is possible, and the genes responsible may affect the development of several CNs. Duane syndrome, associated with numerous congenital anomalies, probably results from dysgenetic events in the middle of the first trimester of pregnancy.31 The frequent association of nonocular abnormalities, such as the Wildervanck syndrome, with Duane retraction syndrome, suggests a teratogenic event that occurs between the fourth and eighth weeks of gestation.32 Acquired Duane retraction syndrome has been reported after palsy of CN VI33 as well as following Kronlein's lateral orbitotomy.34 Entrapment of the medial rectus muscle by a blowout fracture of themedial wall of the orbit, or by metastatic cancer, has been described by Duane and colleagues.35 This “pseudo-Duane retraction syndrome” differs in that there may be a history of trauma, diplopia is common, there is lid retraction on attempted abduction, and the prognosis is good when the entrapment can be reduced surgically. Various other findings and anomalies seen in association with Duane retraction syndrome include Holt-Oram syndrome,36 atrial septal defect and hand anomalies with autosomal dominant transmission; crocodile tears, anomalous lacrimation or paradoxical gustatory-lacrimal reflex, in unilateral37 and bilateral38 case reports; bilateral Duane syndrome, bilateral paroxysmal lacrimation and the Klippel-Feil anomaly39; familial perceptual deafness in five generations and Duane syndrome in two generations40; with myasthenia gravis41; with anisometropia and amblyopia42; with cleft palate43; with nevus of Ota and axial anisometropia44; with optic nerve hypoplasia45; with morning glory syndrome46 an unusual congenital optic nerve dysplasia, characterized by a funnel-shaped optic disc, annulus of surrounding pigment disturbance, and anomalous retinal vessels; with marfanoid hypermobility syndrome47; and with Marcus Gunn jaw-winking,48 fetal alcohol syndrome,31 Rubinstein-Taybi syndrome,49 and with de Morsier syndrome.50 Combined horizontal and vertical retraction syndrome has been reported.51 Duane syndrome has also been described in association with cat-eye syndrome52,53 with a supernumerary chromosome, probably derived from chromosome 22. Postmortem studies54–56 have shown absence of abducens nuclei and nerves from the brain stem, and partial innervation of the lateral rectus muscles by the oculomotor nerves54 and another study55 showed absence of the left abducens nerve and innervation of the left lateral rectus in part by branches from the inferior oculomotor nerve. Parsa and coworkers57 verified the absence of the abducens nerve in vivo in a 36-year-old woman with Duane syndrome type I in her left eye by MRI.
Most patients with Duane syndrome have straight eyes in the primary position, at least during infancy and childhood. The minority gradually develops an increasing esodeviation in the primary position that is offset by the restricted adduction of the involved eye. These patients find it possible to continue their normal binocular status; they adopt a compensatory turn of the head toward the side of the involved eye, thus offsetting the lateral gaze position that the eyes must assume to continue normal binocular vision. Usually, the quantity of abduction of the involved eye is nil; in some patients the eye cannotbe abducted even to the zero straight ahead position. Yet, a few patients with Duane syndrome can abduct the involved eye many degrees; they may have severe restriction of adduction in this eye with retraction on abduction, rather than adduction. This latter variety is designated as “inverse Duane retraction syndrome” by many, or Huber type II.5 The possibility of Duane retraction syndrome having progressive features was documented by Noonan and O'Connor58 in their adult reexaminations of 21 patients with known childhood Duane syndrome. The incidence of severe retraction on adduction, the presence of enophthalmos in the primary position, and the presence of upshoots and downshoots were significantly higher in adults with Duane type I than in children. The awareness that clinical features may become more severe with increasing age is important for future counseling and possible treatment.
Until the advent of electromyography,7–10,19,20 Duane retraction syndrome was attributed to replacement of the normal contractile substance within the lateral rectus muscle with fibrous tissue. This thesis supposedly explained the abduction deficiency, the restricted adduction, the retraction, and the frequently encountered upshoot and downshoot of the adducting eye. Biopsy specimens of the lateral rectus muscle often revealed an increase in fibrous tissue, possibly resulting from a change that occurs secondary to abnormal innervation. Huber5 revealed a paradoxical innervation of the lateral rectus as the pathologic mechanism in all forms of the Duane retraction syndrome through simultaneous electromyographic recordings of the medial and lateral rectus muscles of the affected eye. In 19 of 20 patients studied by Strachan and Brown10 gross abnormalities of the firing pattern of the lateral rectus were noted, varying from paradoxical innervation to innervation with incomplete inhibition in adduction and recruitment in abduction. Thecocontraction of the horizontal rectus muscles onadduction causes the retraction of the globe.9 Thevarious degrees of the paradoxic innervation abnormality determine whether any abduction of the involved eye occurs and also determine the degree of retraction of the eye on adduction. Furthermore, secondary anatomic changes may occur in the abnormally innervated lateral and medial rectus muscles, producing the positive traction test finding and the gradual esodeviation change that occurs in the primary position in some patients.
Auditory brain-stem response testing in one series59 suggested a pontine anomaly in 64%, always ipsilateral to the affected eye, or bilateral when bilateral Duane syndrome was present. Another series,60 however, did not reveal any consistent relationship between the affected eye and the auditory brain-stem response.
Unless the patient has adopted an unsightly compensatory head posture, surgery for Duane syndrome is contraindicated, because it cannot correct the anomalous innervation. Most patients eventually learn to mask the unsightly appearance caused by their motility defect by turning the head rather than the eyes to view in lateral gaze, thus revealing their concomitant esotropia on side gaze only when making a sudden lateral gaze movement when startled. However, surgery is indicated if a compensatory head posture develops to offset a primary position horizontal tropia. Diplopia is occasionally described as a symptom of Duane syndrome.53–56,59–63 Although surgery has been recommended to avoid diplopia in lateral gaze or to reduce asthenopia,63 most authors do not report diplopia and asthenopia as a common complaint, nor as an indication for surgical intervention. MacDonald and associates62 were unable to plot a suppression scotoma with binocular perimetry and concluded that patients with Duane syndrome possess a sensory adaptation to their incomitant strabismus. Some form of suppression must be used to ignore the extra image without producing a scotoma that could be plotted on a screen. The purpose of the surgery is to restore the eyes to a parallel alignment in the primary position, making the unsightly compensatory face turn unnecessary. The prognosis is excellent, provided that the esotropic eye has been surgically aligned to orthophoria in primary position. The literature on surgical correction of Duane syndrome begins in 1955, when Nutt64 recommended a recession of the medial rectus of the involved eye in cases of uncomplicated Duane syndrome with limitation of abduction.
At surgery, the medial rectus muscle of the involved eye is usually hypertrophied and taut. After the medial rectus muscle is disinserted, the positive traction test that manifested resistance to passive abduction becomes negative. Patients receiving surgery for the compensatory head posture resulting from esodeviated retracted eye require maximal recession of the medial rectus muscle. In addition to the recession, if the medial rectus is extremely tight, a Z-tenotomy can be performed on this muscle at the same time that it is maximally recessed. The lateral rectus muscle should not be resected, because an increase in the retraction of the globe follows this procedure. If retraction of the globe is marked and there is extreme narrowing of the palpebral fissure on attempted adduction, the lateral rectus muscle can be recessed at the same time that the medial rectus muscle is weakened. Recessing the lateral rectus muscle does not augment the deficiency of abduction, but it decreases retraction of the globe that occurs on adduction. Whatever surgery is performed to eliminate the primary position esodeviation in Duane retraction syndrome also further aggravates the adduction weakness. Transposition of the muscles,65–67 which entails moving all or portions of the vertical rectus muscles temporally to assist the deficient pull power of the rectus muscle, with the intention of giving the patient abduction beyond the zero straight ahead position, should not be considered, because this causes further embarrassment of adduction. The minimal gain in abduction from such procedures as the Hummelsheim or Jensen or total vertical rectus muscle temporal transpositions is unjustified when it is weighed against the significant adduction loss. However, in one series, Foster68 performed full vertical rectus muscle transpositions for lateral rectus palsies and for 5 patients with Duane syndrome type I. A full tendon transfer of the superior and inferior rectus muscles to the lateral rectus muscle was performed, along with a 5-0 Dacron scleral fixation suture,16 mm from the limbus, placed at the borders of the lateral rectus, and incorporating 2 to 3 mm of the transposed vertical rectus muscles. The medial rectus muscle was not recessed. There was no postoperative limitation of adduction, and 80% (4 of 5) had elimination of the preoperative face turn, whereas 20% had 5 degrees of residual face turn.All patients had a -3 to -3.5 limitation of abduc-tion postoperatively, and four patients had smallhorizontal or vertical deviations. Although aKestenbaum transposition has been advocated for the treatment of Duane syndrome,69 this procedure should be reserved for torticollis and compensatory head posture secondary to nystagmus. Secondary exotropia may be more likely to occur after medial rectus recession in bilateral Duane syndrome,70 and therefore simultaneous medial and lateral rectus recessions may be the treatment of choice to avoid this complication. Treatment of consecutive exotropia after medial rectus recession for Duane syndrome type I has been reported.71 The use of a posterior fixation suture72 has been reported for use in the recession of the medial rectus of the fellow eye in Duane syndrome type I to improve concomitance into the field of action of the involved eye. Despite a large recession of the medial rectus muscle in Duane syndrome with limited abduction, there usually is no significant increase in abduction postoperatively, nor is there a problem with overcorrections, despite large medial rectus recessions, as reported by Pressman and Scott.73
Although ocular alignment in the primary position can generally be achieved by recession of the ipsilateral medial rectus in esotropic patients with Duane type I, and by recession of the ipsilateral lateral rectus muscle in Duane type II,8,17,32,73–75 this further limits horizontal ductions in the operated eye. Therefore, the field of single binocular vision may potentially not improve, and may actually worsen.73,74,76 For esotropia greater than 25 prism diopters in the primary position, Kraft75 has recommended recession of the medial rectus, both eyes, especially when recessing the ipsilateral lateral rectus for globe retraction. Kraft suggests that the recession of the normal medial rectus muscle can stabilize the postoperative results because of Hering's law of equal innervation. For exotropia greater than 25 prism diopters in the primary position, recession of the lateral rectus of both eyes is recommended, for the same reasons.
Saunders and coworkers76 also recommend surgery on the normal eye in patients who lack severe duction deficiencies preoperatively, which not only helps restore primary position alignment but also expands the field of single binocular vision by creating compensatory duction limitations. Sprunger77 described another benefit of recession of both horizontal rectus muscles, by the treatment of significant globe retraction in the primary position in Duane syndrome, and in the improvement of enophthalmos.
Surgery for the leash effect of overelevation and overdepression of the involved eye in adduction is best directed at preventing the tight lateral rectus muscle from snapping around the dorsal surface of the adducted globe (simulating overaction of the inferior oblique muscle) or from leashing around the ventral surface of the adducted eye (simulating an overacting superior oblique muscle). Tenotomizing the superior oblique muscle or recessing the inferior oblique muscle does not reduce the excursion of vertical displacement of the globe caused by the leashing of the tight lateral rectus muscle. The leashing of the lateral rectus is eliminated by placement of a permanent posterior fixation suture on the superior and inferior one third of the muscle width 14 mm posterior to the insertion.78 In addition to the placement of the posterior fixation suture, the lateral rectus also may be recessed to reduce retraction of the globe alone or combined with the restricted adduction that results from the tight muscle. Neither the placement of the posterior fixation suture nor the recession further reduces the already deficient abduction in the involved eye in Duane syndrome. Jampolsky proposed splitting the end of the lateral rectus into a Y configuration, as a means of reducing the upshoot or downshoot in adduction.79 Rogers and Bremer80 demonstrated a marked decrease in upshoot or downshoot in their series of patients who had a Y splitting of the lateral rectus, with or without recession of the ipsilateral medial rectus. In the presence of marked enophthalmos, the authors suggest a recession of the lateral rectus in addition to the Y splitting. Von Noorden81 recommended recession of both horizontal rectus muscles in the presence of both upshoot and downshoot in type III Duane syndrome. The bridle effect was reduced by placing the insertions posteriorly, in relation to the center of the globe.
When surgical intervention is required in Duane retraction syndrome, the approach should not be based purely on classification type, but on the alignment in primary position, the presence and degree of abnormal head position, the severity of retraction, and the pattern of upshoot, downshoot, or accompanying A, V, or X pattern.75