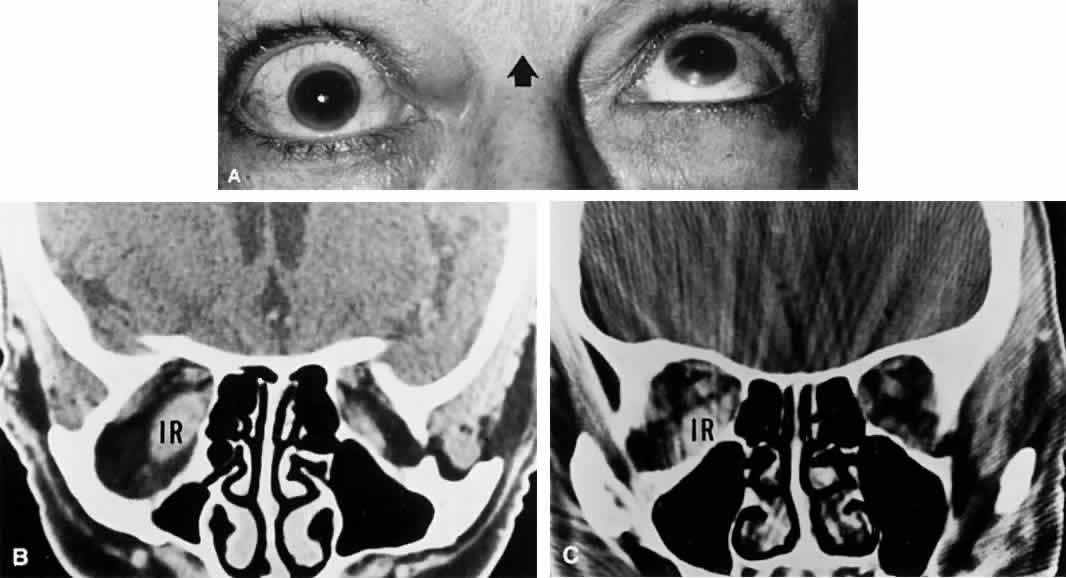
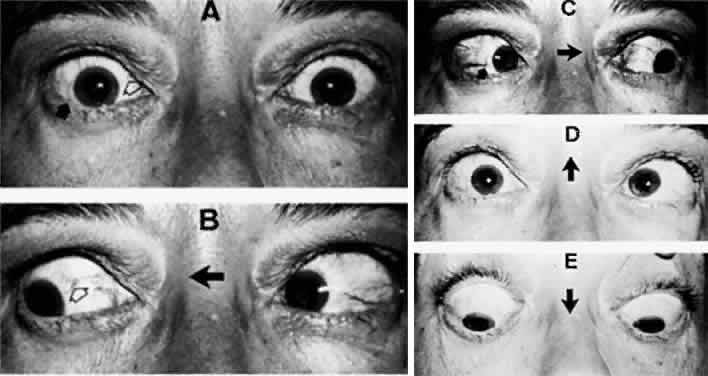
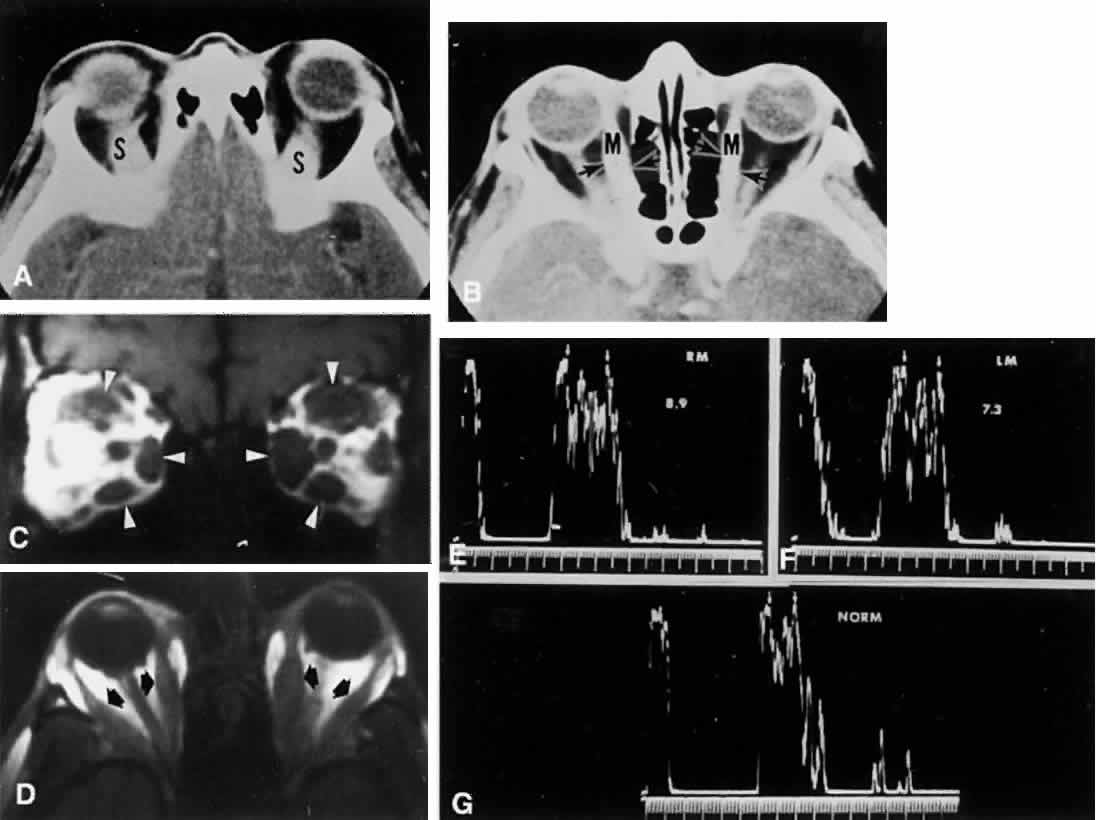
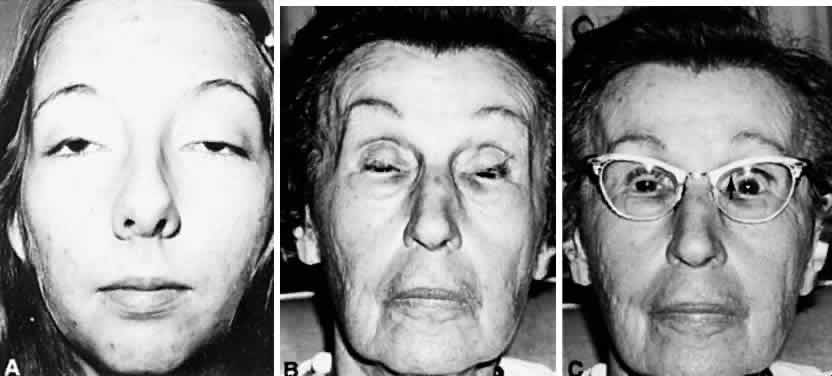
1. Warwick R: Representation of the extra-ocular muscles in the oculomotor complex. J Comp Neurol 98:449, 1953 2. Buttner-Ennever JA, Grob P, Akert K: A transsynaptic autoradiographic study of the pathways controlling the
extraocular muscles using [125l]B-IIb tetanus toxin fragment. Ann NY Acad Sci 374:157, 1981 3. Porter JD, Guthrie BL, Sparks DL: Innervation of monkey extraocular muscles: localization of the sensory
and motor neurons by retrograde transport of horseradish peroxidase. J Comp Neurol 218:208, 1983 4. Burde RM: The visceral nuclei of the oculomotor complex. Trans Am Ophthalmol Soc 81:533, 1983 5. Highstein SM, Baker R: Excitatory termination of abducens internuclear neurons on medial rectus
motoneurons: relationship to syndrome of internuclear ophthalmoplegia. J Neurophysiol 41:1647, 1978 6. Destombes J, Horcholle-Bossavit G, Rouviere A: Données récentes concernant le noyau oculomoteur externe: centre
du mouvement oculaire horizontal. J Fr Ophthalmol 6:605, 1983 7. Henn V, Lang W, Hepp K, Reisine H: Experimental gaze palsies in monkeys and their relation to human pathology. Brain 107:619, 1984 8. Crane TB, Yee RD, Baloh RW, Hepler RS: Analysis of characteristic eye movement abnormalities in internuclear ophthalmoplegia. Arch Ophthalmol 101:206, 1983 9. Gonyea EF: Bilateral internuclear ophthalmoplegia: association with occlusive cerebrovascular
disease. Arch Neurol 31:168, 1974 10. Klintworth GK: The neuro-ophthalmic manifestations of transtentorial herniation. In
Smith JL (ed): Neuro-Ophthalmology: Symposium of the University
of Miami and the Bascom Palmer Eye Institute, Vol 4, pp 113–131. St. Louis, CV
Mosby, 1972 11. Ishikawa H, Inagaki M, Kitano S: The oculomotor nerve in the cavernous sinus and orbit. Orbit 5:91, 1986 12. Kerr FW: The pupil: functional anatomy and clinical correlation. In Smith
JL (ed): Neuro-Ophthalmology: Symposium of the University of Miami
and the Bascom Palmer Eye Institute, Vol 4, pp 49–80. St. Louis, CV
Mosby, 1968 13. Parkinson D, Johnston J, Chaudhuri A: Sympathetic connections of the fifth and sixth cranial nerves. Anat Record 191:221, 1978 14. Milisavljevi M, Marinkovi S, Loli-Dragani V, Kovacevi M: Oculomotor, trochlear, and abducens nerves penetrated by cerebral vessels: microanatomy
and possible clinical significance. Arch Neurol 43:58, 1986 M, Marinkovi S, Loli-Dragani V, Kovacevi M: Oculomotor, trochlear, and abducens nerves penetrated by cerebral vessels: microanatomy
and possible clinical significance. Arch Neurol 43:58, 1986 15. Lapresle J, Lasjaunias P: Cranial nerve ischaemic arterial syndromes: a review. Brain 109:207, 1986 16. Horton JC, Fishman RA: Neurovisual findings in the syndrome of spontaneous intracranial hypotension
from dural cerebrospinal fluid leak. Ophthalmology 101:244, 1994 17. Perlman EM, Barry D: Bilateral sixth-nerve palsy after water-soluble contrast myelography. Arch Ophthalmol 102:1968, 1984 18. Nabors MW, McCrory ME, Fischer BA et al: Delayed abducens nerve palsies associated with cervical spine fractures. Neurology 37:1565, 1987 19. Barr HWK, Blackwood W, Meadows SP: Intracavernous carotid aneurysms: a clinical-pathological report. Brain 94:607, 1971 20. O'Connor PS, Glaser JS: Intracavernous aneurysms and isolated sixth nerve
palsy. In Smith JL (ed): Neuro-Ophthalmology Focus, pp 155–159. New
York, Masson, 1982 21. Newton TH, Hoyt WF: Dural arteriovenous shunts in the region of the cavernous sinus. Neuroradiology 1:71, 1970 22. Gutman I, Levartovski S, Goldhammer Y et al: Sixth nerve palsy and unilateral Horner's syndrome. Ophthalmology 93:913, 1986 23. Keane JR: Bilateral sixth nerve palsy. Arch Neurol 33: 681, 1976 24. Keane JR: Eye movement abnormalities in systemic lupus erythematosus. Arch Neurol 52:1145, 1995 25. Savino PJ, Hiliker JK, Casell GH, Schatz NJ: Chronic sixth nerve palsies: are they really harbingers of serious disease? Arch Ophthalmol 100:1442, 1982 26. Currie J, Lubin JH, Lessell S: Chronic isolated abducens paresis from tumors at the base of the brain. Arch Neurol 40:226, 1983 27. Volpe NJ, Lessell S: Remitting sixth nerve palsy in skull base tumors. Arch Ophthalmol 3:1391, 1993 28. Reisner SH, Perlman M, Ben-Tovim N et al: Transient lateral rectus muscle paresis in the newborn infant. J Pediatr 78:461, 1971 29. Knox DL, Clark DB, Schuster FF: Benign VI nerve palsies in children. Pediatrics 40:560, 1967 30. Nemet P, Ehrlich D, Lazar M: Benign abducens palsy in varicella. Am J Ophthalmol 78:859, 1984 31. Werner DB, Savino PJ, Schatz NJ: Benign recurrent sixth nerve palsies in childhood: secondary to immunization
or viral illness. Arch Ophthalmol 101:607, 1983 32. Straussberg R, Cohen AH, Amir J, Versano I: Benign abducens palsy associated with EBV infection. J Pediatr Ophthalmol Strabismus 30:60, 1993 33. Bixenman WW, von Noorden GK: Benign recurrent VI nerve palsy in childhood. J Pediatr Ophthalmol Strabismus 18:29, 1981 34. Reinecke RD, Thompson WE: Childhood recurrent idiopathic paralysis of the lateral rectus. Ann Ophthalmol 13:1037, 1981 35. Boger WP III, Puliofito CA, Magoon EH et al: Recurrent isolated sixth nerve palsy in children. Ann Ophthalmol 16:237, 1984 36. Sullivan SC: Benign recurrent isolated VI nerve palsy of childhood. Clin Pediatr 24:160, 1985 37. Peatfield RC: Recurrent VI palsy in cluster headache. Headache 25:225, 1985 38. Robertson DM, Hines JD, Rucker CW: Acquired sixth-nerve paresis in children. Arch Ophthalmol 83:574, 1970 39. Harley RD: Paralytic strabismus in children: etiologic incidence and management of
the third, fourth and sixth nerve palsies. Ophthalmology 87:24, 1980 40. Moster ML, Savino PJ, Sergott RC et al: Isolated sixth-nerve palsies in younger adults. Arch Ophthalmol 102: 1328, 1984 41. Shrader EC, Schlezinger NS: Neuro-ophthalmologic evaluation of abducens nerve paralysis. Arch Ophthalmol 63: 84, 1960 42. Rucker CW: The causes of paralysis of the third, fourth and sixth cranial nerves. Am J Ophthalmol 61:1294, 1966 43. Richards BW, Jones FR, Younge BR: Causes and prognosis in 4,278 cases of paralysis of the oculomotor, trochlear, and
abducens cranial nerves. Am J Ophthalmol 113:489, 1992 44. Rose JW, Digre KB, Lynch SG, Marnsberger RH: Acute VIth cranial nerve dysfunction in multiple sclerosis. J Clin Neuro Ophthalmol 12:17, 1992 45. Kirkham TH, Bird AC, Sanders MD: Divergence paralysis with raised intracranial pressure: an electro-oculographic
study. Br J Ophthalmol 56:776, 1972 46. Mays LE: Neural control of vergence eye movements: convergence and divergence neurons
in the midbrain. J Neurophysiol 51:1091, 1984 47. Krohel GB, Tobin DR, Hartnett ME, Barrows NA: Divergence paralysis. Am J Ophthalmol 94:506, 1982 48. Stern RM, Tomsak RL: Magnetic resonance images in a case of “divergence paralysis.” Surv Ophthalmol 30:397, 1986 49. Lewis AR, Kline LB, Sharpe JA: Acquired esotropia due to Arnold-Chiari I malformation. J Neuro Ophthalmol 16:49, 1996 50. Moster ML, Bosley TM, Slavin ML, Rubin SE: Thyroid ophthalmopathy presenting as superior oblique paresis. J Clin Neuro Ophthalmol 12:94, 1992 51. Jacobson DM: Superior oblique palsy manifested during pregnancy. Ophthalmology 98:1874, 1991 52. Astle WF, Rosenblum AL: Familial congenital fourth cranial nerve palsy. Arch Ophthalmol 103:552, 1985 53. Aleksic S, Fudzilovich G, Choy A et al: Congenital ophthalmoplegia in oculoauriculovertebral dysplasia-hemifacial
microsomia (Goldenhar-Gorlin syndrome): a clinicopathologic study and
review of the literature. Neurology 26:638, 1976 54. Cowan WM, Fawcett JW, O'Leary DDM et al: Regressive events in neurogenesis. Science 225:1258, 1984 55. Pinchoff BS, Sandall G: Congenital absence of the superior oblique tendon in craniofacial dysostosis. Ophthalmic Surg 16:375, 1985 56. Helveston EM, Krach D, Plager DA, Ellis FD: A new classification of superior oblique palsy based on congenital variations
in the tendon. Ophthalmology 99:1609, 1992 57. Berlit P: Isolated and combined pareses of cranial nerves III, IV and VI: a retrospective
study of 412 patients. J Neurol Sci 103:10, 1991 58. Keane JR: Fourth nerve palsy: historical review and study of 215 patients. Neurology 43:2439, 1993 59. Grimson BS, Glaser JS: Isolated trochlear nerve palsies in herpes zoster ophthalmicus. Arch Ophthalmol 96:1233, 1978 60. Agostinis C, Caverni L, Moschini L et al: Paralysis of fourth cranial nerve due to superior cerebellar artery aneurysm. Neurology 42:457, 1992 61. Botelho PJ, Giangiacomo JG: Autosomal-dominant inheritance of congenital superior oblique palsy. Ophthalmology 103:1508, 1996 62. Lindenberg R: Significance of the tentorium in head injuries from blunt forces. Clin Neurosurg 12:129, 1966 63. Khawam E, Scott AB, Jampolsky A: Acquired superior oblique palsy. Arch Ophthalmol 77:761, 1967 64. Sydnor CF, Seaber JH, Buckley EG: Traumatic superior oblique palsies. Ophthalmology 89:134, 1982 65. Lee J, Flynn JT: Bilateral superior oblique palsies. Br J Ophthalmol 69:508, 1985 66. Trobe JD: Cyclodeviation in acquired vertical strabismus. Arch Ophthalmol 102:717, 1984 67. Keane JR: Vertical diplopia. Semin Neurol 6:147, 1986 68. Wallace DK, von Noorden GK: Clinical characteristics and surgical management of congenital absence
of the superior oblique tendon. Am J Ophthalmol 118:63, 1994 69. Kushner BJ: ‘V’ esotropia and excyclotropia after surgery for bilateral
fourth nerve palsy. Arch Ophthalmol 110: 1419, 1992 70. Gregersen E, Rindziunski E: Brown's syndrome: a longitudinal long-term study of spontaneous course. Acta Ophthalmol 71:371, 1993 71. Mombaerts I, Koornneef L et al: Superior oblique luxation and trochlear luxation as new concepts in superior
oblique muscle weakening surgery. Am J Ophthalmol 120:83, 1995 72. Clarke WN, Noel LP, Agapitos PJ: Traumatic Brown's syndrome. Am Orthoptic J 37:100, 1987 73. Booth-Mason S, Kyle GM, Rossor M, Bradbury P: Acquired Brown's syndrome: an unusual cause. Br J Ophthalmol 69:791, 1985 74. Hickling P, Beck M: Brown's syndrome: an unusual ocular complication of rheumatoid arthritis. Ann Rheum Dis 50:66, 1991 75. Alonso-Valdivielso JL, Alvarez Lario B et al: Acquired Brown's syndrome in a patient with systemic lupus erythematosus. Ann Rheum Dis 52:63, 1993 76. Christiansen SP, Thomas AH: Postpartum Brown's syndrome. Arch Ophthalmol 112:23, 1994 77. Brazis PW, Miller NR, Henderer JD, Lee AG: The natural history and results of treatment of superior oblique myokymia. Arch Ophthalmol 112:1063, 1994 78. Martin TJ, Corbett JJ, Babikian PV et al: Bilateral ptosis due to mesencephalic lesions with relative preservation
of ocular motility. J Neuro Ophthalmol 16:258, 1996 79. Keane JR, Zaias B, Itabashi HH: Levator-sparing oculomotor nerve palsy caused by a solitary midbrain metastases. Arch Neurol 14:210, 1984 80. Bryan JS, Hamed LM: Levator-sparing nuclear oculomotor palsy: clinical and magnetic resonance
imaging findings. J Clin Neuro Ophthalmol 12:26, 1992 81. Van Dalen JTW, Van Mourek-Noordenbos AM: Isolated inferior rectus paresis: a report of six cases. Neuro Ophthalmology 4:89, 1984 82. Masucci EF: Bilateral ophthalmoplegia in basilar-vertebral artery disease. Brain 88:97, 1965 83. Acers TE: Oculomotor-corpus callosum dysplasia. Trans Am Ophthalmol Soc 80:172, 1982 84. Ksiazek SM, Slamovits TL, Rosen CE et al: Fascicular arrangement in partial oculomotor paresis. J Ophthalmol 118:97, 1994 85. Gauntt CD, Kashii S, Nagata I: Monocular elevation paresis caused by an oculomotor fascicular impairment. J Neuro Ophthalmol 15:11, 1995 86. Castro O, Johnson LN, Mamourian AC: Isolated inferior oblique paresis from brain-stem infarction: perspective
on oculomotor fascicular organization in the ventral midbrain tegmentum. Arch Neurol 47:235, 1990 87. Trobe JD, Glaser JS, Quencer RC: Isolated oculomotor paralysis. Arch Ophthalmol 96:1236, 1978 88. Hyland HH, Barnett HJM: The pathogenesis of cranial nerve palsies associated with intracranial
aneurysms. Proc R Soc Med 47:141, 1956 89. Ross JS, Masaryk TJ, Modic MT et al: Intracranial aneurysms: evaluation by MR angiography. AJNR 11:449, 1990 90. Capó H, Warren F, Kupersmith MJ: Evolution of oculomotor nerve palsies. J Clin Neuro Ophthalmol 12:21, 1992 91. Guy J, Savino PJ, Schatz NJ et al: Superior division paresis of the oculomotor nerve. Ophthalmology 92:777, 1985 92. Heinze J: Cranial nerve avulsion and other neural injuries in road accidents. Med J Aust 2:1246, 1969 93. Eyster EF, Hoyt WF, Wilson CB: Oculomotor palsy from minor head trauma: an initial sign of basal intracranial
tumor. JAMA 220:1083, 1972 94. Neetens A: Extraocular muscle palsy from minor head trauma: initial sign of intracranial
tumor. Neuro Ophthalmology 3:43, 1983 95. Walter KA, Newman NJ, Lessell S: Oculomotor palsy from minor head trauma: initial sign of intracranial aneurysm. Neurology 44:148, 1994 96. Ing EB, Sullivan TJ, Clarke MP, Buncic JR: Oculomotor nerve palsies in children. J Pediatr Ophthalmol Strabismus 29:331, 1992 97. Keane JR: Bilateral ocular motor signs after tentorial herniation in 25 patients. Arch Neurol 43:806, 1986 98. Acierno MD, Trobe JD, Cornblath WT, Gebarski SS: Painful oculomotor palsy caused by posterior-draining dural carotid cavernous
fistulas. Arch Ophthalmol 113:1045, 1995 99. Schultheiss R, Kristol R, Schramm J: Complete removal of an oculomotor nerve neurinoma without permanent functional
deficits. Ger J Ophthalmol 2:228, 1993 100. Kaye-Wilson LG, Gibson R, Bell JE et al: Oculomotor nerve neurinoma. Neuro-ophthalmology 14:37, 1994 101. Jacobson DM, McCanna TD, Layde PM: Risk factors for ischemic ocular motor palsies. Arch Ophthalmol 112:961, 1994 102. Sergott RC, Glaser JS, Buerger LJ: Simultaneous, bilateral diabetic ophthalmoplegia: report of two cases and
discussion of differential diagnosis. Ophthalmology 91:18, 1984 103. Eshbaugh CG, Siatkowski RM, Smith JL, Kline LB: Simultaneous multiple cranial neuropathies in diabetes mellitus. J Neuro Ophthalmol 15:219, 1995 104. Weber RB, Daroff RB, Mackey EA: Pathology of oculomotor nerve palsy in diabetics. Neurology 20:835, 1970 105. Asbury AK, Aldredge H, Hershberg R et al: Oculomotor palsy in diabetes mellitus: a clinicopathological study. Brain 93:555, 1970 106. Breen LA, Hopf HC, Farris BK et al: Pupil-sparing oculomotor nerve palsy due to midbrain infarction. Arch Neurol 48:105, 1991 107. Pratt DV, Orengo-Nania S, Horowitz BL, Oram O: Magnetic resonance imaging findings in a patient with nuclear oculomotor
palsy. Arch Ophthalmol 113:141, 1995 108. Position Statement: Tests of glycemia in diabetes. American Diabetes Association: Clinical
practice recommendations. Diabetes Care, ADA, 1:S18, 1997 109. Sibony PA, Lessell S, Gittenger JW Jr: Acquired oculomotor synkinesis. Surv Ophthalmol 28:382, 1984 110. Sibony PA, Evinger C, Lessell S: Retrograde horseradish peroxidase transport after oculomotor nerve injury. Invest Ophthalmol Vis Sci 27:975, 1986 111. Lepore FE, Glaser JS: Misdirected revisited: a critical appraisal of acquired oculomotor nerve
synkinesis. Arch Ophthalmol 98:2206, 1980 112. Wartenberg R: Associated movements in the oculomotor and facial muscles. Arch Neurol Psychiatry 55:439, 1946 113. Sibony PA, Lessell S: Transient oculomotor synkinesis in temporal arteritis. Arch Neurol 41:87, 1984 114. Schatz NJ, Savino PJ, Corbett JC: Primary aberrant oculomotor regeneration: A sign of intracavernous meningioma. Arch Neurol 34:29, 1977 115. Trobe JD, Glaser JS, Post JD: Meningiomas and aneurysms of the cavernous sinus: neuro-ophthalmologic
features. Arch Ophthalmol 96:457, 1978 116. Cox TA, Wurster JB, Godfrey WA: Primary aberrant oculomotor regeneration due to intracranial aneurysm. Arch Neurol 36:570, 1979 117. Boghen D, Chartrand JP, LaFlamme P et al: Primary aberrant third nerve regeneration. Ann Neurol 6:415, 1979 118. Hepler RS, Cantu RC: Aneurysms and third nerve palsies: ocular status of survivors. Arch Ophthalmol 77:604, 1967 119. Czarnecki JSC, Thompson HS: The iris sphincter in aberrant regeneration of the third nerve. Arch Ophthalmol 96:1606, 1978 120. Miller NR: Solitary oculomotor nerve palsy in childhood. Am J Ophthalmol 83:106, 1977 121. Victor DI: The diagnosis of congenital unilateral third nerve palsy. Brain 99:711, 1976 122. Balkan R, Hoyt CS: Associated neurologic abnormalities in congenital third nerve palsies. Am J Ophthalmol 97:315, 1984 123. Varma R, Miller NR: Primary oculomotor nerve synkinesis caused by an extracavernous intradural
aneurysm. Am J Ophthalmol 118:83, 1994 124. O'Day J, Billson F, King J: Ophthalmoplegic migraine and aberrant regeneration of the oculomotor nerve. Br J Ophthalmol 64:534, 1980 125. Packer AJ, Bienfang DC: Aberrant regeneration involving the oculomotor and abducens nerves. Ophthalmologica 189:80, 1984 126. Krzizok T, Gräf M: Acquired trigemino-abducens synkinesia. Proposed mechanisms and consequences
for eye muscle surgery. Klin Monatsbl Augenheilkd 205:33, 1994 127. Pallini R, Fernandez E et al: Experimental repair of the oculomotor nerve: the anatomical paradigms of
functional regeneration. J Neurosurg 77:768, 1992 128. Marsh RJ, Dulley B, Kelly V: External ocular motor palsies in ophthalmic zoster: a review. Br J Ophthalmol 61:677, 1977 129. Wexler I: Sequence of demyelination-remyelination in Guillain-Barré disease. J Neurol Neurosurg Psychiatry 46:168, 1983 130. Fisher CM: An unusual variant of acute idiopathic polyneuritis (syndrome of ophthalmoplegia, ataxia
and areflexia). N Engl J Med 255:57, 1956 131. Asbury AK, Arnason BG, Adams R: The inflammatory lesion in idiopathic polyneuritis. Medicine 48:173, 1969 132. Grunnet ML, Lubow M: Ascending polyneuritis and ophthalmoplegia. Am J Ophthalmol 74:1155, 1972 133. Berlit P, Rakicky J: The Miller Fisher syndrome. J Clin Neuro Ophthalmol 12:59, 1992 134. Derakhshan I, Lotfi J, Kaufman B: Ophthalmoplegia, ataxia and hyporeflexia (Fisher's syndrome) with
a midbrain lesion demonstrated by CT scanning. Eur Neurol 18:361, 1979 135. Ropper AH: The Guillain-Barré syndrome. N Engl J Med 326:1130, 1992 136. Chalmers AC, Miller RG: Chronic inflammatory polyradiculoneuropathy with ophthalmoplegia. J Clin Neuro Ophthalmol 6:166, 1986 137. Yuki N: Acute paresis of extraocular muscles associated with IgG anti-GQ1b antibody. Ann Neurol 39:668, 1995 138. Friedman AP, Harter DH, Merritt HH: Ophthalmoplegic migraine. Arch Neurol 7:320, 1962 139. Vijayan N: Ophthalmoplegic migraine: ischemic or compressive neuropathy? Headache 20:300, 1980 140. Stigmal EW, Ward TN, Harris RD: MRI findings in a case of ophthalmoplegic migraine. Headache 33:234, 1993 141. Straub A, Bondman O, Butter U, Schmidt H: A contrast enhances lesion of the III nerve on MR of a patient with ophthalmoplegic
migraine as evidence for a Tolosa-Hunt syndrome. Headache 33:446, 1993 142. Østergaard JR, Moller HU, Christensen T: Recurrent ophthalmoplegia in childhood: diagnostic and etiologic considerations. Cephalgia 16:276, 1996 143. Imes RK, Monteiro MLR, Hoyt WF: Ophthalmoplegic migraine with proximal posterior cerebral artery vascular
anomaly. J Clin Neuro Ophthalmol 4:221, 1984 144. Kandt RS, Goldstein GW: Steroid-responsive ophthalmoplegia in a child: diagnostic considerations. Arch Neurol 42:589, 1985 145. Osuntokun O, Osuntokun BO: Ophthalmoplegic migraine and hemoglobinopathy in Nigerians. Am J Ophthalmol 74:451, 1972 146. Sharf B, Hyams S: Oculomotor palsy following varicella. J Pediatr Ophthalmol 9:245, 1972 147. Chan CC, Sogg RL, Steinman L: Isolated oculomotor palsy after measles immunization. Am J Ophthalmol 89:446, 1980 148. Loewenfeld IE, Thompson HS: Oculomotor paresis with cyclic spasms: a critical review of the literature
and a new case. Surv Ophthalmol 20:81, 1975 149. Bourgon P, LaFlamme P: Cited by Thompson HS: 13th Pupil Colloquium. Am J Ophthalmol 96:100, 1983 150. Bateman DE, Saunders M: Cyclic oculomotor palsy: description of a case and hypothesis of the mechanism. J Neurol Neurosurg Psychiatry 46:451, 1983 151. Dudim A, Othman A: Acute orbital swelling: evaluation and protocol. Pediatr Emerg Care 12:16, 1996 152. Bergin DJ, Wright JE: Orbital cellulitis. Br J Ophthalmol 70:174, 1986 153. Del Valle ZA, Suarez RA, Encinas PM et al: Mucormycosis of the sphenoid sinus in an otherwise health patient: case
report and literature review. J Laryngol Otol 110:471, 1996 154. Yohai RA, Bullock JD, Aziz AA, Mirkert RJ: Survival factors in rhino-orbital cerebral mucormycosis. Surv Ophthalmol 39:3, 1994 155. Kennerdell JS, Dresner SC: The nonspecific orbital inflammatory syndromes. Surv Ophthalmol 29:93, 1984 156. Grimson BS, Simmons KB: Orbital inflammation, myositis and systemic lupus erythematosus. Arch Ophthalmol 101:36, 1983 157. Schonder AA, Clift RC, Brophy JW, Dane LW: Bilateral recurrent orbital inflammation associated with retroperitoneal
fibrosclerosis. Br J Ophthalmol 69:783, 1985 158. Rootman J, Nugent R: The classification and management of acute orbital pseudotumors. Ophthalmology 89:1040, 1982 159. Siatkowski RM, Capó H, Byrne SF et al: Clinical and echographic findings in idiopathic orbital myositis. Am J Ophthalmol 118:343, 1994 160. Koorey DJ: Cranial arteritis: a twenty-year review of cases. Aust NZ J Med 14:143, 1984 161. Baricks ME, Traviesa DB, Glaser JS, Levy IS: Ophthalmoplegia in cranial arteritis. Brain 100:209, 1977 162. Cline RA, Rootman J: Enophthalmos: a clinical review. Ophthalmology 91:229, 1984 163. Keane JR: Cavernous sinus syndrome: analysis of 151 cases. Arch Neurol 53:967, 1996 164. Tolosa EJ: Periarteritic lesions of the carotid siphon with clinical features of carotid
infraclinoid aneurysms. J Neurol Neurosurg Psychiatry 17:300, 1954 165. Hunt WE, Meagher JN, Lefever HE, Zeman W: Painful ophthalmoplegia: its relation to indolent inflammation of the cavernous
sinus. Neurology 11:56, 1961 166. Thomas JE, Yoss RE: The parasellar syndrome: problems in determining etiology. Mayo Clin Proc 45:617, 1970 167. Spector RH, Fiandaca MS: The “sinister” Tolosa-Hunt syndrome. Neurology 36:198, 1986 168. Kline LB: The Tolosa-Hunt syndrome. Surv Ophthalmol 27:79, 1982 169. Campbell RJ, Okazaki H: Painful ophthalmoplegia (Tolosa-Hunt variant): autopsy findings in a patient
with necrotizing intracavernous carotid vasculitis and inflammatory
disease of the orbit. Mayo Clin Proc 62:520, 1987 170. Aktan S, Aykut C, Erzen C: Computed tomography and magnetic resonance imaging in three patients with
Tolosa-Hunt syndrome. Eur Neurol 33:393, 1993 171. Takahashi Y, Abe T, Kojima K et al: Tolosa-Hunt syndrome with atypical intrasellar and juxtasellar lesions. Two
case reports. Kurume Med J 43:165, 1996 172. Drevelengas A, Kalaitzoglou I, Tsolaki M: Tolosa-Hunt syndrome with sellar erosion: case report. Neuroradiology 35:451, 1993 173. Hama S, Arita K, Kurisu K et al: Parasellar chronic inflammatory disease presenting as Tolosa-Hunt syndrome, hypopituitarism
and diabetes insipidus: a case report. Endocr J 43:503, 1996 174. Brazis PW, Capobianco DJ et al: Low flow dural arteriovenous shunt: another cause of “sinister” Tolosa-Hunt
syndrome. Headache 34:523, 1994 175. Sanchez Pina C, Pascual-Castroviejo I et al: Burkitt's lymphoma presenting as Tolosa-Hunt syndrome. Pediatr Neurol 9:157, 1993 176. Montecucco C, Caporali R, Pacchetti C, Turla M: Is Tolosa-Hunt syndrome a limited form of Wegener's granulomatosis?: report
of two cases with anti-neutrophil cytoplasmic antibodies. Br J Rheumatol 32:640, 1993 177. Masson C, Henin D, Hauw JJ et al: Cranial pachymeningitis of unknown origin: a study of seven cases. Neurology 17:1329, 1993 178. Vailati A, Marena C et al: Hashimoto's thyroiditis in association with Tolosa-Hunt syndrome: a
case report. Thyroid 3:125, 1993 179. Bills DC, Meyer FB et al: A retrospective analysis of pituitary apoplexy. Neurosurgery 33:602, 1993 180. Vidal E, Cevallos R et al: Twelve cases of pituitary apoplexy. Arch Intern Med 152:1893, 1992 181. Weinberger LM, Adler FH, Grant FC: Primary pituitary adenoma and the syndrome of the cavernous sinus. Arch Ophthalmol 24:1197, 1940 182. Godtfredsen E, Lederman M: Diagnostic and prognostic roles of ophthalmoneurologic signs and symptoms
in malignant nasopharyngeal tumors. Am J Ophthalmol 59:1063, 1965 183. Meadows SP: Intracavernous aneurysms of the carotid artery. Arch Ophthalmol 62:566, 1959 184. Lee AG, Mawad ME, Baskin DS: Fatal subarachnoid hemorrhage from the rupture of a totally intracavernous
carotid artery aneurysm: case report. Neurosurgery 38:596, 1996 185. Keane JR, Talalla A: Post traumatic intracavernous aneurysm: epistaxis with monocular blindness
preceded by chromatopsia. Arch Ophthalmol 87:701, 1972 186. Trobe JD, Glaser JS, Post JD: Meningiomas and aneurysms of the cavernous sinus: neuro-ophthalmologic
features. Arch Ophthalmol 96:457, 1978 187. Rapport R, Murtagh FR: Ophthalmoplegia due to spontaneous thrombosis in a patient with bilateral
cavernous carotid aneurysms. J Clin Neuro Ophthalmol 1:225, 1981 188. Kupersmith MJ, Hurst R, Berenstein A et al: The benign course of cavernous carotid artery aneurysms. J Neurosurg 77:690, 1992 189. Cushing H, Eisenhardt L: Meningiomas: Their Classification, Regional Behavior, Life
History and Surgical End Results. New York, Hofner, 1962 190. Ojemann RG, Thornton AF, Harsh GR: Management of anterior cranial base and cavernous sinus neoplasms with
conservative surgery alone or in combination with fractionated photon
or stereotactic proton radiotherapy. Clin Neurosurg 42:71, 1995 191. Nugent GR, Sprinkle P, Bloor BM: Sphenoid sinus mucoceles. J Neurosurg 32:443, 1970 192. Valvassori GE, Putterman AM: Ophthalmological and radiological findings in sphenoidal mucoceles. Trans Am Acad Ophthalmol Otolaryngol 77:703, 1973 193. Casteels I, DeLoof E, Brock P et al: Sudden blindness in a child: presenting symptoms of sphenoid sinus mucocele. Br J Ophthalmol 76:502, 1992 194. El-Fiki ME, Abdel-Fattah HM, el-Deeb AK: Sphenoid sinus mucopyocele with marked intracranial extension. Surg Neurol 39:115, 1993 195. Post MJD, Mendez DR, Kline LB et al: Metastatic disease to the cavernous sinus: clinical syndrome and CT diagnosis. J Comput Assist Tomogr 9:115, 1985 196. Kline LB, Acker JD, Post JDM: Computed tomographic evaluation of the cavernous sinus. Ophthalmology 89:374, 1982 197. Moore CE, Hoyt WF, North JB: Painful ophthalmoplegia following treated squamous carcinoma of the forehead: orbital
apex involvement from centripetal spread via the supraorbital
nerve. Med J Aust 1:657, 1976 198. Catalano PJ, Sen C, Biller HF: Cranial neuropathy secondary to perineural spread of cutaneous malignancies. Am J Otol 16:772, 1995 199. ten Hove MW, Glaser JS, Schatz NJ: Occult perineural tumor infiltration of the trigeminal nerve. Diagnostic
considerations. J Neuro Ophthalmol 17:170, 1997 200. Hayat G, Ehsan T, Selhorst JB, Manepali A: Magnetic resonance evidence of perineural metastasis. J Neuroimaging 5:122, 1995 201. Olson ME, Cornice NL, Posner JB: Infiltration of the leptomeninges by systemic cancer: a clinical and pathologic
study. Arch Neurol 30:122, 1974 202. Little JR, Dale AJD, Okazaki H: Meningeal carcinomatosis: clinical manifestations. Arch Neurol 30:138, 1974 203. Wasserstrom WR, Glass JP, Posner JB: Diagnosis and treatment of leptomeningeal metastases from solid tumors: experience
with 90 patients. Cancer 49:759, 1982 204. Watanabe M, Tanaka R, Takeda N: Correlation of MRI and clinical features in meningeal carcinomatosis. Neuroradiology 35:512, 1993 205. Stern BJ, Krumholz A, Johns C et al: Sarcoidosis and its neurological manifestations. Arch Neurol 49:909, 1985 206. Oksamen V: Neurosarcoidosis: clinical presentations and course in 50 patients. Acta Neurol Scand 73:283, 1986 207. Post MJD, Chan JC, Hensley GT et al: Toxoplasma encephalitis in Haitian adults with acquired immunodeficiency
syndrome: a clinical-pathologic-CT correlation. Am J Neuroradiol 4:155, 1983 208. Berger JR, Flaster M, Schatz N et al: Cranial neuropathy heralding otherwise occult AIDS-related large cell lymphoma. J Clin Neuro Ophthalmol 13:113, 1993 209. Hamed LM, Schatz, NJ, Galetta SL: Brainstem ocular motility defects and AIDS. Am J Ophthalmol 106:437, 1988 210. Wadia NH, Wadia PN, Katrak SM, Misra VP: A study of the neurological disorder associated with acute hemorrhagic
conjunctivitis due to enterovirus 70. J Neurol Neurosurg Psychiatry 46:559, 1983 211. Katiyar BC, Misra S, Singh RB et al: Adult polio-like syndrome following enterovirus 70 conjunctivitis (natural
history of the disease). Acta Neurol Scand 67:263, 1983 212. Lesser RL: Ocular manifestations of Lyme disease. Am J Med 98:60, 1995 213. Rodriguez M, Gomez MR, Howard FM, Taylor WF: Myasthenia in children: long-term follow-up. Ann Neurol 13:504, 1983 214. Vincent A, Newsom-Davis J: Acetylcholine receptor antibody characteristics in myasthenia gravis: I. Patients
with generalized myasthenia or disease restricted to ocular
muscles. Clin Exp Immunol 49:257, 1982 215. Zimmerman CW, Eblen F: Repertoires of autoantibodies against homologous eye muscle in ocular and
generalized myasthenia gravis. Clin Invest 71:445, 1993 216. Drachman DB, Adams RN, Josifek LF, Self SG: Functional activities of autoantibodies to acetylcholine receptors and
the clinical severity of myasthenia gravis. N Engl J Med 307:769, 1982 217. Weinberg DA, Lesser RL, Vollmer TL: Ocular myasthenia: a protean disorder. Surv Ophthalmol 39:169, 1994 218. Grob D, Arsura EL, Brunner NG, Namba T: The course of myasthenia gravis, and therapies affecting outcome. Ann NY Acad Sci 505:472, 1987 219. Bever CT, Aquino AV, Penn AS et al: Prognosis of ocular myasthenia. Ann Neurol 14:526, 1983 220. Cogan DG: Myasthenia gravis: A review of the disease and a description of lid twitch
as a characteristic sign. Arch Ophthalmol 74:217, 1965 221. Dutton GN, Garson JA, Richardson RB: Pupillary fatigue in myasthenia gravis. Trans Ophthalmol Soc UK 102:510, 1982 222. Glaser JS: Myasthenic pseudo-internuclear ophthalmoplegia. Arch Ophthalmol 75:363, 1966 223. Keane JR, Hoyt WF: Myasthenic (vertical) nystagmus: verification by edrophonium tonography. JAMA 212:1209, 1970 224. Barton JJS, Jama A, Sharpe JA: Saccadic duration and intrasaccadic fatigue in myasthenic and nonmyasthenic
ocular palsies. Neurology 45:2065, 1995 225. Osher RH, Griggs RC: Orbicularis fatigue: the “peek” sign of myasthenia gravis. Arch Ophthalmol 97:677, 1979 226. Osher RH, Glaser JS: Myasthenic sustained gaze fatigue. Am J Ophthalmol 89:443, 1980 227. Retzlaff JA, Kearns TP, Howard FM Jr et al: Lancaster red-green test in evaluation of edrophonium effect in myasthenia
gravis. Am J Ophthalmol 67:13, 1969 228. Seybold ME, Daroff RB: The office Tensilon test for ocular myasthenia. Arch Neurol 43:842, 1986 229. Coll GE, Demer JL: The edrophonium-Hess screen test in the diagnosis of ocular myasthenia
gravis. Am J Ophthalmol 114:489, 1992 230. Siatkowski RM, Shah L, Feuer WJ: The effect of edrophonium chloride on muscle balance in normal subjects
and those with nonmyasthenic strabismus. J Neuro Ophthalmol 17:7, 1997 231. Miller NR, Morris JE, Maguire M: Combined use of neostigmine and ocular motility measurements in the diagnosis
of myasthenia gravis. Arch Ophthalmol 100:761, 1982 232. Oey PL, Wieneke GM, Hoogenraad TU, van Hofgelen AC: Ocular myasthenia gravis: the diagnostic yield of repetitive nerve stimulating
and stimulated single fiber EMG of orbicularis oculi muscle and
infrared reflection oculography. Muscle Nerve 16:142, 1993 233. Odel J, Winterkorn J, Behrens M: The sleep test for myasthenia gravis: a safe alternative. J Clin Ophthalmol 11:288, 1991 234. Janssen RS, Kaye AD, Lisak RP et al: Radiologic evaluation of the mediastinum in myasthenia gravis. Neurology 33:534, 1983 235. Sanders DB, Howard JF: Disorders of neuromuscular transmission. In Bradley
WG, Daroff RB, Fenichel GM, Marsden CD (eds): Neurology in Clinical
Practice, Chap 83, p 1983. Boston, Butterworth-Heinemann, 1996 236. Kupersmith MJ, Moster M, Bhuiyan S et al: Beneficial effects of corticosteroids on ocular myasthenia gravis. Arch Neurol 53:802, 1996 237. McEvoy KM: Diagnosis and treatment of Lambert-Eaton myasthenic syndrome. Neurol Clin 12:387, 1994 238. Molenaar PC, Newson-Davis J, Polak RL, Vincent A: Eaton-Lambert syndrome: acetylcholine and choline acetyltransferase in
skeletal muscle. Neurology 32:1062, 1982 239. Tsuchiya N, Sato M et al: Lambert-Eaton myasthenic syndrome associated with Sjögren's syndrome
and discoid lupus erythematosus. Scand J Rheumatol 22:302, 1993 240. Cruciger MP, Brown B, Denys EH et al: Clinical and subclinical oculomotor findings in the Eaton-Lambert syndrome. J Clin Neuro Ophthalmol 3:19, 1983 241. Dell'Osso LF, Ayyar DR, Daroff RB, Abel LA: Edrophonium test in Eaton-Lambert syndrome: quantitative oculography. Neurology 33:1157, 1983 242. Breen LA, Gutmann L, Birck JF, Riggs JR: Paradoxical lid elevation with sustained upgaze: a sign of Lambert-Eaton
syndrome. Muscle Nerve 14:863, 1991 243. Grisold W, Drlicek M, Liszka-Setinek U, Wondrusch E: Anti-tumour therapy in paraneoplastic neurological disease. Clin Neurol Neurosurg 97:106, 1995 244. Katz LJ, Lesser RL, Merikangas JR, Silverman JP: Ocular myasthenia gravis after D-penicillamine administration. Br J Ophthalmol 73:1015, 1989 245. McQuillen MP, Cantor HE, O'Rourke JR: Myasthenic syndromes associated with antibiotics. Arch Neurol 18:402, 1968 246. Albert DM, Wong VG, Henderson ES: Ocular complications of vincristine therapy. Arch Ophthalmol 78:709, 1967 247. Blanton CL, Sawyer RA: Myasthenia gravis by another name: an elusive impostor [clinical conference]. Surv Ophthalmol 38:219, 1993 248. Hedges TR III, Jones A, Stark L, Hoyt WF: Botulin ophthalmoplegia: clinical and oculographic observations. Arch Ophthalmol 101:211, 1983 249. Schubart P, Kasperski S, Schroder P: Ocular involvement in botulism. Klin Monatsbl Augenheilkd 187:142, 1985 250. Hudson HL, Levin L, Feldon SE: Graves exophthalmos unrelated to extraocular muscle enlargement. Ophthalmology 98:1495, 1991 251. Hermann JS: Paretic thyroid myopathy. Ophthalmology 89:473, 1982 252. Gamblin FT, Galentine PG, Eil C: Intraocular pressure and thyroid disease. In
Gorman CA, Waller RR, Dyer JA (eds): The Eye and Orbit in Thyroid
Disease, pp 155–166. New York, Raven Press, 1984 253. Byrne SF, Glaser JS: Orbital tissue differentiation with standardized echography. Ophthalmology 90:1071, 1983 254. Kung AW, Yau CC, Cheng A: The incidence of ophthalmopathy after radioiodine therapy for Graves' disease: prognostic
factors and the role of methimazole. J Clin Endocrinol Metabol 79:542, 1994 255. Gorman CA, Waller RR, Dyer JA: The Eye and Orbit in Thyroid Eye Disease. New
York, Raven Press, 1984 256. Raikow RB, Tyutyunikov A et al: Correlation of serum immunoglobulin E elevations with clinical states of
dysthyroid orbitopathy. Ophthalmology 99:361, 1992 257. Khuteta A, Mishra YC et al: Mesenchymal chondrosarcoma of orbit with intracranial extension (a rare
case). Ind J Ophthalmol 40:92, 1992 258. Prummel MF, Mourits MP et al: Randomized double-blind trial of prednisone versus radiotherapy in Graves' ophthalmopathy. Lancet 342:949, 1993 259. Wilson WB, Prochoda M: Radiotherapy for thyroid orbitopathy. Arch Ophthalmol 113:1420, 1995 260. Shine CSK, Kendler DL et al: Radiotherapy in the management of thyroid orbitopathy. Arch Ophthalmol 111:819, 1993 261. Lueder GT, Scott WE, Kutschke PJ, Keech RV: Long-term results of adjustable suture surgery for strabismus secondary
to thyroid ophthalmopathy. Ophthalmology 99:993, 1992 262. Kushner BJ: A surgical procedure to minimize lower-eyelid retraction with inferior
rectus recession. Arch Ophthalmol 110:1011, 1992 263. Hudson HL, Feldon SE: Late overcorrection of hypotropia in Graves ophthalmopathy. Ophthalmology 99:356, 1992 264. Johns DR: Mitochondrial DNA and disease. N Engl J Med 333:638, 1995 265. Mitsumoto H, Aprille JR, Wray SH et al: Chronic progressive external ophthalmoplegia (CPEO): clinical, morphologic
and biochemical studies. Neurology 33:452, 1983 266. Hyman BN, Patten BM, Dodson RF: Mitochondrial abnormalities in progressive external ophthalmoplegia. Am J Ophthalmol 83:362, 1987 267. Seibel P, Lauber J et al: Chronic progressive external ophthalmoplegia is associated with a novel
mutation in the mitochondrial tRNA (Asn) gene. Biochem Biophys Res Commun 204:482, 1994 268. Hattori Y, Goto Y et al: Point mutations in mitochondrial tRNA genes: sequence analysis of chronic
progressive external ophthalmoplegia (CPEO). J Neurol Sci 125:50, 1994 269. Laforet P, Lombes A, Eymard B et al: Chronic progressive external ophthalmoplegia with ragged-red fibers: clinical, morphological
and genetic investigations in 43 patients. Neuromuscul Disord 5:399, 1995 270. Mullie MA, Harding AE, Petty RKH et al: Retinal manifestations of mitochondrial myopathy: a study of 22 cases. Arch Ophthalmol 103:1825, 1985 271. Rowland LP: Molecular genetics, pseudogenetics, and clinical neurology. Neurology 33:1179, 1983 272. Ogasahara S, Yorifuji S, Nishikawa Y et al: Improvement of abnormal pyruvate metabolism and cardiac conduction defect
with coenzyme Q10 in Kearns-Sayre syndrome. Neurology 35:372, 1985 273. Coquet M, Vallat JM, Vital C et al: Nuclear inclusions in oculopharyngeal dystrophy: an ultrastructural study
of six cases. J Neurol Sci 60:151, 1983 274. Satoyoshi E, Kinoshita M: Oculopharyngodistal myopathy: report of four families. Arch Neurol 34:89, 1977 275. Ionescu V, Thompson SH, Ionescu R et al: Inherited ophthalmoplegia with intestinal pseudo-obstruction. J Neurol Sci 59:215, 1983 276. Hsu CC, Chuang YH, Tsai JL et al: CPEO and carnitine deficiency overlapping in MELAS syndrome. Acta Neurol Scand 92:252, 1995 277. Flynn JT, Bachynski BN, Rodrigues MM et al: Hyperglycemic acidotic coma and death in Kearns-Sayre syndrome. Trans Am Ophthalmol Soc 83:131, 1985 278. Coulter DL, Allen RJ: Abrupt neurological deterioration in children with Kearns-Sayre syndrome. Arch Neurol 38:247, 1981 279. Litvan I, Agid Y, Caine D et al: Clinical research criteria for the diagnosis of progressive supranuclear
palsy (Steele-Richardson-Olszewski syndrome). Neurology 47:1, 1996 280. Jackson JA, Jankovic J, Ford J: Progressive supranuclear palsy: clinical features and response to treatment
in 16 patients. Ann Neurol 13:273, 1983 281. Dehaene J: Apraxia of lid opening in progressive supranuclear palsy. Ann Neurol 15:115, 1984 282. Cole DG, Growdon JH: Therapy for progressive supranuclear palsy: past and future. J Neural Transm 42(suppl): 283, 1994 283. DiMonte DA, Harati Y et al: Muscle mitochondrial ATP production in progressive supranuclear palsy. J Neurochem 62:1631, 1994 284. Savoiardo M, Girotti F, Strada L, Ciceri E: Magnetic resonance imaging in progressive supranuclear palsy and other
parkinsonian disorders. J Neural Transm 42(suppl):93, 1994 285. Calabrese VP, Hadfield MG: Parkinsonism and extraocular motor abnormalities with unusual neuropathological
findings. Mov Disord 6:257, 1991 286. Knox DL, Green WR, Troncoso JC et al: Cerebral ocular Whipple's disease: a 62-year odyssey from death to
diagnosis. Neurology 45:617, 1995 287. Rabiah PK, Bateman JB, Demer JL, Perlman S: Ophthalmologic findings in patients with ataxia. Am J Ophthalmol 123:108, 1997 288. Shelbourne P, Johnson K: Myotonic dystrophy: another case of too many repeats? Hum Mutat 1:183, 1992 289. Lessell S, Coppeto J, Samet S: Ophthalmoplegia in myotonic dystrophy. Am J Ophthalmol 71:1231, 1971 290. Burian HM, Burns CA: Ocular changes in myotonic dystrophy. Am J Ophthalmol 63:22, 1967 291. Thompson HS, Van Allen MW, von Noorden GK: The pupil in myotonic dystrophy. Invest Ophthalmol 3:325, 1964 292. Hayasaka S, Kiyosawa M, Katsumata S et al: Ciliary and retinal changes in myotonic dystrophy. Arch Ophthalmol 102:88, 1984 293. Pryse-Phillips W, Johnson GJ, Larsen B: Incomplete manifestations of myotonic dystrophy in a large kinship in Labrador. Ann Neurol 11:582, 1982 294. Ter Bruggen JP, Bastiaensen LA, Tyssen CC, Gielen G: Disorders of eye movement in myotonic dystrophy. Brain 113(pt 2):463, 1990 295. Ono S, Kanda F, Takahashi K et al: Neuronal loss in the medullary reticular formation in myotonic dystrophy: a
clinicopathological study. Neurology 46:228, 1996 296. Schults WT, Hoyt WF, Behrens M et al: Ocular neuromyotonia: a clinical description of six patients. Arch Ophthalmol 104:1028, 1986 297. Lessell S, Lessell IM, Rizzo JF: Ocular neuromyotonia after radiation therapy. Am J Ophthalmol 102:766, 1986 298. Newman SA: Gaze-induced strabismus (clinical conference). Surv Ophthalmol 38:303, 1993 299. Barroso L, Hoyt WF: Episodic exotropia from lateral rectus neuromyotonia: appearance and remission
after radiation therapy for a thalamic glioma. J Pediatr Ophthalmol Strabismus 30:56, 1993 300. Frohman EM, Zee DS: Ocular neuromyotonia clinical features, physiological mechanisms, and response
to therapy. Ann Neurol 37:620, 1995 301. Ezra E, Spalton D, Sanders MD et al: Ocular neuromyotonia. Br J Ophthalmol 80:350, 1996 302. Seybold ME, Yoss RE, Hollenhorst RW, Moyer NJ: Pupillary abnormalities associated with tumors of the pineal region. Neurology 21:232, 1971 303. Gressel MG, Hanson MR, Tomsak RL: Reversible Sylvian aqueduct syndrome
following jejunoileal bypass. In Smith JL (ed): Neuro-ophthalmology Focus, pp 231–233. New
York, Masson, 1981 304. Daras M, Koppel BS, Samkoff L, Marc J: Brain stem toxoplasmosis in patients with acquired immunodeficiency syndrome. J Neuroimaging 4:85, 1994 305. Paulus W, Straube A, Bauer W, Harding AE: Central nervous system involvement in Leber's optic neuropathy. J Neurol 240:251, 1993 306. Neville BGR, Lake BD, Stephens R, Sanders MD: A neurovisceral storage disease with vertical supranuclear ophthalmoplegia, and
its relationship to Niemann-Pick disease: a report of nine patients. Brain 96:97, 1973 307. Yan-Go FL, Yanagihara T, Pierre RV, Goldstein NP: A progressive neurologic disorder with supranuclear vertical gaze paresis
and distinctive bone marrow cells. Mayo Clin Proc 59:404, 1984 308. Shulman LM, David NJ, Weiner WJ: Psychosis as the initial manifestation of adult-onset Niemann-Pick disease
type C. Neurology 45:1739, 1995 309. Palmer M, Green WR, Maumenee IH et al: Niemann-Pick disease—type C. Arch Ophthalmol 103:817, 1985 310. Kirkham TH, Kamin DF: Slow saccadic eye movements in Wilson's disease. J Neurol Neurosurg Psychiatry 37:191, 1974 311. Hoyt CS, Bilson FA, Alpins N: The supranuclear disturbances of gaze in kernicterus. Ann Ophthalmol 10:1487, 1978 312. Edis RH, Mastaglia FL: Vertical gaze palsy in barbiturate intoxication. Br Med J 1:144, 1977 313. Hamed LM, Mancuso A: Inferior rectus muscle contracture syndrome after retrobulbar anesthesia. Ophthalmology 98:1506, 1991 314. Hamilton SM, Elsas FJ, Dawson TL: A cluster of patients with inferior rectus restriction following local
anesthesia for cataract surgery. J Pediatr Ophthalmol Strabismus 30:288, 1993 315. Hunter DG, Lam GC, Guyton DL: Inferior oblique muscle injury from local anesthesia for cataract surgery. Ophthalmology 102:501, 1995 316. Muñoz M: Letter to the Editor: Inferior rectus muscle overaction after cataract
extraction. Am J Ophthalmol 118:664, 1994 317. Capó H, Roth E, Johnson T et al: Vertical strabismus after cataract surgery. Ophthalmology 103:918, 1996 318. Muñoz M, Parrish RK II: Strabismus following implantation of Baerveldt drainage devices. Arch Ophthalmol 111: 1096, 1993 319. Smith SL, Starita RJ, Fellman RL, Lynn JR: Early clinical experience with the Baerveldt 350-mm2 glaucoma implant and associated extraocular muscle imbalance. Ophthalmology 100:914, 1993 320. Cornblath WT, Elner V, Rolfe M: Extraocular muscle involvement in sarcoidosis. Ophthalmology 100:501, 1993 321. Patel AS, Kelman SE, Duncan GW, Rismondo V: Painless diplopia caused by extraocular muscle sarcoid. Arch Ophthalmol 112:879, 1994 322. Hakin KN, McNab AA, Sullivan TJ: Spontaneous hemorrhage within the rectus muscle. Ophthalmology 101:1631, 1994 323. Harris GJ, Murphy ML et al: Orbital myositis as a paraneoplastic syndrome. Arch Ophthalmol 112:380, 1994 324. Leib ML, Odel JG, Cooney MJ: Orbital polymyositis and giant cell myocarditis. Ophthalmology 101:950, 1994 325. Atabay C, Tyutyunikov A et al: Serum antibodies reactive with eye muscle membrane antigens are detected
in patients with nonspecific orbital inflammation. Ophthalmology 102: 145, 1995 | 
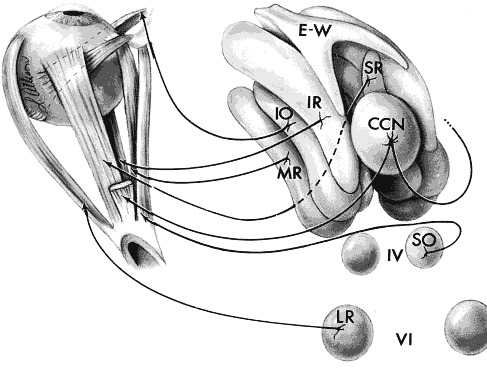
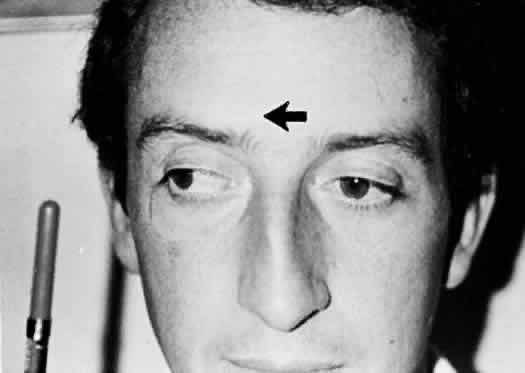
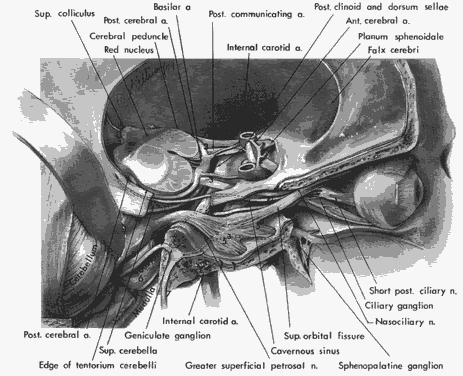
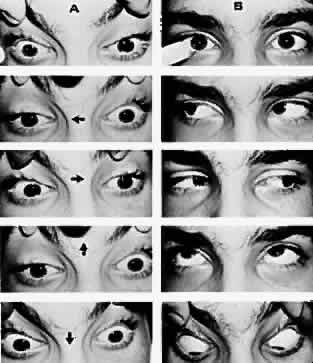
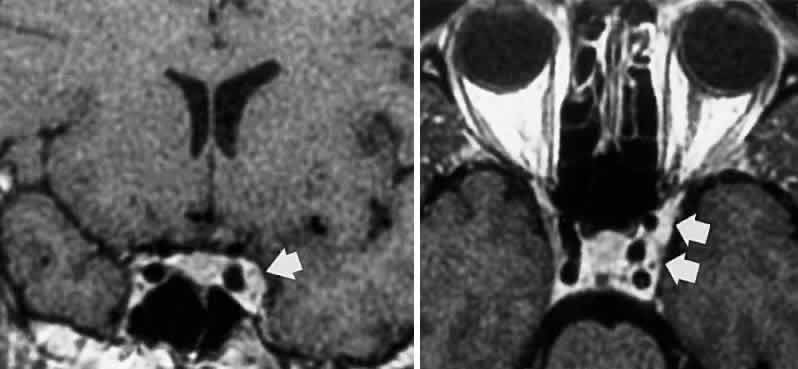
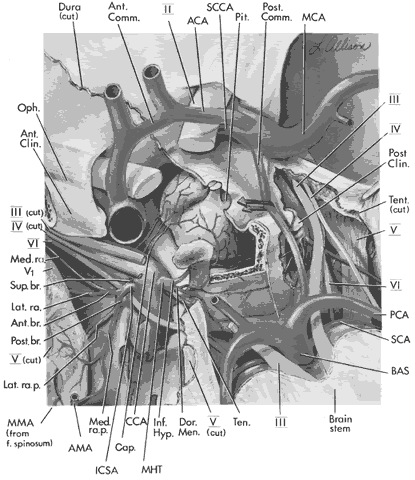
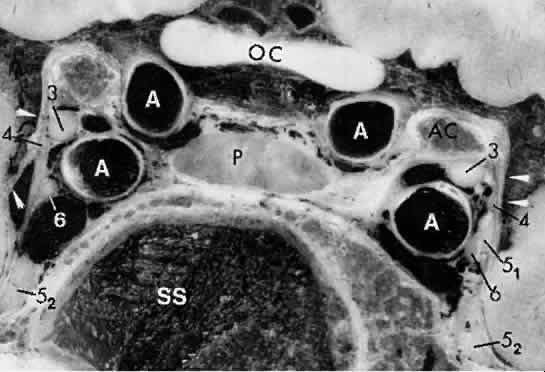
 and associates14 demonstrated penetration of oculomotor trunks by circumflex mesencephalic
arteries or branches of the posterior cerebral perforating vessels, but
clinical implications of such anatomic anomalies are not clear.
and associates14 demonstrated penetration of oculomotor trunks by circumflex mesencephalic
arteries or branches of the posterior cerebral perforating vessels, but
clinical implications of such anatomic anomalies are not clear.