The prevalence of tuberous sclerosis in the general population is at least 1 in 10,000.6 About one third of cases are familial, whereas two thirds are sporadic. There is no recognized racial predilection, and the sexes are affected equally. Signs and symptoms of tuberous sclerosis usually begin before the patient is 6 years old.
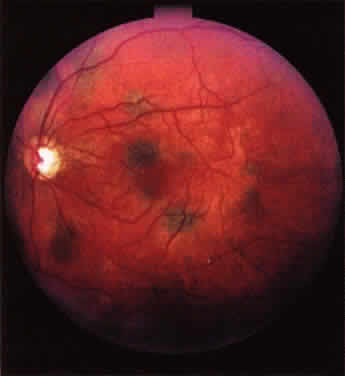
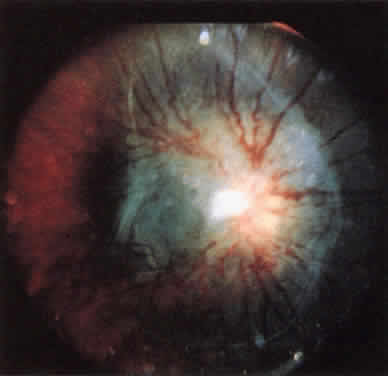
The classic ophthalmoscopic feature of tuberous sclerosis is the retinal astrocytoma (astrocytic hamartoma).7 This lesion arises within the nerve fiber layer of the retina. It appears more frequently in the posterior fundus than in the periphery. Small lesions commonly appear as translucent intraretinal patches with minimal thickness. Slightly larger lesions usually appear more opalescent or even opaque white. Large lesions tend to be opaque white, substantially elevated, and frequently multilobulated (Fig. 1). Intralesional calcification develops within some larger lesions. Multiple lesions can be present in one eye, and many affected patients have binocular involvement. The retinal blood vessels associated with these lesions tend not to be dilated or tortuous in most patients. If lesion growth occurs, it tends to be extremely slow. Approximately one half of patients with tuberous sclerosis have at least one typical astrocytic retinal hamartoma.8
|
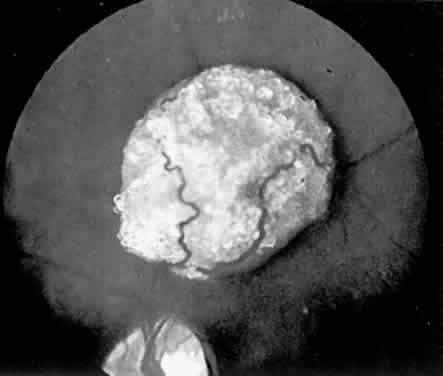
Lesions similar to those that occur in the retina commonly develop in the cerebrum and less commonly develop in the basal ganglion, brain stem, and cerebellum. Both retinal and central nervous system (CNS) lesions can be calcified and identified radiologically.9 Epilepsy and mental deficiency are common in severely affected individuals, and early death occurs in occasional patients as a result of progressive CNS tumor expansion or status epilepticus.5
A variety of unusual tumors develop in the heart, kidney, lungs, thyroid, and other visceral organs in some patients with tuberous sclerosis.5 The classic cardiac tumor is the rhabdomyoma, which can lead to conduction abnormalities, heart failure, and premature death. Angiomyofibromas tend to develop in the kidney, and these lesions can lead to death from renal failure.
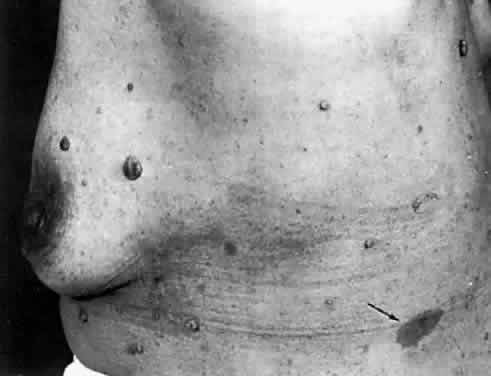
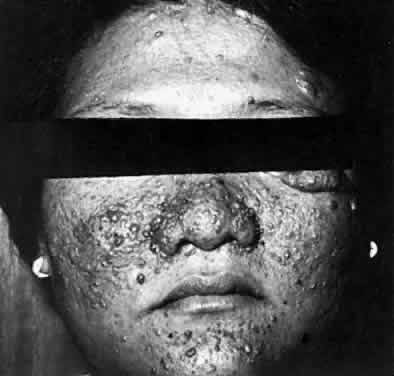
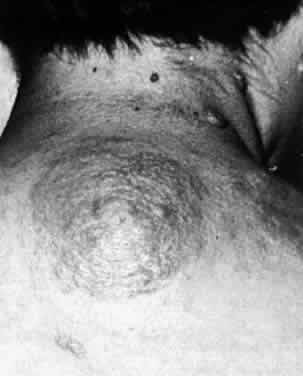
Several cutaneous lesions are prominent features of tuberous sclerosis. Adenoma sebaceum is an unusual facial dermatologic eruption characterized by pinhead- to pea-sized yellowish to reddish-brown papules distributed in butterfly fashion over the nose, cheeks, and nasolabial folds (Fig. 2). Histopathologically, the individual skin lesions are angiofibromas.10 Ash leaf spots are congenital white or hypomelanotic skin macules ranging from about 1 mm to several centimeters in diameter and having a configuration resembling an ash leaf (Fig. 3). These lesions are most easily demonstrated under ultraviolet light (Wood's lamp).11 The shagreen patch is a thickened patch of skin with the texture of pigskin or sharkskin, usually occurring over the lower back (Fig. 4). Subungual fibromas are benign fibrous tumors that develop at the sides of the nail beds in some patients.
|
|

|
Treatment of affected individuals is purely symptomatic at present. Death tends to occur in many severely affected individuals by the third decade as a result of the CNS lesions or visceral tumors.
Recent molecular biologic studies have identified loci on the long arm of chromosome 9 (9q32-34), on the long arm of chromosome 11, on the short arm of chromosome 16 (16p13.3), and on the long arm of chromosome 12 (12q22-24.1) as tuberous sclerosis genes in different multigenerational families that have been studied.6 Of these loci, the 9q32-34 locus has been the most consistent, being associated with one third to one half of all familial cases.
Systemic evaluation of individuals suspected of having tuberous sclerosis should probably include fundus examination, dermatologic evaluation to identify characteristic skin lesions, magnetic resonance imaging (MRI) of the central nervous system,12 and computed tomography (CT) or MRI of the abdominal viscera. Examination of family members to look for a familial pattern is also appropriate.