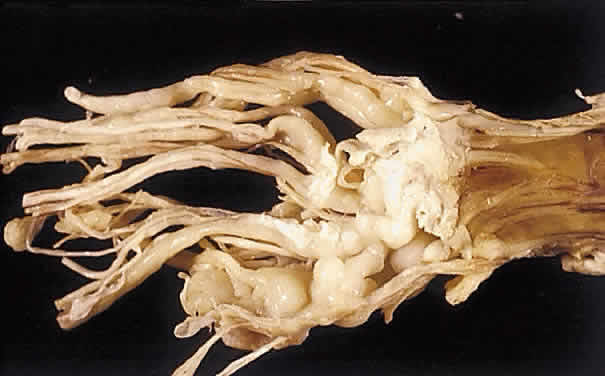
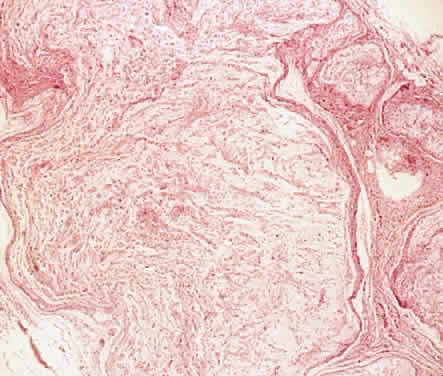
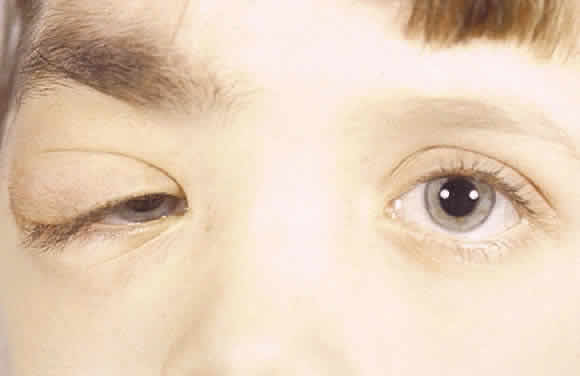
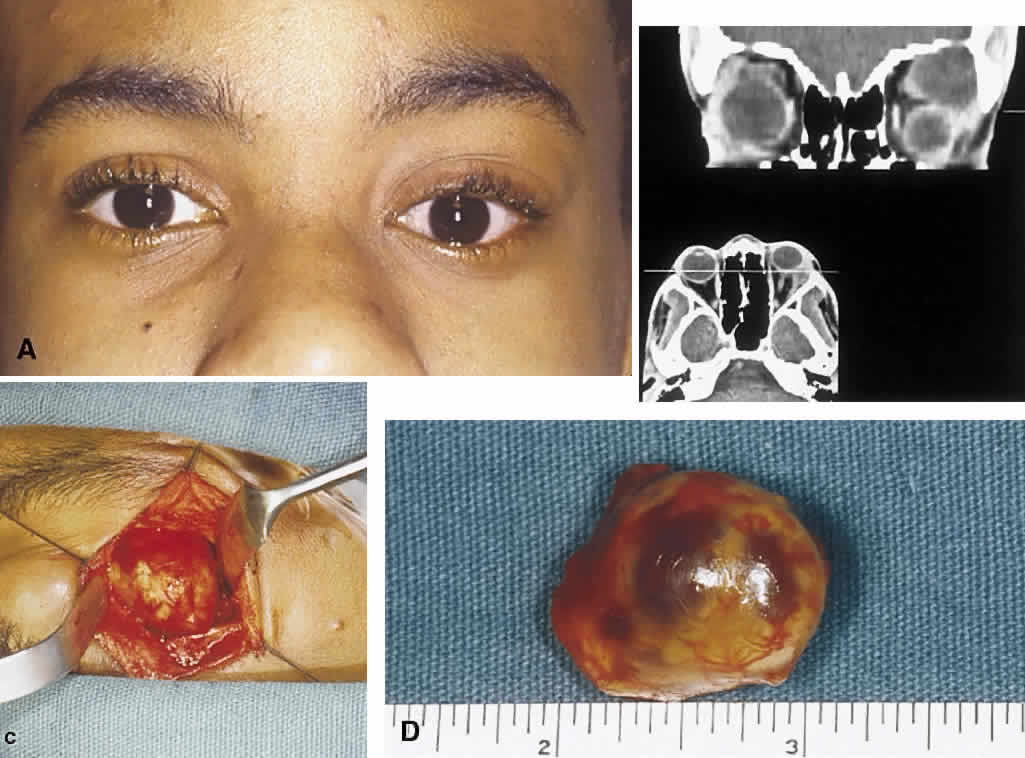
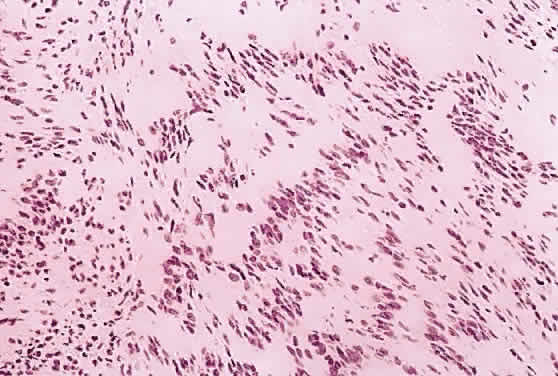
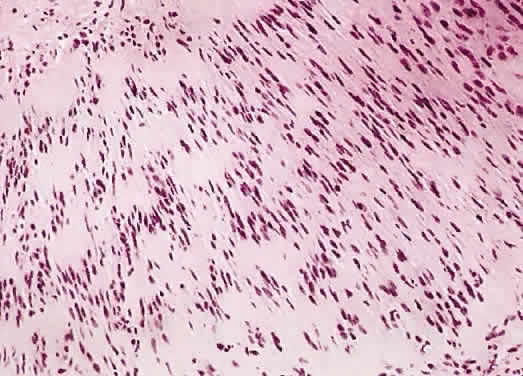
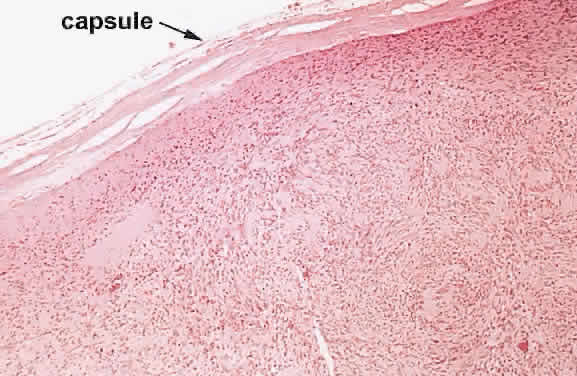
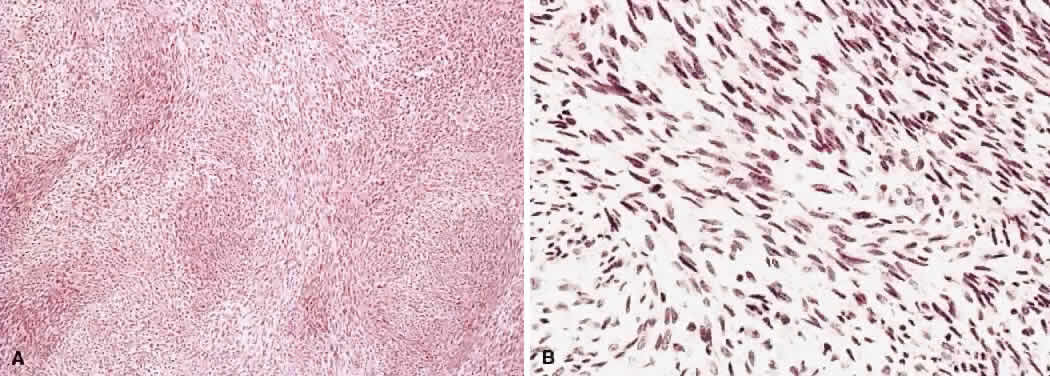
1. Levin LA, Jakobiec FA: Peripheral nerve sheath tumors of the orbit. In
Albert DM, Jakobiec FA (eds): Principles and Practice of Ophthalmology: Clinical
Practice, p 1978. Philadelphia: WB Saunders, 1994 2. Jakobiec FA, Bilyk JR, Font RL: Orbit. In Spencer WH (ed): Ophthalmic Pathology: An
Atlas and Text, p 2438. 4th ed. Philadelphia: WB Saunders, 1996 3. Shields JA, Shields CL, Lieb WE: Multiple orbita neurofibromas unassociated with von Recklinghausen's
disease. Arch Ophthalmol 108:80, 1990 4. Krohel GB, Rosenberg PN, Wright JE: Localized orbital neurofibromas. Am J Ophthalmol 100:458, 1985 5. Cantore G, Cipetta P, Raco A, et al: Orbital schwannomas: Report of nine cases and review of the literature. Neurosurgery 19:583, 1986 6. Enzinger FM, Weiss SW: Soft Tissue Tumors. 3rd ed. St. Louis: Mosby, 1995 7. Wanebo JE, Malik JM, Vandenberg SR et al: Malignant peripheral nerve sheath tumors. Cancer 71:1247, 1997 8. Jakobiec FA, Font RL, Zimmerman LE: Malignant peripheral nerve sheath tumors of the orbit: A clinicopathologic
study of eight cases. Trans Am Ophthalmol Soc 83:332, 1985 9. Meis JM, Enzinger FM, Martz KL et al: Malignant peripheral nerve sheath tumors (malignant schwannomas) in children. Am J Surg Pathol 16:694, 1992 10. Lack E, Cubilla AL, Woodruff JM et al: Paragangliomas of the head and neck. Cancer 39:397, 1977 11. Netland PA, Font R, Jakobiec FA: Rare intraosseous and primary orbital
tumors, in Albert DM, Jakobiec FA (eds): Principles and Practice of Ophthalmology: Clinical
Practice, p 2051. Philadelphia: WB Saunders, 1994 12. Amemiya T, Kadoya M: Paraganglioma of the orbit. J Cancer Res Clin Oncol 96:169, 1980 13. Goldstein BG, Font RL, Alper MG: Granular cell tumor of the orbit: a case report including electron microscopic
observations. Ann Ophthalmol 14:231, 1982 14. Rosai J: Adrenal gland and paraganglia. In Rosai J (ed): Ackerman's
Surgical Pathology, p 1015. 8th ed. St. Louis: Mosby Year Book Inc, 1996 15. Dehner LP: Peripheral and central primitive neuroectodermal tumors: a nosologic concept
seeking consensus. Arch Pathol Lab Med 110:997, 1986 16. Singh AD, Husson M, Shields CL: Primitive neuroectodermal tumor of the orbit. Arch Ophthalmol 112:217, 1994 17. Arora R, Sarkar C, Betharia SM: Primary orbital primitive neuroectodermal tumour with immunohistochemical
and electron microscopy confirmation. Orbit 12:7, 1993 18. Folberg R, Bernardino VB, Aguilar GL et al: Amputation neuroma mistaken for recurrent melanoma in the orbit. Ophthalmic Surg 12:275, 1981 19. Katenkamp D, Sullivan TJ: Ultrastructure of perineurial cells during peripheral nerve regeneration: Electron
microscopical investigations on the so-called amputation neuroma. Exp Pathol 16:5, 1978 20. Abrikossoff A: Uber myome ausgehend von der guergestreiften willkurlichen muskulatur. Virchows Arch A Pathol Anat Histopathol 260:215, 1926 21. Jaeger MJ, Green WR, Miller NR et al: Granular cell tumor of the orbit and ocular adnexa. Surv Ophthalmol 31:417, 1987 22. Karcioglu ZA, Hemphill GL, Wool BM: Granular cell tumor of the orbit: case report and review of the literature. Ophthalmic Surg 14:125, 1987 23. Morgan G: Granular cell myoblastoma: Report of a case. Arch Ophthalmol 94:2135, 1976 24. Drummond JW, Hall DL, Steen WH et al: Granular cell tumor (myoblastoma) of the orbit. Arch Ophthalmol 97:1492, 1979 25. Charles NC, Fox DM, Glasberg SS et al: Epibulbar granular cell tumor: report of a case and review of the literature. Ophthalmology 104:1454, 1997 26. Ferry AP: Granular cell tumor (myoblastoma) of the palpebral conjunctiva causing
pseudoepitheliomatous hyperplasia of the conjunctival epithelium. Am J Ophthalmol 91:234, 1981 27. Pe'er J, Schwartzenberg T, Kopolovic J: Granular cell tumor (myoblastoma) of the eyebrow. Ophthalmology 194:1, 1987 |