RHABDOMYOSARCOMA Rhabdomyosarcoma is the most common soft-tissue sarcoma in patients younger
than 15 years of age and the most common primary orbital malignancy
in childhood. These facts should not imply its frequent occurrence. Including
all body sites, the annual incidence of childhood rhabdomyosarcoma
in the United States is approximately 225 cases.2 The orbit is the site of origin in 5% to 25% of cases.3,4 However, site distribution varies with age. In children 5 to 9 years of
age, for example, approximately 40% of primary rhabdomyosarcomas involve
the orbit or eyelid.2 Although relatively rare, the tumor has a devastating natural history
and demands a high index of suspicion in all cases of pediatric proptosis. Orbital rhabdomyosarcomas are slightly more common in females, with a 0.79 to 1 male-to-female
ratio.2 The average age of presentation is 7.8 years, but the tumor may be present
at birth and has been reported in patients as old as 78 years.5 A positive family history and associated anomalies have at times been
identified, but these are exceptions rather than the rule. Classically, orbital
rhabdomyosarcoma presents in an abrupt manner, with rapid progression
of proptosis over days to weeks. A somewhat more indolent course
does not exclude the diagnosis, however. Vigilance also should be
exercised when rapidly expanding eyelid lesions are encountered. Rhabdomyosarcoma
may present as ptosis or an eyelid mass rather than with
proptosis.4 An eyelid rhabdomyosarcoma can occur as a congenital lesion.6 Within the orbit, rhabdomyosarcoma occurs most often, but not exclusively, in
the superior nasal quadrant, with downward and outward displacement
of the globe. CT scans show the topography of the orbital mass (Fig. 1A), as well as the possible extension into adjacent bone, paranasal sinuses, or
the intracranial cavity. The circumscription that may be noted
on CT is relative, because the lesion is not encapsulated and microscopically
infiltrates normal tissue. Echography shows internal echoes of
low-to-medium amplitude. Because the cellular tumor absorbs acoustic
energy, the amplitude of the spikes falls off somewhat through the lesion (see Fig. 1B and C). MRI can help define the tumor's relationship to extraocular muscles (Fig. 2). 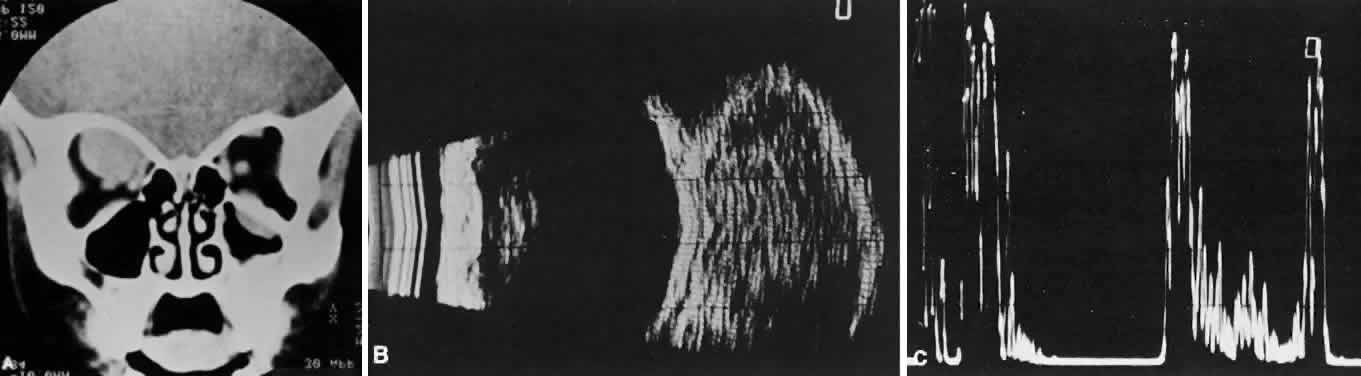 Fig. 1. A. Proptosis and downward, outward globe displacement developed over 2 days
in a 3-year-old girl. A homogeneous mass fills the superomedial orbit. B. Contact B-scanning shows a relatively well-circumscribed mass with uniform
internal echoes. C. Contact A-scanning shows the internal reflectivity to be of low to medium
amplitude, consistent with a sarcomatous lesion. Biopsy results confirmed
the diagnosis of rhabdomyosarcoma. Fig. 1. A. Proptosis and downward, outward globe displacement developed over 2 days
in a 3-year-old girl. A homogeneous mass fills the superomedial orbit. B. Contact B-scanning shows a relatively well-circumscribed mass with uniform
internal echoes. C. Contact A-scanning shows the internal reflectivity to be of low to medium
amplitude, consistent with a sarcomatous lesion. Biopsy results confirmed
the diagnosis of rhabdomyosarcoma.
|
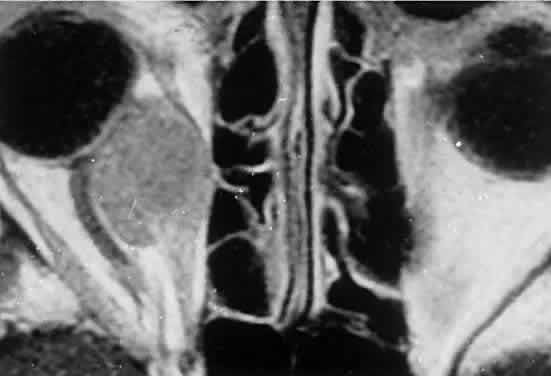 Fig. 2. MRI shows an intraconal tumor of lower intensity than the medial rectus
muscle. The proximal muscle is splayed rather than compressed, suggesting
that the lesion originated within the medial rectus. The diagnosis
was alveolar rhabdomyosarcoma. Fig. 2. MRI shows an intraconal tumor of lower intensity than the medial rectus
muscle. The proximal muscle is splayed rather than compressed, suggesting
that the lesion originated within the medial rectus. The diagnosis
was alveolar rhabdomyosarcoma.
|
The clinical diagnosis must be confirmed by biopsy. Because of the risk
of seeding the biopsy tract, a transcranial approach should be avoided. If
possible, the periosteum should not be violated because it presents
a relative barrier to tumor invasion. Depending on its location, the
lesion should be approached transconjunctivally or with an eyelid crease
incision/transseptal dissection. The surgeon must balance the benefit
of complete gross tumor resection with the risks of functional impairment
and tumor dissemination that may accompany that effort. Tissue
samples should be fixed in formaldehyde solution and glutaraldehyde
for light and electron microscopic study. In addition, the value of immunohistochemical
differentiation has been established for some time, and
the potential uses of molecular genetic studies are rapidly emerging. Consequently, the
procurement of fresh or frozen tissue, or both, has
been given the highest priority by the Biopathology Discipline within
the Intergroup Rhabdomyosarcoma Study Group (IRSG).2 These techniques can facilitate the diagnosis of poorly differentiated
tumors, and they may refine diagnostic and prognostic classifications, identify
candidate genes, and contribute to potential gene therapies. Since the inception of IRSG-I in 1972, the multicenter collaboration has
enrolled the overwhelming majority of patients diagnosed with rhabdomyosarcoma
in the United States and has contributed significantly to enhanced
patient survival. Patients with orbital tumors had a 96% versus 83% failure-free
survival in IRSG-IV compared with those in the IRSG-III.2 As of the year 2000, with the IRSG-V study underway, the overall (all
primary sites) 5-year survival of children and adolescents with nonmetastatic
and metastatic tumors was approaching 80%. This progress reflects
advances in diagnostic imaging and multimodal treatment, including
chemotherapy (e.g., agents, combinations, timing), radiation therapy (e.g., doses, fractionation, timing), and surgery (e.g., diagnostic biopsy, local
staging, salvage procedures). Therapeutic protocols have evolved over the past 30 years, but they also
have not been uniform at any given point in time. Rather, they have
been tailored to the patient's level of risk, as determined by multiple
prognostic factors (Table 3). The concept of “risk-appropriate therapy”7 recognizes, for example, that a 6-year-old child with an embryonal rhabdomyosarcoma
confined to the orbit might do well with a relatively simple
chemotherapy protocol, avoiding the late adverse effects of high-dose
radiation. Conversely, an 18-year-old patient with an alveolar rhabdomyosarcoma
arising in the retroperitoneum, with metastases at presentation, needs
aggressive, complex chemotherapy and radiation, and may
still do poorly. Prognostic factors considered by the multidisciplinary
team include the presence of gross or microscopic residual tumor, and
this determination currently is being redefined with molecular techniques
that may show residual disease even without microscopic evidence2; whether tumor is confined to the anatomic site of origin or invades surrounding
tissues; tumor size, with 5 cm considered a breakpoint; regional
lymph node involvement; and distant metastasis. Body site plays
a role, and the orbit is relatively favored. The age of the patient at
diagnosis is a strong independent predictor of outcome.7 The current pathologic classification for childhood rhabdomyosarcomas
by prognosis2 is as follows:
 - Superior prognosis: botryoid, spindle cell
- Intermediate prognosis: embryonal
- Poor prognosis: alveolar, undifferentiated, anaplastic (formerly pleomorphic)
- Indeterminate prognosis: rhabdomyosarcoma with rhabdoid features
Although no single regimen is appropriate for every child with orbital
rhabdomyosarcoma, a sample protocol might include multiple 3-week cycles
of chemotherapy, each beginning with intravenous vincristine, actinomycin-D
and cyclophosphamide, with vincristine repeated on the eighth
and fifteenth days of each cycle. The regimen might include external
radiation to a total dosage of 5040 cGy. For poor prognosis cases (e.g., metastatic
alveolar rhabdomyosarcoma), newer agents under investigation
include ifosfamide, etoposide, and topotecan.2 Having made the diagnosis and contributed to local staging at the time
of presentation, the orbital surgeon continues to follow the patient along
with the pediatric oncology team. In cases of treatment failure, “salvage” surgery may take the form of orbital exenteration8 or excision of residual tumor combined with brachytherapy.9 Rhabdomyosarcoma underscores the importance of clinical suspicion when
dealing with acute proptosis in childhood. Prompt referral to a tertiary
center after appropriate imaging is the responsibility of the primary
ophthalmologist, family practitioner, or pediatrician who may first
encounter the patient. OTHER PRIMARY SARCOMAS Other primary orbital sarcomas may have a relatively abrupt onset of proptosis, although
their progression generally is less explosive than that
of rhabdomyosarcoma. As with rhabdomyosarcoma, a prompt biopsy is
critical for appropriate management. Alveolar soft part sarcoma, an example of this group of lesions, is a rare
tumor that may affect the pediatric orbit. In an extensive review
of the literature, Sullivan and colleagues10 identified about 50 orbital cases. They noted that the tumor tends to
involve the extremities of young adults or the head and neck region of
children, with a predilection for the orbit and tongue. A myogenic origin
is favored, but there also is evidence for a neural derivation. The
findings of imaging studies are nonspecific. Diagnosis depends on the
light and elec-tron microscopic demonstration of periodic acid-Schiff-positive, diastase-resistant crystals within the cytoplasm of large
polyhedral tumor cells.11 Pediatric patients appear to have a better prognosis than do adults. The
currently recommended treatment is local excision of circumscribed
primary lesions, with exenteration reserved for diffuse orbital involvement
or local recurrence. Radiation therapy may have adjunctive value. Epithelioid sarcoma is a rare tumor that can occur in older children and
young adults. Most lesions originate in the distal upper extremities. White
and coworkers12 identified two patients; one was a 17-year-old girl with primary epithelioid
sarcoma of the orbit. The tumors have both mesenchymal and epithelial
histologic qualities and grow in tendon sheaths in a grossly nodular
pattern. Treatment strategies have yet to be defined for this rare
lesion. NEUROBLASTOMA Neuroblastoma is the most common metastatic orbital lesion in children.13 It represents 10% to 15% of all pediatric malignancies, ranking behind
only leukemia and solid central nervous system tumors in frequency. It
is a tumor of primitive neuroblastic tissue and, in some respects, is
the autonomic nervous system counterpart of retinoblastoma. It usually
originates in the adrenal medulla or other retroperitoneal sites but
also may arise from any of the sympathetic ganglia in the mediastinum
or neck. Neuroblastoma typically afflicts children from 18 months to 3 years
of age, although it may be present at birth or may not appear
until the midteens. In a review of more than 400 cases of neuroblastoma, Musarella and colleagues14 found the incidence of ophthalmologic signs to be 20%. In almost half
of these cases, the ocular symptoms were the presenting complaints. The
most common eye signs were related to orbital metastasis. Orbital involvement
was bilateral in approximately half of the cases. Characteristic
eye findings include proptosis and periorbital ecchymosis. The latter
results from hemorrhagic necrosis within a rapidly growing tumor
that has outstripped its blood supply. Other eye signs may reflect more
distant tumor involvement. Horner's syndrome can result from a
primary neuroblastoma in the sympathetic ganglia of the neck or mediastinum
or from metastases to either of these regions.13–15 An infantile Horner's syndrome is characterized by hypochromia of
the ipsilateral iris. Ocular signs may include opsoclonus, a wild conjugate
oscillation of both eyes that may be associated with myoclonus, and
truncal ataxia.14 It has been proposed that this complex results from an antibody directed
against neuroblastoma antigen, which may cross-react with cerebellar
tissue, producing damage in that area.16 Neuroblastomas manufacture catecholamines, which, in sufficient quantity, can
produce flushing, systemic hypertension, and diarrhea. Diagnosis
is aided by the demonstration of catecholamine metabolites in the urine.17 In cases of suspected neuroblastoma metastatic to the orbit, the primary
tumor may be shown by abdominal or thoracic imaging studies. A histologic
diagnosis generally is required, however. Orbital soft tissue involvement
usually follows extension from bony metastasis (Fig. 3). Therefore, orbital biopsies should be performed extraperiosteally, because
the periorbita may still be intact and constitute a relative barrier
to tumor extension. Because the histologic differential diagnosis
includes other poorly differentiated tumors of childhood, speci-mens
should be fixed in both formalin and glutaraldehyde, and fresh tissue
also should be submitted. 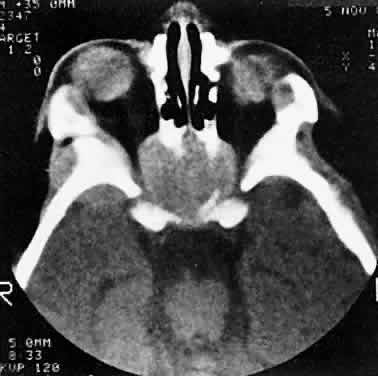 Fig. 3. A large metastatic focus of neuroblastoma has destroyed the body of the
sphenoid bone and has extended into both orbital apices. A second site
involves the outer portion of the right sphenoid wing and extends into
the orbit and the middle cranial and temporal fossas. The tumor originated
in the right adrenal gland. Fig. 3. A large metastatic focus of neuroblastoma has destroyed the body of the
sphenoid bone and has extended into both orbital apices. A second site
involves the outer portion of the right sphenoid wing and extends into
the orbit and the middle cranial and temporal fossas. The tumor originated
in the right adrenal gland.
|
Histologically, neuroblastomas display features commensurate with their
degree of differentiation. At the more primitive end of the spectrum
are tumors comprising small round cells with minimal cytoplasm. At the
other extreme are lesions consisting of large, cytoplasm-rich elements
resembling ganglion cells. It has been proposed that neuroblastomas
having undergone spontaneous clinical regression have evolved into benign
ganglioneuromas.18 Homer-Wright pseudorosettes characteristically present in well-differentiated
primary neuroblastomas and are rarely, if ever, found within orbital
metastases. In poorly differentiated tumors, electron microscopy
may be required to show neurosecretory granules containing catecholamines. The
rapidly advancing field of immunohistochemistry also has contributed
to diagnosis and prognostic assessment in neuroblastoma.19 As with rhabdomyosarcoma, the choice of treatment protocols for neuroblastoma
is based on tumor staging and the multiple prognostic factors that
have been identified in large cooperative trials. Approximately 25% of
children with newly diagnosed neuroblastoma present with nonmetastatic
and localized disease.19 This group has a 98% survival with surgery alone as primary therapy. However, children
with localized disease who have amplification of the
MYCN oncogene or who are 2 years of age or older with either unfavorable
histopathology or positive lymph nodes are at greater risk of death. Other
negative factors include elevated serum ferritin and elevated
serum neuron-specific enolase. The majority of children with neuroblastoma have metastatic disease at
diagnosis.20 In order of frequency, the most common sites are bone marrow, bone, lymph
nodes, liver, intracranial and orbital sites, lung, and central nervous
system. The metastatic pattern differs with age. Patients younger
than 1 year are more likely to have liver or skin metastases at diagnosis
and less likely to have bone and bone marrow metastases at diagnosis
than do patients age 1 year or older. Among children with metastases
at diagnosis, event-free survival is decreased in patients with bone, bone
marrow, central nervous system, intracranial/orbital, lung and
pleural metastases, and improved in those with liver and skin metastases. Depending on tumor staging and risk factors, treatment protocols may include
surgical resection of the primary tumor, combination chemotherapy (cisplatin, cyclophosphamide, doxorubicin, and etoposide) of varying
dose-intensity, and myeloablative therapy with autologous purged bone
marrow transplantation.19 OTHER METASTATIC LESIONS Orbital metastases from other solid pediatric tumors are less common. Of
these, Ewing's sarcoma accounts for the majority, and Wilms' tumor
is responsible for an extremely small number of cases.13,21 Ewing's sarcoma accounts for approximately 10% of tumors that metastasize
to the pediatric orbit. Albert and colleagues13 noted orbital involvement in five of 12 patients with Ewing's sarcoma. In
all patients, the orbital metastases were unilateral and were
clinically noted several months after diagnosis of the primary lesion. Ewing's
sarcoma usually arises within the medullary canals of the
bones of the trunk or extremities. Unlike neuroblastoma, its peak incidence
is in late childhood and adolescence. Ewing's sarcoma and
malignant peripheral neuroectodermal tumor (PNET) are closely related, but
are distinguished by the microscopic and immunohistochemical findings
of greater neuroectodermal differentiation (e.g., rosette formation) in
the latter lesion.22,23 Although earlier studies suggested a poorer prognosis in PNET than in
Ewing's sarcoma, more recent work showed no difference in clinical
outcome.24 Radiation therapy, surgery, and chemotherapy are used in management. The
principal adverse prognostic factor is metastasis at diagnosis. In
a European study of 975 patients enrolled from 1977 to 1993, the 5-year
relapse-free survival of patients without metastases at diagnosis was 55% compared
to 22% for patients with metastases at diagnosis.25 During the second 8 years of the study, these figures were 60% and 30%, respectively, indicating
continued outcome improvement. Wilms' tumor (nephroblastoma) arises from embryonic elements within the
kidney. Although it affects children almost as frequently as neuroblastoma
and can metastasize extensively to other sites, orbital lesions
have been rarely described.21,26 Reports have concerned children younger than 3 years of age. As of 1999, the
overall survival rate for Wilms' tumor was 90%.27 Most patients have favorable histology (nonanaplastic or focally anaplastic
tumors), and survive after preoperative chemotherapy and nephrectomy.27–29 However, poor outcomes are associated with diffuse anaplasia, chromosomal
loss on 1p and 16q, diploidy, lung or liver metastases, major tumor
spillage during resection, remote lymph node involvement, and bilateral
tumors. ACUTE LEUKEMIA Leukemia is the most common malignancy in childhood, and nearly all pediatric
leukemias are acute rather than chronic. The lymphoblastic variety
is approximately four times more common than the myelogenous form. Leukemic
cells frequently lodge in the eye and adnexa, and bilateral
involvement is common.30 Proptosis occurs less often than intraocular or optic nerve complications
and may result from a combination of local tumefaction and hemorrhage. Most
ophthalmic complications of leukemia are associated with the
acute lymphoblastic rather than the myelogenous form of disease. Orbital
infiltration may occur in either condition but is disproportionately
more common in acute myelogenous leukemia (AML).31 In the latter disorder, orbital tumefactions (myeloid or granulocytic
sarcomas) may curiously precede bone marrow and peripheral blood evidence
of leukemia by several months.32–34 For this reason, ophthalmologists may be the first to diagnose this systemic
disease. The correct early diagnosis is important, because chemotherapy
may be more effective if initiated before the leukemic phase
develops. Extramedullary deposits of primitive myeloblasts may occur at any time
in the course of acute myeloid leukemia. The growths have been termed chloromas because of a greenish hue imparted by the enzyme myeloperoxidase within
the tumor cells. This discoloration fades on exposure to the air and
is therefore an inconstant finding at the time of histopathologic preparation
and diagnosis.35 Granulocytic or myeloid sarcoma is considered a more appropriate term and is preferred. Favored sites
of involvement are the bones and periosteum of the skull, including those
of the orbit. Granulocytic sarcomas also may occur in the orbital
soft tissues and the eyelids. Zimmerman and Font32 studied 33 granulocytic sarcomas of the orbit and eyelids, 29 of which
were examined by biopsy before a diagnosis of leukemia had been established. In
most of their cases, hematologic investigation confirmed the
systemic process soon after the sarcomas were identified. In some cases, however, intervals
of 4 to 15 months elapsed before leukemia was
diagnosed by bone marrow and peripheral blood examination. Because of
these delays and because the lesions can be histologically ambiguous, granulocytic
sarcomas may be misdiagnosed as independent, primary sarcomas
or as histiocytic lymphomas. Granulocytic sarcomas of the orbit may be bilateral in 10% to 45% of cases.32,33 Hemorrhage occurs frequently, and eyelid ecchymosis may be a pre-senting
sign. Most patients are affected in the first decade of life, and males
are involved more often than females. The majority of cases in the
series cited were derived from Asia, Africa, or the South Pacific, and, interestingly, granulocytic sarcoma was the second most common cause
of proptosis in Uganda after Burkitt's lymphoma. Findings on imaging studies are nonspecific. The diagnosis depends on biopsy
results.36 As in other pediatric tumors, light microscopy may yield ambiguous findings, and
immunohistochemical stains and electron microscopy are helpful. Granulocytic
sarcomas are composed of large mononuclear cells that
resemble histiocytes. Diagnosis may be difficult when immature myeloblasts
dominate the histologic picture, and evidence of granulocytic differentiation
is minimal. Diagnosis is aided in these cases by the Leder
stain, which indicates esterase activity and cellular differentiation
in a myelocytic direction. In addition, the immunohistochemical stain
for lysozyme is positive in 60% to 89% of cases. In patients in whom
both the Leder and lysozyme stains are negative, the monoclonal antibody
MAC387 may establish the diagnosis.34 Electron microscopy may show early granule formation. Compared to progress in pediatric acute lympho-blastic leukemia during
the past 2 decades, improvement in the cure rate of children with AML
has been modest.37 Among children treated with chemotherapy alone, about 40% are long-term
survivors. Prognosis improves with bone marrow transplants from histocompatible
sibling donors early in the first remission. Molecular genetic
advances are expected to improve therapeutic strategies. BURKITT'S LYMPHOMA Although it is a rare cause of proptosis in the Western hemisphere, Burkitt's
lymphoma deserves attention because of its distinctive epidemiologic
and clinical features. The tumor occurs endemically within
certain geographic and climatic boundaries in East Africa. It is the most
common pediatric orbital tumor in Uganda, accounting for almost 50% of
cases.38 The average age of presentation is 7 years, with a range from 3 to 15 years. Large
extranodal tumors occur in the bones of the jaw and the abdominal
viscera. Unilateral or bilateral proptosis is present in 20% of
cases and usually results from maxillary extension. The progression
of proptosis may be explosive. Burkitt's lymphoma has a doubling
time that may be as brief as 24 hours, ranking it as the fastest-growing
tumor in humans.39 Endemic African cases have been linked to the Epstein-Barr virus and to
a t(8;14q) chromosomal translocation, suggesting an interaction between
environmental factors and host susceptibility. Sporadic North American cases have a less-definitive viral association. These
patients differ clinically in their age of presentation (mean, 11 years) and
in the usual site of tumor origin (intra-abdominal lymphoid
tissue).40,41 Involvement of the facial bones and orbit is less common in the North
American cases, but invasion of the orbit from the sinuses may occur42,43 (Fig. 4). 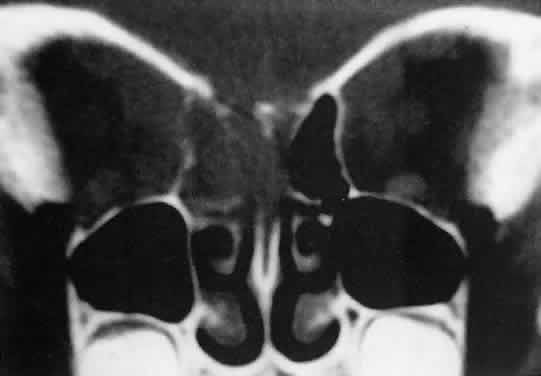 Fig. 4. Burkitt's lymphoma involving the posterior ethmoids, skull base, and
both orbital apices in a 5-year-old boy. Fig. 4. Burkitt's lymphoma involving the posterior ethmoids, skull base, and
both orbital apices in a 5-year-old boy.
|
Biopsy is necessary to establish the diagnosis. The characteristic microscopic
picture is that of a “starry sky,” made up of a homogeneous
background of neoplastic lymphocytes and interspersed larger
histiocytes with more abundant cytoplasm. Burkitt's lymphoma was
considered a small noncleaved cell lymphoma in the Working Formulation
and was identified as a peripheral B-cell neoplasm in the Revised European-American
Lymphoma Classification. Burkitt's lymphoma subsequently
was subdivided into endemic, sporadic, immunodeficiency-associated, and
atypical forms in the World Health Organization scheme.44 In children, survival rates of 80% to 90% are being achieved with intensive, short
duration chemotherapeutic protocols.45 EOSINOPHILIC GRANULOMA Unifocal eosinophilic granuloma, or unifocal Langerhans' cell histiocytosis (LCH), is
a relatively benign, probably reactive lesion composed
of histiocyte-type cells and an inflammatory infiltrate of eosinophils
and neutrophils.46 Of the disorders traditionally grouped under the histiocytosis X rubric (acute
disseminated LCH or Letterer-Siwe disease; unifocal LCH; and
multifocal LCH or Hand-Schuller-Christian disease), unifocal eosinophilic
granuloma is the most frequent cause of orbital disease. It generally
affects the superotemporal quadrant as an extension from an osteolytic
lesion47,48 (Fig. 5). There is a male predominance with onset in the first or second decade. Symptoms
include bone pain, tenderness, and local swelling. The differential
diagnosis, based on initial CT studies, includes other primary
causes of bone erosion in this region, such as dermoid cysts and lacrimal
neoplasms, as well as tumors metastatic to orbital bone, such as
Ewing's sarcoma. Diagnosis ultimately requires microscopic analysis
of tissue. Percutaneous fine-needle aspiration, with or without core-needle
biopsy, has been used for lesions of the extremities49 and can be considered for orbital lesions. However, the need for general
anesthesia in children, the risk of uncontrollable hemorrhage, and
the frequent extension of tumor to the dura all weigh toward open biopsy. Expression
of CD1a by immunohistochemistry, which requires frozen
sections or fresh cytologic preparations, is considered diagnostic of
LCH.46 Electron microscopic studies show “Langerhans” or “Birbeck
granules,” with a characteristic tennis-racket shape. 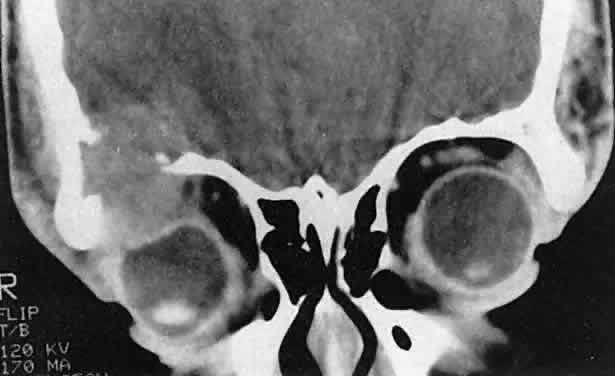 Fig. 5. Eosinophilic granuloma in a 16-year-old boy. The growth pattern suggests
an intraosseous origin. (Harris GJ, Beatty RL: Acute proptosis in childhood. In Linberg JV [ed]: Oculoplastic
and Orbital Emergencies. Norwalk, CT: Appleton & Lange, 1989:93.) Fig. 5. Eosinophilic granuloma in a 16-year-old boy. The growth pattern suggests
an intraosseous origin. (Harris GJ, Beatty RL: Acute proptosis in childhood. In Linberg JV [ed]: Oculoplastic
and Orbital Emergencies. Norwalk, CT: Appleton & Lange, 1989:93.)
|
Treatment options include surgical curettage, low-dose irradiation (400 to 800 cGy), and
intralesional steroids. CT-guided steroid injection48 offers a safer alternative than blind injection, but some of the concerns
about diagnostic needle aspiration still apply. Interestingly, spontaneous
resolution after diagnostic biopsy has been observed.50 Our preferred approach in suspected cases includes an eyelid crease incision
for access, frozen sections for immediate provisional diagnosis, and
gentle curettage and instillation of methylprednisolone for treatment. Multifocal
disease is excluded by an x-ray bone survey or bone
scanning, chest x-ray, liver function studies, dental evaluation, urinalysis, and
water-deprivation test.51 Multifocal eosinophilic granuloma (multifocal LCH or Hand-Schuller-Christian
disease) occurs in children younger than 5 years of age and preferentially
affects males. Multifocal osteolytic lesions may be detected
at the time of presentation, or the patient may present with a single
bone lesion and others may emerge within the ensuing 6 to 12 months. Hepatosplenomegaly
and lymphadenopathy occur in 25% to 50% of patients.52 Nonspecific systemic symptoms include malaise, anorexia, and fever. A
minority of patients show the classic triad of exophthalmos, diabetes
insipidus, and bone destruction. Orbital involvement usually follows extension
from adjacent bony foci.53 Histopathologically, the lesions are similar to those of the unifocal
disease. Treatment also is similar, but chemotherapy in modest doses often
is used to shorten the course and diminish morbidity. The prognosis
generally is favorable, with recovery the rule. SICKLE CELL DISEASE Children in the midst of sickle cell crises may experience acute orbital
pain, proptosis, eyelid edema, and fever.54–58 In some cases, the rapid increase in orbital pressure is extreme, causing
vision loss.58 The proposed mechanism is infarction of orbital bone with adjacent subperiosteal
hemorrhage or effusion (Fig. 6). Infarction may be diagnosed by radionuclide imaging, which also should
differentiate the condition from the osteomyelitis that can occur in
sickle cell disease. In infarction, bone and bone marrow uptake usually
are decreased. In osteomyelitis, tracer accumulation generally is
increased. Infarction also may be detected by MRI.58 Children with sickle cell disease are prone to other infections as well. If
the periosteal elevation abuts an opacified paranasal sinus, subperiosteal
abscess secondary to bacterial sinusitis should enter the differential
diagnosis.59 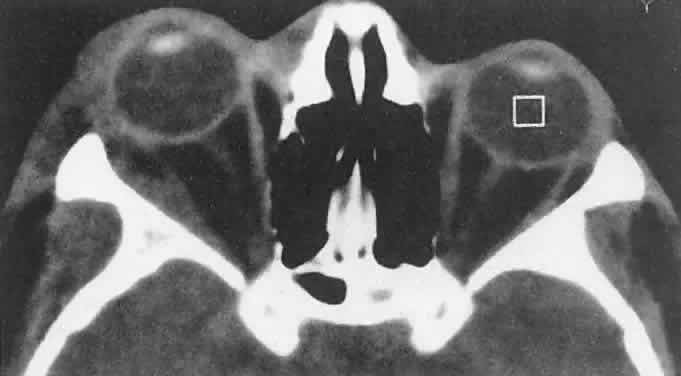 Fig. 6. An 18-year-old boy had onset of right proptosis during a sickle cell crisis. Abnormal
radiodensity is seen along the lateral wall of the orbit
with a similar process in the intratemporal fossa. Bone scanning showed
decreased uptake consistent with a sphenoid bone infarct. (Harris GJ, Beatty RL: Acute proptosis in childhood. In Linberg JV [ed]: Oculoplastic
and Orbital Emergencies. Norwalk, CT: Appleton & Lange, 1989:94.) Fig. 6. An 18-year-old boy had onset of right proptosis during a sickle cell crisis. Abnormal
radiodensity is seen along the lateral wall of the orbit
with a similar process in the intratemporal fossa. Bone scanning showed
decreased uptake consistent with a sphenoid bone infarct. (Harris GJ, Beatty RL: Acute proptosis in childhood. In Linberg JV [ed]: Oculoplastic
and Orbital Emergencies. Norwalk, CT: Appleton & Lange, 1989:94.)
|
Curran and colleagues58 reported one case of subperiosteal orbital hemorrhage in sickle cell disease
and identified 16 others in a literature review. The median patient
age was 12.8 years. Ten of 12 patients tested had bone marrow infarctions. Thirteen
patients were treated conservatively, and four underwent
surgical evacuation. Sixteen of 17 patients recovered without sequelae. One
patient, treated conservatively, showed mild visual impairment. Kersten60 offered an alternative explanation for this clinical condition. He described
an 11-year-old girl with hemoglobin SS and bilateral acute subperiosteal
orbital hematomas. Radionuclide bone and bone marrow scans and
MRI study results were negative for infarction, but the serum vitamin
C level was abnormally low. Hemorrhage was attributed to vascular fragility
related to subclinical scurvy (which can result from hemoglobinopathy-induced
hemosiderosis), combined with vascular wall damage caused
by sickle cells. In general, children with subperiosteal hematoma or effusion secondary
to sickle cell disease can be treated conservatively. However, surgical
evacuation should be performed in cases of compromised vision. CAPILLARY HEMANGIOMA The orbit and eyelids are common sites of capillary hemangioma (benign
hemangioendothelioma, infantile hemangioma, strawberry nevus). These tumors
differ from other developmental vascular anomalies in having a growth
potential that is disproportionate to overall body growth. Their
essential elements are endothelial cells that actively replicate, as
shown by the incorporation of tritiated thymidine in experimental studies.61 Vascular malformations, including port-wine stains, lymphangiomas, primary
varices, and arteriovenous malformations, have stable populations
of endothelial cells, do not incorporate labeled precursors, and proliferate
in proportion to total body growth. The expansion of these other
lesions occurs by hemodynamic dilation of vascular channels or by intrinsic
hemorrhage rather than by cellular replication.62,63 Capillary hemangioma also should be distinguishedfrom cavernous hemangioma
of adulthood, which is both clinically and morphologically a separate
entity.64 Capillary hemangiomas are characteristically noted within the first 2 weeks
of life, with the overwhelming majority apparent by 6 weeks of age.65 They are more common in females than males by a 3:2 ratio. They may affect
the eyelids, the orbit, or both. In a series of 101 cases reviewed
by Haik and colleagues,65 there was a 25% incidence of additional, nonperiocular hemangiomas. The
serious systemic complications of large visceral hemangiomas, such as
the Kasabach-Merrit syndrome of consumption coagulopathy or shunt-induced
high-output cardiac failure, are rarely seen in isolated orbital
lesions.66 Growth of the lesions is most active during the first 4 months after their
appearance, but continues for 8 to 12 months.65 Stabilization usually occurs by 12 to 30 months of age, followed by gradual
spontaneous involution. Histologically, endothelial cell proliferation
slows, fibrous tissue deposition begins, and an initially abundant
population of mast cells decreases.66 In later stages of involution, cytoplasmic bridges form between mast cells
and fibroblasts. Early concepts of thrombosis and infarction have
been rejected. Seventy-five percent of lesions resolve totally by 7 years
of age.65 In some cases, the growth rate may be remarkable, with a doubling of volume
in a matter of days. Because of its potential for rapid growth, capillary
hemangioma must be included in the differential diagnosis of
acute proptosis in infancy. The depth of involvement determines the clinical appearance. The most superficial
lesions occur in the dermis or superficial subcutaneous tissues, producing
the typical cherry or strawberry appearance. These lesions
represented 25% of cases in the series of Haik and coworkers.65 Because elastin fibers are disrupted over the lesion, skin changes may
be irreversible, leaving a crepe-paper appearance after tumor regression. Lesions
of moderate depth, which accounted for 68% of cases in the
same series, appear more bluish than red. Tumor expansion with crying
or other elevation of venous pressure averages approximately 50% and
is rarely as dramatic as that noted with orbital varices, which communicate
more patently with the systemic venous circulation. Deep orbital
tumors (7% of cases) may occur without an anterior, obviously vascular
component (Fig. 7A). Rapid growth in these cases raises the specter of a malignant orbital
tumor, and diagnostic studies may be required for differentiation. 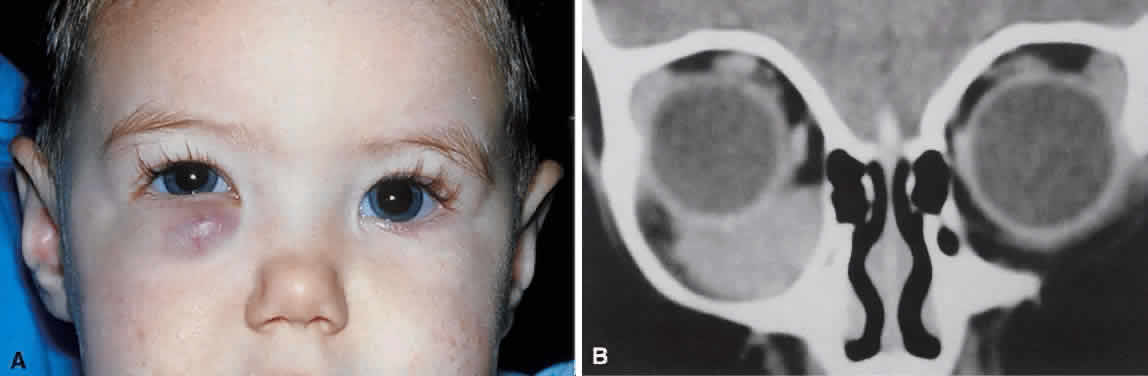 Fig. 7. A. A 10-month-old girl presenting with an inferior orbital tumor that proved
to be a capillary hemangioma. B. CT of this patient shows an inferior orbital tumor mass that has caused
generalized expansion of the bony orbit, suggesting chronicity. Fig. 7. A. A 10-month-old girl presenting with an inferior orbital tumor that proved
to be a capillary hemangioma. B. CT of this patient shows an inferior orbital tumor mass that has caused
generalized expansion of the bony orbit, suggesting chronicity.
|
CT (see Fig. 7B) shows a homogeneous mass that is not particularly distinct from normal
orbital structures. Depending on the growth rate, there may be enlargement
of the bony orbit in a smooth, symmetric pattern, a nonspecific
finding produced by any lesion that has slowly expanded in the first
few years of life. Capillary hemangiomas are soft, compressible lesions
that grow without indenting the globe. Intravenous contrast agent enhances
the tumor's radiodensity, but this feature does not distinguish
the lesion from malignant tumors that may be in the differential
diagnosis. The high contrast sensitivity of MRI allows better delineation of capillary
hemangioma from normal structures than does CT. The tumor is hyperintense
in T2-weighted images with gadolinium and fat suppression (Fig. 8). Low-intensity streaks represent flow voids within higher flow draining
and feeding vessels. 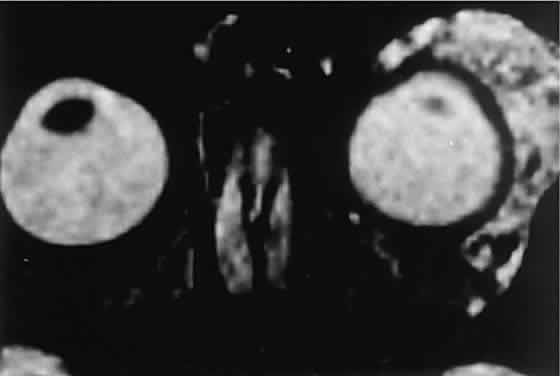 Fig. 8. Gadolinium-enhanced, fat-suppressed, T2-weighted MRI shows the extent of a large capillary hemangioma that infiltrates
the lacrimal gland. Dark streaks represent flow voids within
feeding and draining vessels. Fig. 8. Gadolinium-enhanced, fat-suppressed, T2-weighted MRI shows the extent of a large capillary hemangioma that infiltrates
the lacrimal gland. Dark streaks represent flow voids within
feeding and draining vessels.
|
Standardized echography can help differentiate capillary hemangioma from
rhabdomyosarcoma. The high-amplitude spikes reflected from the vessel
lumen/cell cluster interfaces within the tumor (Fig. 9) are in contrast to the relatively low-amplitude spikes seen in densely
cellular tumors (see Fig. 1C). Compressibility of the lesion also is a valuable echographic finding. 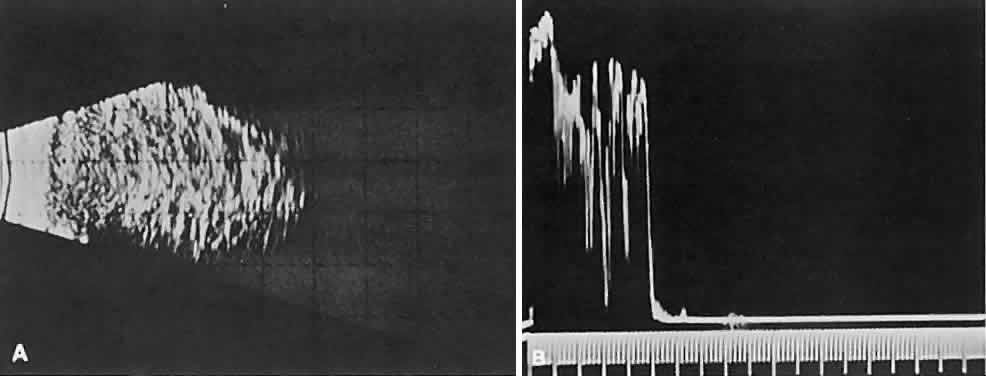 Fig. 9. A. B-scan echography shows marked internal acoustic heterogeneity and lack
of circumscription. B. Quantitative A-scanning shows a corresponding pattern. High-amplitude
spikes are reflected from the vessel-lumen/cell-cluster interfaces within
the tumor. Fig. 9. A. B-scan echography shows marked internal acoustic heterogeneity and lack
of circumscription. B. Quantitative A-scanning shows a corresponding pattern. High-amplitude
spikes are reflected from the vessel-lumen/cell-cluster interfaces within
the tumor.
|
Additional diagnostic methods include technetium-99m—labeled erythrocyte
scintigraphy,67 Doppler studies,68 and arteriography,65 all of which yield more strikingly positive results in capillary hemangioma
than in rhabdomyosarcoma. On rare occasions, histologic examination
may be necessary for definitive diagnosis. As noted, the histology
of capillary hemangioma evolves with its natural history. At initial
presentation, the lesion consists of lobular proliferations of plump endothelial
cells that circumscribe small vascular spaces (Fig. 10). Electron microscopy shows pericytes about the endothelial cells, but
smooth muscle is lacking.69 Involuted lesions show diminished endothelial cellularity and islands
of fibrofatty infiltration.61 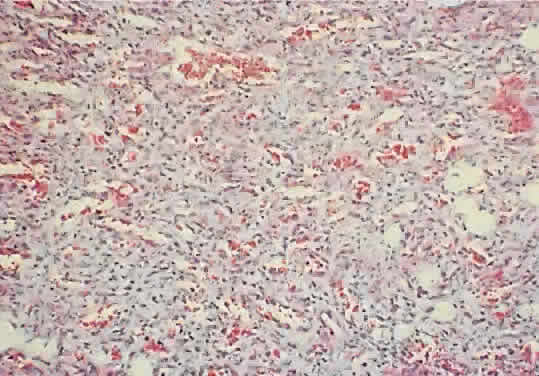 Fig. 10. A capillary hemangioma consists of sheets of plump endothelial cells that
surround small blood-filled channels (hematoxylin-eosin; × 96). Fig. 10. A capillary hemangioma consists of sheets of plump endothelial cells that
surround small blood-filled channels (hematoxylin-eosin; × 96).
|
A conservative approach to therapy is favored, because the majority of
lesions regress spontaneously. However, irreversible functional and cosmetic
changes may occur while the tumor is present. Haik and colleagues65 noted an 80% complication rate, which included residual proptosis, ptosis, strabismus, skin
abnormalities, and a 60% incidence of amblyopia. Amblyopia
results more commonly from anisometropia than from stimulus
deprivation.70,71 Upper eyelid lesions are more responsible for amblyopia than lower lid
lesions, with the axis of the correcting plus cylinder pointing toward
the lesion.66 The indications for treatment include threatened or established amblyopia
and massive proptosis that may be compromising visual function by
optic nerve compression or corneal exposure. There are several treatment
methods available for capillary hemangioma; however, the risk/benefit
ratio and suitability of each method for each patient must be considered.62 No single treatment is appropriate for all. Corticosteroids may sensitize terminal vascularbeds to circulating catecholamines, leading
to constriction, sluggish flow, and coagulation.72,73 Steroids can be administered orally in the form of prednisone, 2 to 4 mg/kg/day. The
major risks are adrenal suppression and growth retardation, and
treatment should be directed by the infant's pediatrician. Intralesional
corticosteroid injection has been used often, with generally
good results.71,74 Treatment consists of triamcinolone 1 ml (40 mg/ml) and betamethasone 1 ml (6 mg/ml). Although
this route is designed to avoid systemic complications, cases
of adrenal suppression have been documented.75,76 Of additional concern are rare embolic complications, including ipsilateral
and bilateral vision loss.77,78 When the tumor's histology (see Fig. 10) and hemodynamic continuity are considered, it would seem that any intralesional
injection is, to some degree, an intravascular one, regardless
of needle size. However, retrograde arterial embolization might be
avoidable by limiting injection force. Corticosteroids can be administered
topically to relatively superficial hemangiomas in the form of clobetasol
propionate cream, 0.05%.79,80 Some systemic absorption should be anticipated. Among lasers in current use, the Candela dye laser may be the most selective
for these vascular lesions. However, penetration is limited, and
its utility may be restricted to superficial, bright red tumors. Interferon-α-2A (1 to 3 million units/m2/day) generally has been reserved for lesions that are life- or sight-threatening
or cause severe facial distortion.81 Significant risks include bone marrow suppression, liver damage, and neurotoxicity. Deans and associates82 described surgical dissection for carefully selected cases. Tumors must
be relatively circumscribed and deep enough that a surgical plane can
be developed without causing necrosis of overlying tissues. To minimize
the risk of hemorrhage, the entire surface of the lesion must be dissected, with
pinpoint bipolar cautery of draining veins and feeding
arteries. Direct incisions into the lesion are avoided. LYMPHANGIOMA A lymphangioma consists of an interanastomosing network of channels that
are each defined by thin septa lined by endothelial cells (Fig. 11). The lumens contain proteinaceous material that probably represents local
transudation. This fluid has the appearance, if not the physiologic
function, of lymph. The designation of these lesions as lymphatic malformations
also derives from the variable presence of lymphoid follicles. Conflicts
of terminology regarding developmental vascular malformations
of the orbit, particularly lymphangiomas and primary varices, have
largely derived from strict histopathologic definitions. As a step
toward uniform nomenclature, a classification based on hemodynamic relationships
has been recommended.63,83,84 The septa of a lymphangioma do contain small nutrient arteries and veins, but
there is little communication between the systemic circulation
and the actual channels of the tumor. Neither arteriography nor venography
causes filling of these spaces, and increases in orbital venous
pressure, as with crying, do not cause the twofold to threefold increases
in size observed with orbital varices. The fragility of the septa, with
their intrinsic vascular supply, may explain the characteristic
hemorrhages that occur in lymphangiomas. Bleeding into a lumen may produce
a single hemorrhagic cyst or a mass that is multilobulated because
of the intercommunication of vascular channels (Fig. 12). The sparse stroma allows dramatic expansion of blood cysts. 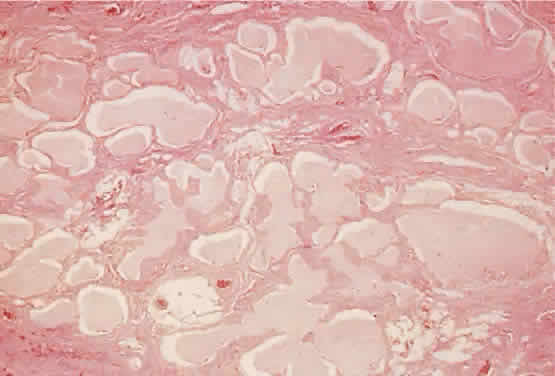 Fig. 11. The labyrinthine structure of a lymphangioma appears as multiple microcysts
in histologic sections. The lumens contain pale staining lymphlike
fluid. The channels extend into surrounding normal tissue without circumscription
or encapsulation (hematoxylin-eosin; × 40). Fig. 11. The labyrinthine structure of a lymphangioma appears as multiple microcysts
in histologic sections. The lumens contain pale staining lymphlike
fluid. The channels extend into surrounding normal tissue without circumscription
or encapsulation (hematoxylin-eosin; × 40).
|
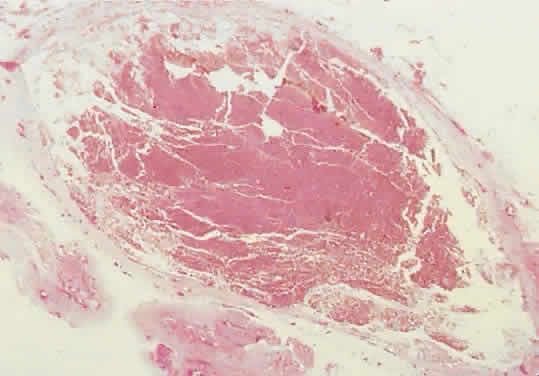 Fig. 12. Bleeding into a lumen produces a hemorrhagic macrocyst. The sparse stroma
and absence of a tumor capsule permit dramatic expansion, and proptosis
may appear abruptly in a previously unrecognized case (hematoxylin-eosin; × 15). Fig. 12. Bleeding into a lumen produces a hemorrhagic macrocyst. The sparse stroma
and absence of a tumor capsule permit dramatic expansion, and proptosis
may appear abruptly in a previously unrecognized case (hematoxylin-eosin; × 15).
|
Unlike capillary hemangiomas, lymphangiomas have a stable population of
endothelial cells.61 Proliferation does not exceed the rate of overall body growth, and enlargement
of the basic lesion ceases after adolescence. Regression does
not occur. However, proptosis may be intermittent and variable because
of recurrent intrinsic hemorrhage and blood resorption. Variations in
proptosis may parallel upper respiratory tract infections and are attributed
to lymphoid hyperplasia in response to immune challenge.85 A history of such variation often cannot be elicited, however. Although all orbital lymphangiomas are probably congenital, they often
do not become clinically manifest until the first hemorrhagic episode (Fig. 13). Most orbital cases are apparent within the first decade of life, with
an average age of presentation of 6 years.86 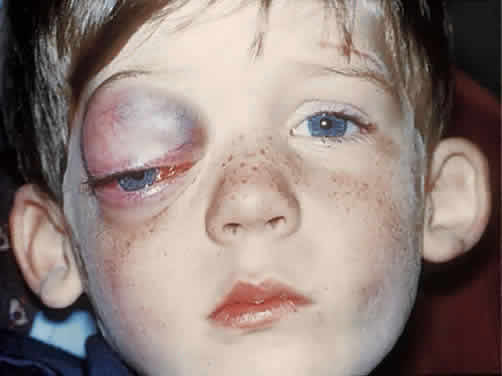 Fig. 13. This 4-year-old boy had rapid progression of inferior globe displacement, proptosis, pain, and
diplopia. Massive blood cysts had formed in an
underlying lymphangioma. Fig. 13. This 4-year-old boy had rapid progression of inferior globe displacement, proptosis, pain, and
diplopia. Massive blood cysts had formed in an
underlying lymphangioma.
|
Acute blood cyst formation in this age group makes the distinction between
a pre-existent but clinically silent lymphangioma and a rapidly emerging
rhabdomyosarcoma a common orbital diagnostic problem. Evidence
suggesting an orbitallymphangioma includes the variable finding of conjunctival
or eyelid components of the malformation.86 Conjunctival lesions appear as ectatic channels filled with clear or hemorrhagic
fluid. Eyelid ecchymosis may result from the seepage of blood
out of the thin-walled orbital cysts. Additional developmental anomalies
of the eye and adnexa may be present. Other head and neck involvement
may be manifest as local hypertrophy (e.g., of the cheek or lips), and
cystic palatal lesions may be seen. CT discloses a single or multilobulated mass, which represents only the
blood cyst portion of the tumor (Fig. 14). Individual lobules may have different radiodensities depending on the
presence of clots or liquefied blood within each cyst (Fig. 15). A generalized increase in orbital dimensions suggests a long-standing, probably
congenital process. Echography may help differentiate the
cystic components of lymphangioma from cellular rhabdomyosarcoma. Echography
shows the blood cysts to be acoustically inactive spaces, with
extremely low internal reflectivity (Fig. 16). Clots within the cysts can increase internal heterogeneity, however. MRI
has virtually eliminated the need for diagnostic biopsy in this condition, because
of its ability to show differing magnetic properties
of suspended, degrading blood products (Fig. 17). 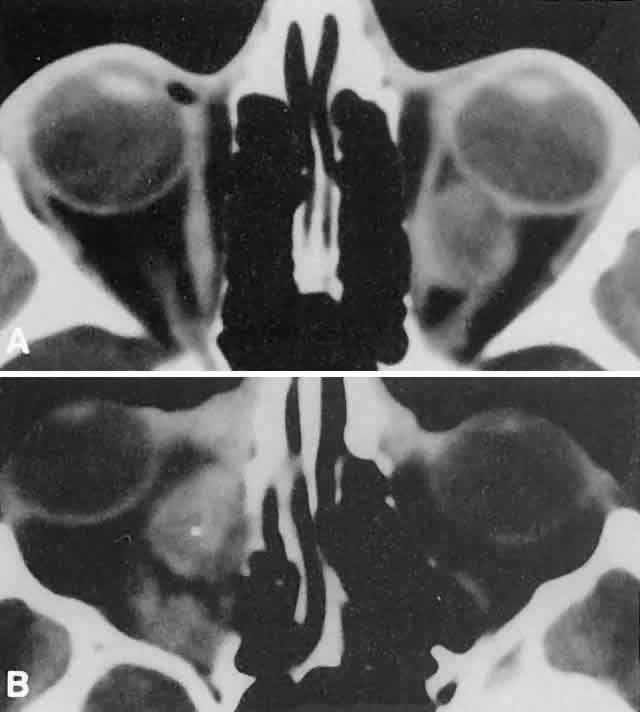 Fig. 14. CT appearance of hemorrhagic cysts in two different cases. A. A single intraconal cyst compressing the optic nerve. B. Multilobulated contiguous cysts. Intervening, nonexpanded segments are
microscopic and not detectable with imaging studies. Fig. 14. CT appearance of hemorrhagic cysts in two different cases. A. A single intraconal cyst compressing the optic nerve. B. Multilobulated contiguous cysts. Intervening, nonexpanded segments are
microscopic and not detectable with imaging studies.
|
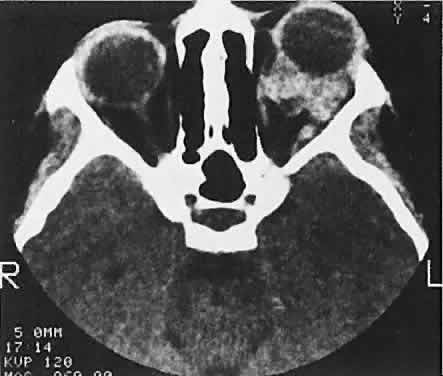 Fig. 15. Lymphangioma with heterogeneous radiodensities. Clots were found in the
denser anterior blood cysts, whereas the most posterior cyst had liquid
contents. Fig. 15. Lymphangioma with heterogeneous radiodensities. Clots were found in the
denser anterior blood cysts, whereas the most posterior cyst had liquid
contents.
|
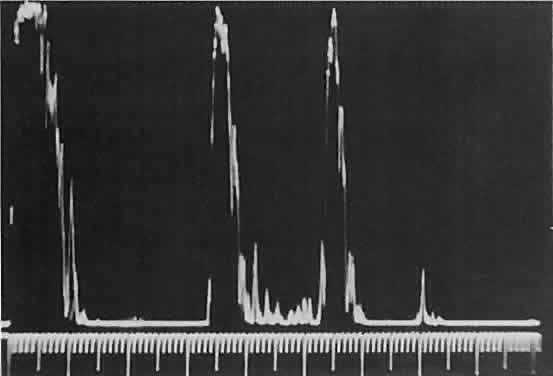 Fig. 16. Standardized A-scan echography shows low internal reflectivity and no decrement
in sound energy transmission, which is consistent with a fluidlike
cystic structure. Fig. 16. Standardized A-scan echography shows low internal reflectivity and no decrement
in sound energy transmission, which is consistent with a fluidlike
cystic structure.
|
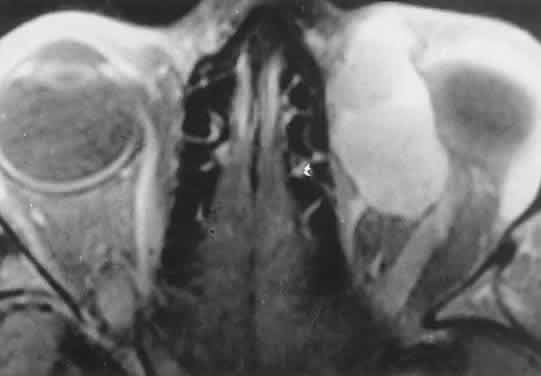 Fig. 17. MRI scan of a 17-year-old girl with abrupt-onset proptosis. Multilobulated
cystic spaces with fluid-fluid levels suggest recent hemorrhage within
a previously unrecognized lymphangi-oma. Fig. 17. MRI scan of a 17-year-old girl with abrupt-onset proptosis. Multilobulated
cystic spaces with fluid-fluid levels suggest recent hemorrhage within
a previously unrecognized lymphangi-oma.
|
The intimate association of orbital lymphangiomas with structures critical
to normal vision makes their complete excision almost impossible without
incurring vision loss. Because their vascular components do not
actively proliferate, the response to radiation therapy is limited and
probably is proportionate to whatever lymphoid tissue is present. The
presence of a blood cyst is not in itself an indication for treatment
if vision is not impaired. In many cases, the blood resorbs during several
weeks without residual problems. Frequently, however, vision is
compromised by the sudden expansion of multilobulated cysts that surround
the optic nerve, and simple observation may result in permanent deficits. Treatment
requires evacuation of the offending cysts in a conservative
manner consistent with preservation of vision.84 Because the channels of a lymphangioma are hemodynamically isolated from
the systemic circulation (“no flow anomalies”),63 their surgical decompression does not produce brisk new bleeding from
within them. Rather, the hemorrhagic risk of surgery involves intraoperative
and, more often, postoperative, intrinsic bleeding, creating new
blood-filled macrocysts.62 Conservative surgery restricts intraorbital manipulation, involves evacuation
of offending blood cysts, and avoids disturbance of nonexpanded
portions of the lymphangioma. Extensive dissection for cosmetic purposes
should be undertaken with the same respect for the fragility of nonexpanded
channels. TRAUMATIC ORBITAL HEMATOMA Although the diagnosis of a traumatic orbital hematoma would seem obvious
on the basis of history alone, some element of trauma within a few
days of the onset of proptosis is such a common historical finding among
small children that it may have little differential value. Conversely, a
history of culpable trauma may not always be forthcoming, as in
cases of child abuse. In penetrating orbital injuries, the entry wounds suggest the diagnosis. Retained
foreign bodies should be ruled out. In blunt injuries, other
diagnostic clues are helpful. Ecchymosis may be present but also may
be a feature of granulocytic sarcoma, neuroblastoma, or lymphangioma
with recent bleeding. CT may show an associated fracture. Most orbital
hematomas that result from blunt injury occur in the potential subperiosteal
space (Fig. 18). The lack of adjacent sinus opacification and the absence of systemic
toxicity differentiate this entity from a subperiosteal abscess, which
can have a similar appearance.59 Echography shows the low acoustic reflectivity characteristic of fluid-filled
spaces. 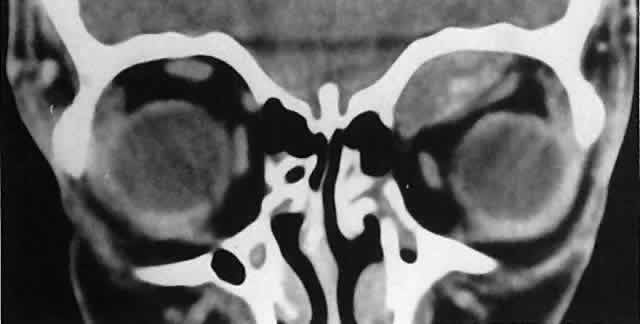 Fig. 18. Left superior subperiosteal hematoma without associated fracture. The denser
areas represent clots. (Harris GJ, Beatty RL: Acute proptosis in childhood. In Linberg JV [ed]: Oculoplastic
and Orbital Emergencies. Norwalk, CT: Appleton & Lange, 1989:97.) Fig. 18. Left superior subperiosteal hematoma without associated fracture. The denser
areas represent clots. (Harris GJ, Beatty RL: Acute proptosis in childhood. In Linberg JV [ed]: Oculoplastic
and Orbital Emergencies. Norwalk, CT: Appleton & Lange, 1989:97.)
|
If a traumatic orbital hematoma has compromised vision by acutely elevating
orbital pressure, the pressure should be reduced promptly with a
lateral canthotomy and cantholysis. If, conversely, vision is compromised
because of extreme globe displacement and optic nerve attenuation, the
hematoma should be evacuated.87 This is a relatively simple procedure if the blood is compartmentalized
in the subperiosteal space. We favor a lid crease incision, with dissection
between orbicularis muscle and orbital septum to the orbital rim. The
subperiosteal space is then entered, and the hematoma is evacuated. If
vision is not compromised, patients can be treated conservatively. Spontaneous
absorption generally follows, but hematomas occasionally
enlarge with osmotic imbibition. DERMOID CYST Dermoid and epidermoid cysts often occur in the orbit and paraorbital region. Epidermoid
cysts are lined by stratified squamous epithelium and
are filled with desquamated keratin. The walls of dermoid cysts include
dermal appendages that contribute sebum, sweat, and hair shafts to
the cyst contents. Both forms probably result from abnormal invagination
of surface ectoderm during fetal development. Differences may relate
to the depth of tissue that has been sequestered or to the degree of
ectodermal differentiation at the time of inclusion.88 Most dermoid cysts are closely related to bone suture lines, suggesting
that the surface ectoderm has been trapped between fusing mesodermal
processes. Dermoid cysts are most often encountered at the frontozygomatic articulation
but can occur at other suture lines, including those deep in the
orbit. Most lesions are anterior and paraorbital (Fig. 19), located between the orbicularis muscle and the periosteum overlying
the orbital rim, and have a fibrous stalk to the suture line. Anterior
cysts produce minimal bone change. Other lesions may be entirely intraorbital, causing
proptosis and globe displacement. Their expansion produces
an overall increase in orbital volume as well as local bone changes (Fig. 20). Dermoid and epidermoid cysts also may be largely intradiploic, with
expansion into the anterior cranial fossa, the temporal fossa, or the
orbit. Dumbbell lesions may be present with narrow intraosseous components. 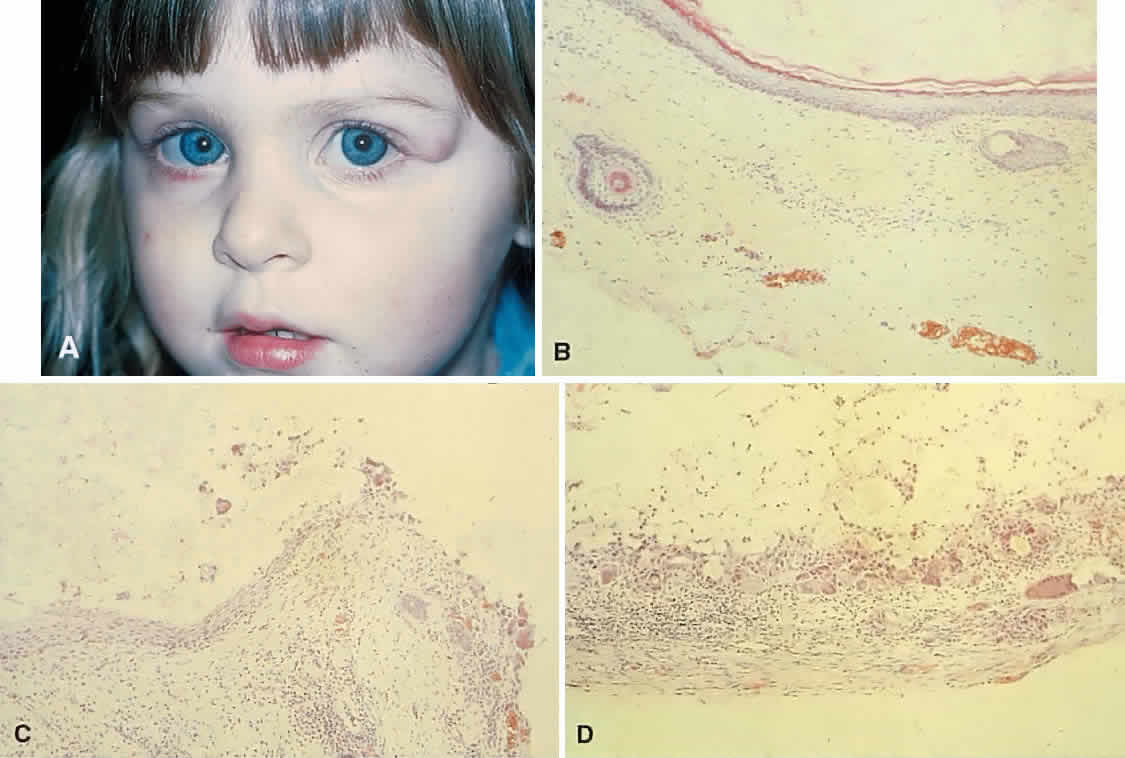 Fig. 19. A. This 2-year-old girl had a mass overlying the superotemporal orbital rim
since birth. The area had enlarged and had become red and tender in
the preceding few weeks. B. The recent clinical changes are explained by rupture of the cyst wall
and a granulomatous inflammatory response to the expelled contents. This
section shows typical stratified squamous epithelium, hair shafts in
the wall, and keratin in the lumen. C. Transitional zone between the dermoid cyst wall and an encapsulated granulomatous
response. D. The wall of the cyst beyond the point of rupture. Note the multinucleated
giant cells and fibrous capsule. (B-D, hematoxylin-eosin; × 96.) Fig. 19. A. This 2-year-old girl had a mass overlying the superotemporal orbital rim
since birth. The area had enlarged and had become red and tender in
the preceding few weeks. B. The recent clinical changes are explained by rupture of the cyst wall
and a granulomatous inflammatory response to the expelled contents. This
section shows typical stratified squamous epithelium, hair shafts in
the wall, and keratin in the lumen. C. Transitional zone between the dermoid cyst wall and an encapsulated granulomatous
response. D. The wall of the cyst beyond the point of rupture. Note the multinucleated
giant cells and fibrous capsule. (B-D, hematoxylin-eosin; × 96.)
|
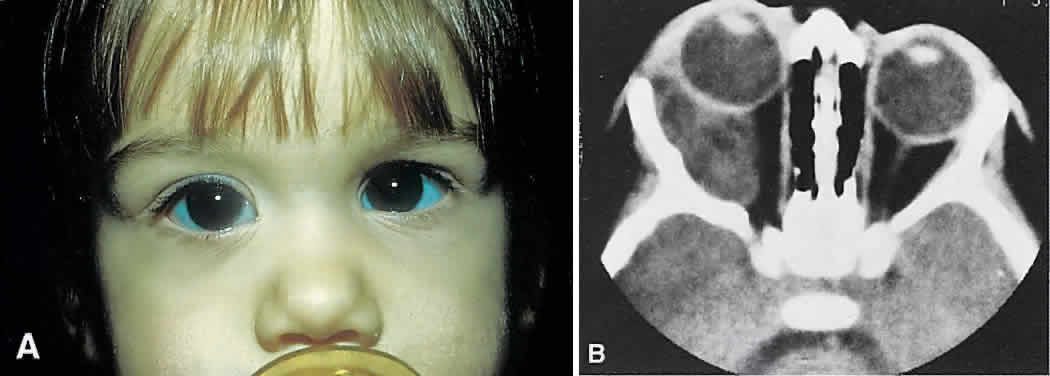 Fig. 20. A. Right proptosis was noted only a few weeks before presentation in this 3-year-old
girl. B. A long-standing process is suggested by overall expansion of the bony
orbit and local fossas on the orbital faces of the zygomatic and sphenoid
bones. Keratin clumps and glandular products within the dermoid cyst
have different radiodensities. Fig. 20. A. Right proptosis was noted only a few weeks before presentation in this 3-year-old
girl. B. A long-standing process is suggested by overall expansion of the bony
orbit and local fossas on the orbital faces of the zygomatic and sphenoid
bones. Keratin clumps and glandular products within the dermoid cyst
have different radiodensities.
|
Anterior, paraorbital dermoid cysts usually are evident soon after birth. Deeper
lesions may not declare themselves until mid- or late childhood, or
even the adult years. Expansion of the cysts generally is slow
and linear, reflecting continuous desquamation of keratinizing epithelium. There
may be a point at which the pressure within the cyst inhibits
further proliferation and sloughing of epithelial cells, accounting
for the clinically observed stability of many lesions. Sporadic enlargement
may be caused by hormonally influenced sebaceous gland secretion
or by rupture of the cyst wall with a granulomatous inflammatory response
to the cyst contents (see Figs. 19B-D). Such episodic change in an otherwise gradual growth pattern places intraorbital
dermoid cysts into the current differential diagnosis. Anterior lesions generally are diagnosed and removed without difficulty, although
their occasional occurrence near the lacrimal excretory system
can complicate treatment.89 Surgeons should strive for excision of an intact cyst, because residual
epithelial elements can lead to recurrence. CT examination of deeper
lesions discloses a cystic mass with some internal heterogeneity caused
by the different radiodensities of keratin clumps and oily secretions (see Fig. 20B). Bone changes, from shallow fossas to spherical defects, are smooth, with
a sclerotic margin and a punched-out appearance. Based on the CT
findings, the differential diagnosis includes cholesterol granuloma and
unifocal eosinophilic granuloma. Superomedial orbital dermoid cysts
must be distinguished from meningoencephaloceles before surgical intervention. Most intraorbital cysts can be removed through a lateral or anterior orbitotomy. The
walls of deep lesions may be intimately attached to adjacent
bone and may not peel off intact. Gentle use of a high-speed steel
burr can facilitate complete removal of the cyst lining. ANEURYSMAL BONE CYST Aneurysmal bone cyst is an uncommon, benign tumor-like lesion of unknown
origin.90 Most lesions present in the second decade with pain and swelling. Any
bone may be involved, but the long bones and vertebrae are most often
affected. Aneurysmal bone cyst of the orbital roof is an unusual cause
of rapidly progressive proptosis.91 In one case, a 16-month-old boy was affected.92 The lesions both erode and expand cancellous and cortical bone.90 They are surrounded by a shell of periosteal new bone that prevents their
extension into soft tissue. MRI may show fluid-fluid levels indicative
of hemorrhage.92 In some cases, aneurysmal bone cyst appears to be a pathophysiologic change
superimposed on a pre-existing lesion, such as a giant-cell tumor.90 In most cases, however, the bone cyst is considered a distinct pathologic
and radiologic entity. Treatment of facial lesions with intralesional
resection or curettage has a substantial rate of recurrence. Recurrence
can be reduced with marginal resection or cryotherapy. INFLAMMATORY PSEUDOTUMOR Idiopathic inflammatory pseudotumor (IIPT) is a general term applied to
those orbital inflammations without an identified inciting agent and
with a sparsely cellular, mixed inflammatory infiltrate that does not
suggest a systemic disease. Despite efforts to replace the pseudotumor designation, the term remains entrenched in the literature.93 However, the spectrum of clinical and pathologic conditions included under
the rubric has been narrowed and refined since the term was first
applied a century ago. IIPT can occur in the first 2 decades of life
as well as in adulthood, and it may affect children as young as 3 years
of age.94 There appears to be no sex predilection. The condition can be subdivided
topographically into myositis, dacryoadenitis, episcleritis/tenonitis/perineuritis, and
a localized mass. However, combined forms are common, and
even when the process is centered in one structure, inflammatory
changes appear microscopically and in imaging studies to spill into
adjacent tissues. Among these variants, orbital myositis and dacryoadenitis
are the most common forms of IIPT encountered in children. Local
tumefactions may occur anywhere in the orbit. When they involve the
crowded orbital apex or superior orbital fissure, they can produce the
Tolosa-Hunt syndrome of painful ophthalmoplegia. The typical patient with IIPT has an abrupt onset of pain, proptosis, eyelid
edema, chemosis, and conjunctival vascular engorgement.94 The left orbit is affected twice as often as the right, but bilateral
orbital involvement, either simultaneous or separated by variable intervals, occurs
in almost half of the pediatric cases. Among children with
IIPT, there is a higher incidence of iritis than among adults with
this disorder. Optic nerve head edema is noted in one-third of cases. Systemic
complaints are variable but may include fever, malaise, anorexia, and
nausea. Orbital symptoms may follow an upper respiratory tract
infection. In pediatric cases, laboratory abnormalities may include
peripheral blood eosinophilia and elevations of the erythrocyte sedimentation
rate, complement level, and antinuclear antibody titer.95 The absence of a marked leukocytosis with a left shift should help differentiate
this condition from bacterial orbital cellulitis. Orbital myositis may represent a greater proportion of cases of IIPT in
childhood than in adulthood, and involvement of multiple extraocular
muscles may occur more frequently in children than inadults. In orbital
myositis, early diplopia and increased discomfort with attempted eye
movement are typical symptoms. CT may show enlargement of one or more
extraocular muscles in one or both orbits (Figs. 21 and 22). When a single muscle is involved, the specter of a primary or metastatic
neoplasm within the muscle may be raised. However, external inflammatory
signs, considerable pain and limited motility, and an explosive
onset of symptoms within 24 hours all suggest orbital myositis. The
uniform enlargement of the muscle, including its tendinous insertion (see Fig. 22), also helps distinguish the process from a neoplasm, which might be expected
to produce a more focal, globular expansion. Echography may support
the diagnosis of inflammation by showing edema in the episcleral
space as a relative sonolucency between the scleral and orbital fat echoes (Fig. 23). Its CT counterpart is an increase in the radiodensity and thickness
of the ocular tunica. 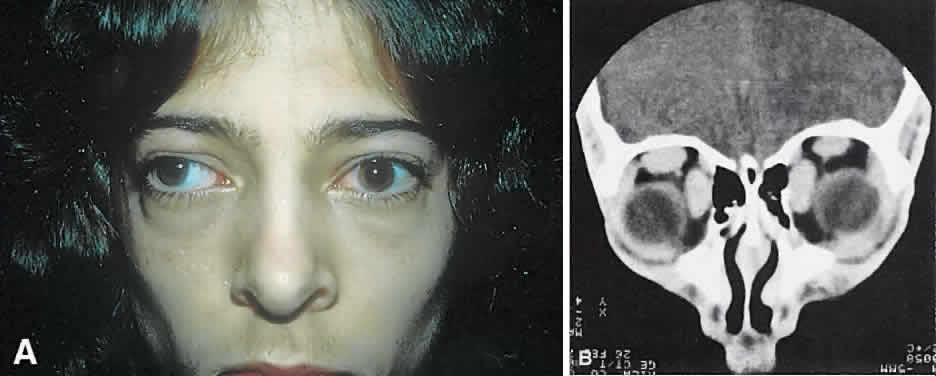 Fig. 21. A. This 16-year-old boy had acute onset of bilateral proptosis, pain, diplopia, chemosis, and
conjunctival injection. B. Bilateral enlargement of the superior and medial rectus and inferior oblique
muscles. Other sections showed similar involvement of other extraocular
muscles. Fig. 21. A. This 16-year-old boy had acute onset of bilateral proptosis, pain, diplopia, chemosis, and
conjunctival injection. B. Bilateral enlargement of the superior and medial rectus and inferior oblique
muscles. Other sections showed similar involvement of other extraocular
muscles.
|
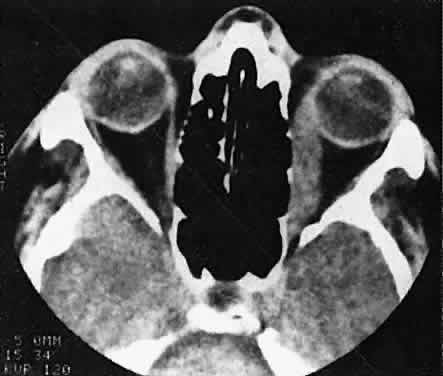 Fig. 22. The uniform enlargement of the left medial rectus muscle, including its
tendinous insertion, is characteristic of orbital myositis. Fig. 22. The uniform enlargement of the left medial rectus muscle, including its
tendinous insertion, is characteristic of orbital myositis.
|
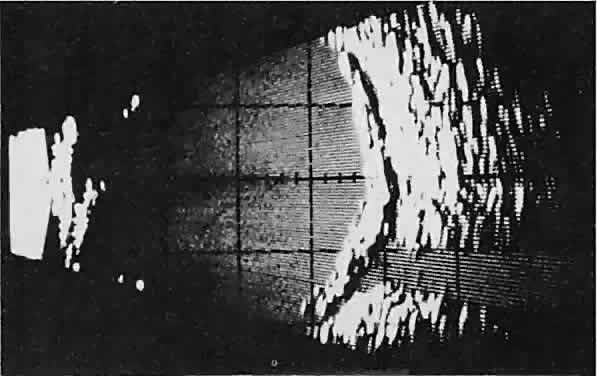 Fig. 23. Acoustic discontinuity between the globe and the orbital fat indicates
inflammatory edema in Tenon's space. Fig. 23. Acoustic discontinuity between the globe and the orbital fat indicates
inflammatory edema in Tenon's space.
|
In dacryoadenitis, external inflammatory signs are localized to the superotemporal
quadrant, and CT shows enlargement of the lacrimal gland (Fig. 24). Lacrimal gland inflammation may be bacterial, viral, or a variant of
IIPT. It is possible, however, that many cases of “idiopathic” dacryoadenitis
represent unidentified viral infections. In bacterial
dacryoadenitis, a leukocytosis with a left shift may be present.96 In questionable cases, a 1-week course of oral antibiotics can be administered
to these patients. Among children, the probability that an enlarged
lacrimal gland represents neoplasia rather than inflammation is
lower than among adults, although epithelial lacrimal gland tumors occasionally
may occur in the pediatric population and can produce external
inflammatory signs. If the general signs and symptoms of IIPT are
lacking, a biopsy should be performed. 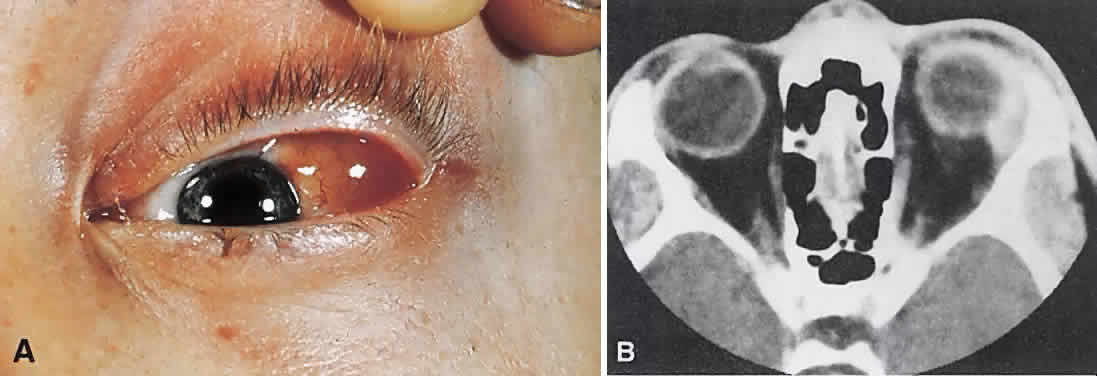 Fig. 24. A. Nonbacterial dacryoadenitis may be unilateral or bilateral. External inflammatory
signs are maximal in the superotemporal quadrant. B. The left lacrimal gland is enlarged, with a shape molded by the globe
and orbital walls. A neoplasm usually can be ruled out by analysis of
the history, CT findings, and echographic characteristics, but a biopsy
may be required in equivocal cases. Fig. 24. A. Nonbacterial dacryoadenitis may be unilateral or bilateral. External inflammatory
signs are maximal in the superotemporal quadrant. B. The left lacrimal gland is enlarged, with a shape molded by the globe
and orbital walls. A neoplasm usually can be ruled out by analysis of
the history, CT findings, and echographic characteristics, but a biopsy
may be required in equivocal cases.
|
Histopathologically, IIPT is characterized by a sparse, mixed inflammatory
cell infiltration of the tissues primarily involved (i.e., extraocular
muscle, lacrimal gland, Tenon's fascia). The predominant cell
is a mature lymphocyte, but there are significant numbers of polymorphonuclear
neutrophils, plasma cells, and eosinophils.95 As the inflammatory process evolves, fibrosis becomes a prominent feature. In
the variant of IIPT termed sclerosing pseudotumor, collagen deposition is an early finding, and the fibroblast may be the
primary mediator of the inflammatory process rather than the lymphocyte.97,98 If the histopathologic findings include true vasculitis (i.e., destruction
of vessel wall intima and muscularis) or granulomatous inflammation (i.e., epithelioid
and giant cells), Wegener's granulomatosis
and other systemic diseases should be excluded. By definition, if a systemic
process is confirmed, the IIPT designation no longer applies. If
the microscopic picture is dominated by a highly cellular population
of uniform lymphocytes, the spectrum of reactive and neoplastic lymphoid
lesions should be suspected rather than IIPT. IIPT usually shows a dramatic response to high doses of oral corticosteroids. Clinical
improvement occurs within several days, but treatment
should be tapered slowly to prevent recrudescence of the inflammation. A
pediatrician should collaborate in the treatment of young children
with corticosteroids because of the risks of growth retardation and other
complications. Although the etiology remains unknown, this exquisite
treatment response adds credence to an immunologic basis. Patients
tend to follow one of three long-term clinical patterns: single unilateral
episodes; recurrent unilateral episodes; or recurrent bilateral episodes, usually
alternating from one orbit to the other.94 Among children with IIPT, bilateral involvement and an anterior uveal
component prognosticate a more severe course in terms of multiple recurrences
and permanent vision loss. Sclerosing pseudotumor, which may involve a distinctly different pathogenesis, generally
carries a poorer prognosis with a higher likelihood
of cicatricial entrapment of orbital structures.97 Early aggressive treatment with corticosteroids is indicated.Surgical
debulking may be necessary, and immunosuppressive agents may be needed
in refractory cases. ORBITAL CELLULITIS Orbital cellulitis and its variants are the most common causes of rapidly
progressive proptosis in childhood. The term orbital cellulitis often is applied broadly to an anatomic spectrum of bacterial infection, including
preseptal cellulitis, diffuse orbital cellulitis, subperiosteal
abscess (SPA), intraorbital abscess and, in rare complicated or
neglected cases, cavernous sinus thrombosis. In children, possible etiologies
include penetrating trauma, extension of local periocular infection (e.g., impetigo
or dacryocystitis), and hematogenous seeding from
a distant site (e.g., otitis media).99 However, orbital cellulitis most often results from bacterial infection
of the paranasal sinuses, which share insubstantial bony walls and an
extensive valveless venous system with the orbits.59,100 The clinical profile includes eyelid edema and erythema. If there is true
orbital involvement, there may be proptosis, chemosis, and diminished
motility. Diagnosis is aided by a history of antecedent respiratory
tract infection and signs of systemic toxicity, including fever and
leukocytosis. The CT findings of sinus opacification and an orbital abnormality
suggest the diagnosis. However, rhabdomyosarcomas and neuroblastomas
may affect the orbits and sinuses simultaneously. In these cases, destructive
bone change might be expected. The initial recognition of orbital infection generally is not problematic. However, appropriate
management requires other early determinations, including
proper staging and risk assessment. If CT scans show only
preseptal or orbital cellulitis, the prompt administration of appropriate
intravenous antibiotics to these well-perfused tissues should be
curative. If, however, infection is sequestered in the relatively avascular
subperiosteal space (Fig. 25), concerns are raised about achieving therapeutic drug levels.59 An SPA also may have visual implications. The rapid accumulation and extension
of purulent material within this potential space can increase
orbital pressure, compromising optic nerve or retinal perfusion. There
also are well-documented age-related variations in both the bacteriology
and clinical response of the SPA/sinusitis complex.101,102 Children younger than 9 years of age are more likely to improve without
surgical drainage of the sinuses or orbit, to have negative cultures
if drained, or to have cultures positive for single aer-obes if drained
within the first 3 days of treatment. Patients 15 years of age or older
are more likely to have refractory infections, with positive cultures
after more than 3 days of antibiotics usually effective in vitro, and to harbor multiple pathogens, including mixed aerobes and anaerobes. The 9- to 14-year-old
age group shows a transition from simple to complex
infections. 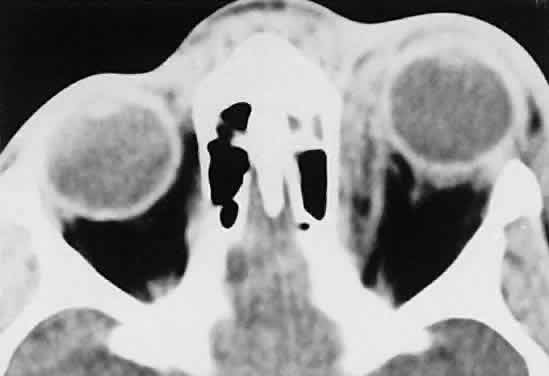 Fig. 25. A 6-year-old boy with a clinical diagnosis of left orbital cellulitis. A
medial sub-periosteal abscess is present, secondary to a seemingly minor
infection of the anterior ethmoid complex. (Harris GJ, Beatty RL: Acute proptosis in childhood. In Linberg JV [ed]: Oculoplastic
and Orbital Emergencies. Norwalk, CT: Appleton & Lange, 1989:100.) Fig. 25. A 6-year-old boy with a clinical diagnosis of left orbital cellulitis. A
medial sub-periosteal abscess is present, secondary to a seemingly minor
infection of the anterior ethmoid complex. (Harris GJ, Beatty RL: Acute proptosis in childhood. In Linberg JV [ed]: Oculoplastic
and Orbital Emergencies. Norwalk, CT: Appleton & Lange, 1989:100.)
|
Management includes otolaryngology consultation: nasal decongestion promotes
nonsurgical sinus drainage; if surgery is needed, the orbit and
sinuses should be drained simultaneously. Antibiotics are given intravenously. At
present, appropriate choices include ampicillin/sulbactam
for all age groups or a third-generation cephalosporin for children younger
than 9 years of age, with the addition of clindamycin for patients 9 years
of age or older. Because the inventory of available drugs is
continually changing, consultation with infectious disease specialists
may be appropriate. Surgical drainage of an SPA and the responsible
sinuses is performed as soon as possible if optic nerve or retinal function
is impaired by the mass effect (any age). Surgical drainage within 24 hours
of presentation is recommended for large SPAs causing pain, for
those along the superior or inferior orbital walls, for patients
with frontal sinusitis, and in cases in which anaerobic pathogens are
suspected (e.g., infections of known dental origin, chronic sinusitis, patients 9 years
of age or older). In the absence of these surgical
criteria, expectant observation, with inpatient antibiotic administration, is
elected for children younger than 9 years of age with small- to
moderate-sized medial SPAs.101,102 This approach requirescareful monitoring, and conservatively treated patients
still default to surgery if a prompt clinical response is not
noted. This judgment should not be made on the basis of serial CT scans
alone, since SPAs may enlarge during the first few days of antibiotic
therapy that ultimately proves effective.101,103 Garcia and Harris104 prospectively applied this protocol to a cohort of 37 patients younger
than 9 years of age. Eight children met criteria for surgical treatment
and underwent prompt drainage. Of the 29 patients for whom initial
nonsurgical treatment was recommended, 27 (93%) recovered with antibiotics
alone, and two defaulted to surgery. All patients had successful
clinical outcomes. |





