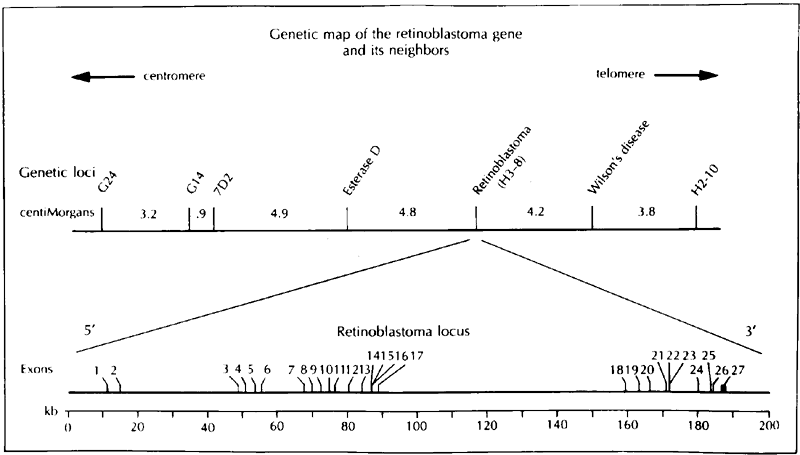
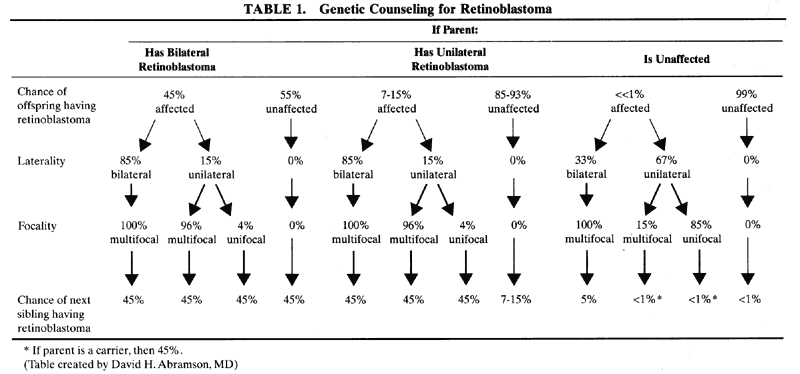
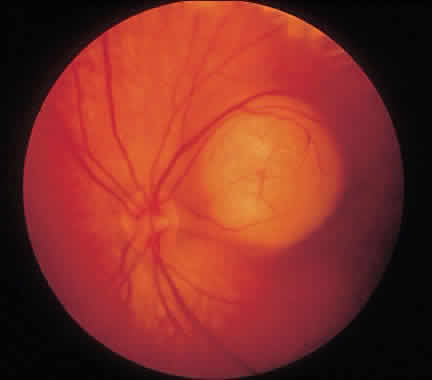
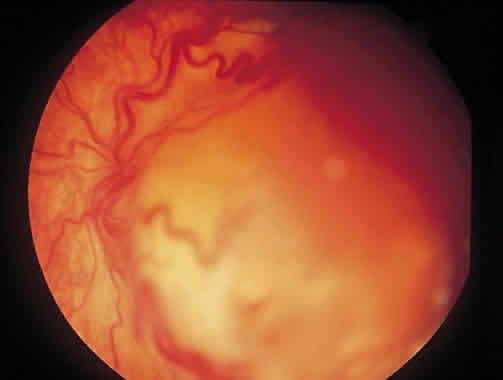
1. Francois J, Matton-Van Leuven MT: Recent data on the heredity of retinoblastoma. In
Boniuk M (ed): Ocular and Adnexal Tumors: New and Controversial
Aspects. St Louis: CV Mosby, 1964 2. Freedman J, Goldberg L: Incidence of retinoblastoma in the Bantu of South Africa. Br J Ophthalmol 60:655–656, 1976 3. Koch E, Naeser P: Retinoblastoma in Sweden 1958-1971: A clinical and histopathological study. Acta Ophthalmol 57:344–350, 1979 4. Suckling RD, Fitzgerald PH, Stewart J, Wells E: The incidence and epidemiology of retinoblastoma in New Zealand: A 30-year
survey. Br J Cancer 46:729–736, 1982 5. Sang DN, Albert DM: Recent advances in the study of retinoblastoma. In
Peyman GA, Apple DJ, Sanders DR (eds): Intraocular Tumors, pp 285–329. New
York: Appleton-Century-Crofts, 1977 6. Suckling RD, Fitzgerald PH: An epidemiological study of retinoblastoma in New Zealand. Trans Ophthalmol Soc NZ 24:17–21, 1972 7. Albert DM, Lahav M, Lesser R, Craft J: Recent observations regarding retinoblastoma: Ultrastructure, tissue culture
growth, incidence, and animal models. Trans Ophthalmol Soc UK 94:909–928, 1974 8. Pendergrass TW, Davis S: Incidence of retinoblastoma in the United States. Arch Ophthalmol 98:1204–1210, 1980 9. Abramson DH, Mendelsohn ME, Servodidio CA et al: Familial retinoblastoma: Where and when? Acta Ophthalmol Scand 76:334–338, 1998 10. Murphree AL, Benedict WF: Retinoblastoma: Clues to human oncogenesis. Science 223:1028–1033, 1984 11. Cavenee WK, Dryja TP, Phillips RA et al: Expression of recessive alleles by chromosomal mechanisms in retinoblastoma. Nature 305:779–784, 1983 12. Cavenee WK, Murphree AL, Skull MM: Prediction of familial predisposition to retinoblastoma. N Engl J Med 19: 1201, 1986 13. Dryja TP, Rapaport JM, Joyce JM, Petersen RA: Molecular detection of deletions involving band q14 of chromosome 13 in
retinoblastomas. Proc Natl Acad Sci USA 83:7391–7394, 1986 14. Hong FD, Huang HJ, To H et al: Structure of the human retinoblastoma gene. Proc Natl Acad Sci USA 86:5502–5506, 1989 15. Toguchida J, McGee TL, Paterson JC et al: Complete genomic sequence of the human retinoblastoma susceptibility gene. Genomics 17:535–543, 1993 16. Chellappan SP, Hiebert S, Mudryj M et al: The E2F transcription factor is a cellular target for the retinoblastoma
protein. Cell 65:1053–1061, 1991 17. Sellers WR, Kaelin WG Jr: Role of the retinoblastoma protein in the pathogenesis of human cancer. J Clin Oncol 15: 3301–3312, 1997 18. Knudson AG Jr: Mutation and cancer: Statistical study of retinoblastoma. Proc Natl Acad Sci USA 68:820–823, 1971 19. Li Y, Qiu J: Alterations of retinoblastoma gene and its protein expression in aggressive
bone tumors. Chung Hua Ping Li Hsueh Tsa Chih 26:262–265, 1997 20. Cryns VL, Thor A, Xu HJ et al:Loss of the retinoblastoma tumor-suppressor gene in parathyroid carcinoma. N Engl J Med 330:757–761, 1994 21. Cohen JA, Geradts J: Loss of retinoblastoma and MTS1. CDKN2 expression in human sarcomas. Hum Pathol 28:893–898, 1997 22. Maat-Kievit JA, Oepkes D, Hartwig NG et al: A large retinoblastoma detected in a fetus at 21 weeks of gestation. Prenat Diagn 13:377–384, 1993 23. Abramson DH, Servodidio CA: Retinoblastoma in the first year of life. Ophthalmic Pediatr Genet 13:191–203, 1992 24. Gallie BL, Ellsworth RM, Abramson DH, Phillips RA: Retinoma: Spontaneous regression of retinoblastoma or benign manifestation
of the mutation? Br J Cancer 45:513–521, 1982 25. Eagle RC Jr, Shields JA, Donoso L, Milner RS: Malignant transformation of spontaneously regressed retinoblastoma, retinoma, retinocytoma
variant. Ophthalmology 96:1389–1395, 1989 26. Abramson DH, Frank CM, Susman M et al: Presenting signs of retinoblastoma. J Pediatr 132:505–508, 1998 27. Zilelioglu G, Gunduz K: Ultrasonic findings in intraocular retinoblastoma and correlation with
histopathologic diagnosis. Int Ophthalmol 19:71–75, 1995 28. Basta LL, Israel CW, Gourley RD, Acers TE: Which pathologic characteristics influence echographic patterns of retinoblastoma? Ann Ophthalmol 13:585–588, 1981 29. Rehurek J, Snopkova J: Lactate dehydrogenase activity in the diagnosis of retinoblastoma. Cesk Oftalmol 51:14–18, 1995 30. Wang CY: Significance of the lactate dehydrogenase level in aqueous humor in the
diagnosis of retinoblastoma. Chung Hua Yen Ko Tsa Chih 25:289–291, 1989 31. Wu Z, Yang H, Pan S: Electrophoretic determination of aqueous and serum neuron-specific enolase (NSE) in
the diagnosis of retinoblastoma. Chung Hua Yen Ko Tsa Chih 32:219–223, 1996 32. Palazzi M, Abramson DH, Ellsworth RM: Endophytic vs. exophytic unilateral retinoblastoma: Is there any real difference? J Pediatr Ophthalmol Strabismus 27:255–258, 1990 33. Chantada GL, de Davila MT, Fandino A et al: Retinoblastoma with low risk for extraocular relapse. Ophthalmic Genet 20:133–140, 1999 34. Khelfaoui F, Validire P, Auperin A et al: Histopathologic risk factors in retinoblastoma: A retrospective study of 172 patients
treated in a single institution. Cancer 77:1206–1213, 1996 35. Shields CL, Shields JA, Baez KA et al: Choroidal invasion of retinoblastoma: metastatic potential and clinical
risk factors. Br J Ophthalmol 77:544–548, 1993 36. Howard GM: Invasion of choroid and sclera by retinoblastoma following photocoagulation. Trans Am Acad Ophthalmol Otolaryngol 70:984–989, 1966 37. Magramm I, Abramson DH, Ellsworth RM: Optic nerve involvement in retinoblastoma. Ophthalmology 96:217–222, 1989 38. Namouni F, Doz F, Tanguy ML et al: High-dose chemotherapy with carboplatin, etoposide and cyclophosphamide
followed by a haematopoietic stem cell rescue in patients with high-risk
retinoblastoma: A SFOP and SFGM study. Eur J Cancer 33:2368–2375, 1997 39. Decaussin M, Boran MD, Salle M et al: Cytological aspiration of intraocular retinoblastoma in an 11-year-old
boy. Diagn Cytopathol 19:190–193, 1998 40. Karcioglu ZA, Gordon RA, Karcioglu GL: Tumor seeding in ocular fine needle aspiration biopsy. Ophthalmology 92: 1763–1767, 1985 41. Dass AB, Trese MT: Surgical results of persistent hyperplastic primary vitreous. Ophthalmology 106:280–284, 1999 42. Pivetti-Pezzi P: Uveitis in children. Eur J Ophthalmol 6:293–298, 1996 43. Carden SM, Colville DJ, Gonis G, Gilbert GL: Kingella kingae endophthalmitis in an infant. Aust NZ J Ophthalmol 19:217–220, 1991 44. Chien SY, Sung TC, Mu SC, Hu CC: Endophthalmitis as a complication of meningococcal meningitis: Report of
one case. Chung Hua Min Kuo Hsiao Erh Ko I Hsueh Hui Tsa Chih 40:116–118, 1999 45. Saint-Blancat P, Morand I, Clabaut FX et al: Toxocara canis infection: 2 cases of peripheral granuloma in adults. J Fr Ophtalmol 20:252–257, 1997 46. Mets MB, Holfels E, Boyer KM et al: Eye manifestations of congenital toxoplasmosis. Am J Ophthalmol 123:1–16, 1997 47. Paul M: Immunoglobulin G avidity in diagnosis of toxoplasmic lymphadenopathy and
ocular toxoplasmosis. Clin Diagn Lab Immunol 6:514–518, 1999 48. Wallon M, Dunn D, Slimani D et al: Diagnosis of congenital toxoplasmosis at birth: What is the value of testing
for IgM and IgA? Eur J Pediatr 158:645–649, 1999 49. Anteby II, Blumenthal EZ, Zamir E, Waindim P: The role of preoperative ultrasonography for patients with dense cataract: A
retrospective study of 509 cases. Ophthalmic Surg Lasers 29:114–118, 1998 50. Neppert B: Measles retinitis in an immunocompetent child. Klin Monatsbl Augenheilkd 205:156–160, 1994 51. Du LT, Coats DK, Kline MW et al: Incidence of presumed cytomegalovirus retinitis in HIV-infected pediatric
patients. J AAPOS 3:245–249, 1999 52. Slusher MM, Hutton WE: Familial exudative vitreoretinopathy. Am J Ophthalmol 87:152–156, 1979 53. Sauer CG, Gehrig A, Warneke-Wittstock R et al: Positional cloning of the gene associated with X-linked juvenile retinoschisis. Nat Genet 17:164–170, 1997 54. Daufenbach DR, Ruttum MS, Pulido JS, Keech RV: Chorioretinal colobomas in a pediatric population. Ophthalmology 105:1455–1458, 1998 55. Eustis HS, Sanders MR, Zimmerman T: Morning glory syndrome in children: Association with endocrine and central
nervous system anomalies. Arch Ophthalmol 112:204–207, 1994 56. O'Shea WF, Powers JE: Solitary retinal astrocytoma. J Am Optom Assoc 62:519–524, 1991 57. Sharma A, Ram J, Gupta A: Solitary retinal astrocytoma. Acta Ophthalmol 69:113–116, 1991 58. Mullaney PB, Jacquemin C, Abboud E, Karcioglu ZA: Tuberous sclerosis in infancy. J Pediatr Ophthalmol Strabismus 34:372–375, 1997 59. Kim HY, Yu YS: Retinopathy of prematurity-mimicking retinopathy in full-term babies. Korean J Ophthalmol 12:98–102, 1998 60. Kasmann-Kellner B, Jurin-Bunte B, Ruprecht KW: Incontinentia pigmenti (Bloch-Sulzberger syndrome): Case report and differential
diagnosis to related dermato-ocular syndromes. Ophthalmologica 213:63–69, 1999 61. Filous A, Raskova D, Kodet R: Retinal detachment in an infant with the ring chromosome 13 syndrome. Acta Ophthalmol Scand 76:739–741, 1998 62. Ghiasvand NM, Shirzad E, Naghavi M, Vaez Mahdavi MR: High incidence of autosomal recessive nonsyndromal congenital retinal nonattachment (NCRNA) in
an Iranian population. Am J Med Genet 78:226–232, 1998 63. Shamamian P, Mancini M, Kawakami Y et al: Recognition of neuroectodermal tumors by melanoma-specific cytotoxic T
lymphocytes: Evidence for antigen sharing by tumors derived from the neural
crest. Cancer Immunol Immunother 39:73–83, 1994 64. Provis JM, Leech J, Diaz CM et al: Development of the human retinal vasculature: Cellular relations and VEGF
expression. Exp Eye Res 65:555–568, 1997 65. Arora R, Betharia SM: Retinoblastoma: A histologic and immunohistologic study. Indian J Pathol Microbiol 40:37–46, 1997 66. Bogenmann E, Mark C: Routine growth and differentiation of primary retinoblastoma cells in culture. J Natl Cancer Inst 70:95–104, 1983 67. Gonzalez-Fernandez F, Lopes MB, Garcia-Fernandez JM et al: Expression of developmentally defined retinal phenotypes in the histogenesis
of retinoblastoma. Am J Pathol 141:363–375, 1992 68. Shuangsgoti Sj, Chaiwum B, Kasantikul V: A study of 39 retinoblastomas with particular reference to morphology, cellular
differentiation and tumour origin. Histopathology 15:113–124, 1989 69. Abramson DH, Niksarli K, Ellsworth RM, Servodidio CA: Changing trends in the management of retinoblastoma: 1951-1965 vs. 1966-1980. J Pediatr Ophthalmol Strabismus 31:32–37, 1994 70. Shields CL, Shields JA: Recent developments in the management of retinoblastoma. J Pediatr Ophthalmol Strabismus 36:8–18, 1999 71. Abramson DH, Frank CM: Second nonocular tumors in survivors of bilateral retinoblastoma: A possible
age effect on radiation-related risk. Ophthalmology 105:573–580, 1998 72. Pui CH, Boyett JM, Hughes WT et al: Human granulocyte colony-stimulating factor after induction chemotherapy
in children with acute lymphoblastic leukemia. N Engl J Med 336:1781–1787, 1997 73. Murphree AL, Villablanca JG, Deegan WF 3rd et al:Chemotherapy plus local treatment in the management of intraocular retinoblastoma. Arch Ophthalmol 114:1348–1356, 1996 74. Levy C, Doz F, Quintana E et al: Role of chemotherapy alone or in combination with hyperthermia in the primary
treatment of intraocular retinoblastoma: Preliminary results. Br J Ophthalmol 82:1154–1158, 1998 75. Lueder GT, Goyal R: Visual function after laser hyperthermia and chemotherapy for macular retinoblastoma. Am J Ophthalmol 121:582–584, 1996 76. Mullaney PB, Abboud EB, Al-Mesfer SA: Retinal detachment associated with type III retinoblastoma regression after
cryotherapy and external-beam radiotherapy. Am J Ophthalmol 123:140–142, 1997 77. Blach LE, McCormick B, Abramson DH: External beam radiation therapy and retinoblastoma: Long-term results in
the comparison of two techniques. Int J Radiat Oncol Biol Phys 35:45–51, 1996 78. Fortney JT, Halperin EC, Hertz CM, Schulman SR: Anesthesia for pediatric external beam radiation therapy. Int J Radiat Oncol Biol Phys 44:587–591, 1999 79. Abramson DH, Ellsworth RM, Kitchin FD, Tung G: Second nonocular tumors in retinoblastoma survivors: Are they radiation-induced? Ophthalmology 91:1351–1355, 1984 80. Smith LM, Donaldson SS, Egbert PR et al: Aggressive management of second primary tumors in survivors of hereditary
retinoblastoma. Int J Radiat Oncol Biol Phys 17:499–505, 1989 81. Marcus DM, Brooks SE, Leff G et al: Trilateral retinoblastoma: Insights into histogenesis and management. Surv Ophthalmol 43:59–70, 1998 82. Paulino AC: Trilateral retinoblastoma: Is the location of the intracranial tumor important? Cancer 86:135–141, 1999 83. Schwartz AM, Ghatak NR, Laine FJ: Intrasellar primitive neuroectodermal tumor (PNET) in familial retinoblastoma: A
variant of trilateral retinoblastoma. Clin Neuropathol 9:55–59, 1990 84. Bejjani GK, Donahue DJ, Selby D et al: Association of a suprasellar mass and intraocular retinoblastoma: A variant
of pineal trilateral retinoblastoma? Pediatr Neurosurg 25: 269–275, 1996 85. De Potter P, Shields CL, Shields JA: Clinical variations of trilateral retinoblastoma: A report of 13 cases. J Pediatr Ophthalmol Strabismus 31:26–31, 1994 86. Skulski M, Egelhoff JC, Kollias SS et al: Trilateral retinoblastoma with suprasellar involvement. Neuroradiology 39:41–43, 1997 87. Amoaku WM, Willshaw HE, Parkes SE et al: Trilateral retinoblastoma. A report of five patients. Cancer 78:858–863, 1996 88. Bagley LJ, Hurst RW, Zimmerman RA et al: Imaging in the trilateral retinoblastoma syndrome. Neuroradiology 38:166–170, 1996 |