1. Anderson DR: Glaucoma: The damage caused by pressure. XLVI Edward Jackson Memorial Lecture. Am J Ophthalmol 108:485, 1989 2. David R, Livingston DG, Luntz MH: Ocular hypertension: A long-term follow-up of treated and untreated patients. Br J Ophthalmol 61:668, 1977 3. Hollows FC, Graham PA: Intraocular pressure, glaucoma, and glaucoma suspects in a defined population. Br J Ophthalmol 50:570, 1966 4. Sommer A, Tielsch JM, Katz J et al: Relationship between intraocular pressure and primary open angle glaucoma
in black and white Americans: The Baltimore Eye Study. Arch Ophthalmol 109:1090, 1991 5. Sommer A: Intraocular pressure and glaucoma. Am J Ophthalmol 107:186, 1989 6. Armaly MF, Krueger DE, Maunder LR et al: Biostatistical analysis of the collaborative glaucoma study. I. Summary
report of the risk factors for glaucomatous visual-field defects. Arch Ophthalmol 98:2163, 1980 7. Tielsch JM et al: Racial variations in the prevalence of primary open angle glaucoma. The
Baltimore Eye Survey. JAMA 266:369, 1991 8. Kolker AE: Glaucoma family study: ten-year follow-up (preliminary report). Isr J Med Sci 8:1357, 1972 9. Cole DF: Secretion of the aqueous humor. Exp Eye Res 25(Suppl):161, 1977 10. Cole DF: Evidence for active transport of chloride in ciliary epithelium of the
rabbit. Exp Eye Res 8:5, 1969 11. Friedenwald JS: Formation of the intraocular fluid. Am J Ophthalmol 32:9, 1949 12. Bill A: Conventional and uveoscleral drainage of aqueous humor in the cynomolgus
monkey (Macaca irus) at normal and high intraocular pressures. Exp Eye Res 5:45, 1966 13. Bill A: Physiology of the outflow mechanism. In Drance SM (ed): Applied
Pharmacology in the Medical Treatment of Glaucomas. Orlando: Grune & Stratton, 1984:111 14. Maepae O, Bill A: The pressures in the episcleral veins, Schlemm's canal and trabecular
meshwork in monkeys: Effects of changes in intraocular pressure. Exp Eye Res 49:645, 1989 15. Kaufman P, Mittag TW: Medical therapy of glaucoma. In Podos SM, Yanoff
M (eds): Textbook of Ophthalmology. London: Mosby-Year Book, 1988:9.7 16. Bill A: Aqueous humor dynamics in monkeys (Macaca irus and Cercopithecus aethiops). Exp Eye Res 11:195, 1971 17. Bill A, Phillips C: Uveoscleral drainage of aqueous humor in human eyes. Exp Eye Res 12:275, 1971 18. Ahlquist RP: A study of the adrenotropic receptors. Am J Physiol 153:586, 1948 19. Mittag TW: Adrenergic and dopaminergic drugs in glaucoma. In Ritch R, Shields
MB, Krupin T (eds): The Glaucomas. 2nd ed. St Louis: Mosby-Year
Book, 1996:1409–1424. 20. Stryer L, Bourne HR: G-proteins: A family of signal transducers. Annu Rev Cell Biol 2:391, 1986 21. Gregory DS et al: Intraocular pressure and aqueous flow are decreased by cholera toxin. Invest Ophthalmol Vis Sci 20:371, 1981 22. Caprioli J et al: Forskolin lowers intraocular pressure by reducing aqueous outflow. Invest Ophthalmol Vis Sci 25:268, 1984 23. Erickson KA: Basic aspects of parasympathetic pharmacology. In Ritch R, Shields
MB, Krupin T (eds): The Glaucomas. 2nd ed. St Louis: Mosby-Year
Book, 1996:1385–1389. 24. Bonner TI: New subtypes of muscarinic acetylcholine receptors. In Levine
RR, Birdsall NJM (eds): Subtypes of Muscarinic Receptors IV. Cambridge, UK: Elsevier, 1989 25. Lefkowitz RJ, Hoffman BB, Taylor P: Drug actions at synaptic and neuroeffector
junctional sites. In Gilman AG et al (eds): Goodman and Gilman's
the Pharmacological Basis of Therapeutics. 8th ed. New York: Pergamon
Press, 1990 26. True-Gabelt B, Kaufman PL: Muscarinic receptor subtype antagonist inhibition of pilocarpine-stimulated
outflow facility, accommodation, and pupil constriction in rhesus
monkeys. Invest Ophthalmol Vis Sci 32(Suppl):1256, 1991 27. Bankes JL, Perkins ES, Tsolakis S et al: Bedford Glaucoma Survey. BMJ 1:791, 1968 28. Leibowitz HM, Krueger DE, Maunder LR et al: The Framingham Eye Study monograph: An ophthalmological and epidemiological
study of cataract, glaucoma, diabetic retinopathy, macular degeneration, and
visual acuity in a general population of 2631 adults, 1973-1975. Surv Ophthalmol 24(Suppl):335, 1980 29. Anderson DR: The management of elevated intraocular pressure with normal optic discs
and visual fields. I. Therapeutic approach based on high risk factors. Surv Ophthalmol 21:479, 1977 30. Milton RC, Ganley JP: Risk of glaucoma in myopia: A population study. Invest Ophthalmol Vis Sci 16(Suppl): 85, 1977 31. Perkins ES, Phelps CD: Open angle glaucoma, ocular hypertension, low-tension glaucoma, and refraction. Arch Ophthalmol 100:1464, 1982 32. Migdal C, Gregory W, Hitchings RA: Long-term functional outcome after early surgery compared with laser and
medicine in open-angle glaucoma. Ophthalmology 101:1651, 1994 33. Palmberg P: The rationale and effectiveness of glaucoma therapy. Presented
at the annual meeting of the American Glaucoma Society, Miami, December 1988 34. Hart WM Jr, Becker B: The onset and evolution of glaucomatous visual field defects. Ophthalmology 89:268, 1982 35. Mikelberg FS, Schulzer M, Drance SM et al: The rate of progression of scotomas in glaucoma. Am J Ophthalmol 101:1, 1986 36. Epstein DL et al: A long-term clinical trial of timolol therapy versus no treatment in the
management of glaucoma suspects. Ophthalmology 96:1460, 1989 37. Kass MA, Gordon MO, Hoff MR et al: Topical timolol administration reduces the incidence of glaucomatous damage
in ocular hypertensive individuals: A randomized, double-masked, long-term
clinical study. Arch Ophthalmol 107:1590, 1989 38. Schulzer M, Drance SM, Douglas GR: A comparison of treated and untreated glaucoma suspects. Ophthalmology 98:301, 1991 39. Bito L: Glaucoma: A physiologic perspective with darwinian overtones. J Glaucoma 1:193, 1992 40. Martinez-Bello C, Chauhan BC, Nicolela MT et al: Intraocular pressure and progression of glaucomatous visual field loss. Am J Ophthalmol 129:302, 2000 41. Asrani S, Zeimer R, Wilensky J et al: Large diurnal fluctuations in intraocular pressure are an independent risk
factor in patients with glaucoma. J Glaucoma 9;134, 2000 42. Chandler PA: Long-term results in glaucoma therapy. Am J Ophthalmol 49:221, 1960 43. Grant WM, Burke JF Jr: Why do some people go blind from glaucoma? Ophthalmology 89:991, 1982 44. Mao LK, Steward WC, Shields MB: Correlation between intraocular pressure control and progressive glaucomatous
damage in primary open-angle glaucoma. Am J Ophthalmol 111:51, 1991 45. Odberg T: Visual field prognosis in advanced glaucoma. Acta Ophthalmol 182(Suppl 65):27, 1987 46. Kolker AE: Visual prognosis in advanced glaucoma: A comparison of medical and surgical
therapy for retention of vision of 101 eyes with advanced glaucoma. Trans Am Ophthalmol Soc 75:539, 1977 47. Quigley HA, Maumenee AF: Long-term follow-up of treated open-angle glaucoma. Am J Ophthalmol 87:519, 1979 48. Cartwright MJ, Anderson DR: Correlation of asymmetric damage with symmetric intraocular pressure in
normal-tension glaucoma (low-tension glaucoma). Arch Ophthalmol 106:898, 1988 49. Crichton A, Drance SM, Douglas GR et al: Unequal intra-ocular pressure and its relation to asymmetric visual field
defects in low-tension glaucoma. Ophthalmology 96:1213, 1989 50. Hitchings RA, Wu J, Poinoosawmy D et al: Surgery for normal tension glaucoma. Br J Ophthalmol 79:402, 1995 51. The Advanced Glaucoma Intervention Study Investigators: Advanced Glaucoma
Intervention Study: 2. Visual field test scoring and reliability. Ophthalmology 101:1445, 1994 52. Hodapp E, Parrish RK, Anderson DR: The asymptomatic patient with elevated
pressure. In: Clinical Decisions in Glaucoma. St Louis: CV Mosby, 1993:53 53. Alward PD, Wilensky JT: Determination of acetazolamide compliance in patients with glaucoma. Arch Ophthalmol 99:1973, 1981 54. Kass MA, Meltzer DW, Gordon MO: A miniature compliance monitor for eyedrop medication. Arch Ophthalmol 102:1550, 1984 55. Kass MA et al: Compliance with topical pilocarpine treatment. Am J Ophthalmol 101:515, 1986 56. Schuman JS: Effects of systemic beta-blocker therapy on the efficacy and safety of
topical brimonidine and timolol. Brimonidine study groups 1 and 2. Ophthalmology 107:1171, 2000 57. Linnér E, Prijot E: Cervical sympathetic ganglionectomy and aqueous flow. Arch Ophthalmol 54:831, 1955 58. Powell CE, Slater IH: Blocking of inhibitory adrenergic receptors by a dichloro analog of isoproterenol. J Pharmacol Exp Ther 122:480, 1958 59. Sears ML, Bárány EH: Outflow resistance and adrenergic mechanisms. Arch Ophthalmol 64:839, 1960 60. Phillips CI, Howitt G, Rowlands DL: Propranolol as ocular hypotensive agent. Br J Ophthalmol 56:770, 1972 61. Musini A, Fabbri B, Bergamaschi M et al: Comparison of the effect of propranolol, lignocaine, and other drugs on
normal and raised intraocular pressure in man. Am J Ophthalmol 72:773, 1971 62. Coakes RL, Brubaker RF: The mechanisms of timolol in lowering intraocular pressure. Arch Ophthalmol 96:2045, 1978 63. Yablonski ME, Zimmerman TJ, Waltman SR et al: A fluorophotometric study of the effect of topical timolol on aqueous humor
dynamics. Exp Eye Res 27:135,1978 64. Dailey RA, Brubaker RF, Bourne WM: The effects of timolol maleate and acetazolamide on the rate of aqueous
formation in normal human subjects. Am J Ophthalmol 93:232, 1982 65. Zimmerman TJ, Harbin R, Pett M et al: Timolol: Facility of outflow. Invest Ophthalmol 16:623, 1977 66. Brubaker RF, Nagataki S, Bourne WM: Effect of chronically administered timolol on aqueous humor flow in patients
with glaucoma. Ophthalmology 89:280, 1982 67. Zimmerman TJ, Kaufman HE: Timolol: A beta adrenergic blocking agent for the treatment of glaucoma. Arch Ophthalmol 95:601, 1977 68. Zimmerman TJ, Kass MA, Yablonski ME et al: Timolol maleate: Efficacy and safety. Arch Ophthalmol 97:656, 1979 69. Sonntag JR, Brindley GO, Shields MB et al: Timolol and epinephrine: Comparison of efficacy and side effects. Arch Ophthalmol 97:273, 1979 70. Radius RL, Diamond GR, Pollack IP et al: Timolol: A new drug for management of chronic simple glaucoma. Arch Ophthalmol 96:1003, 1978 71. IMS National Disease Therapeutic Index, year ending June 1985 72. Bohn RL et al: Patterns of glaucoma medication use in the elderly: 1980-1990. J Glaucoma 3:259, 1994 73. Bromberg BB, Gregory DS, Sears ML: ss-adrenergic receptors in ciliary processes in the rabbit. Invest Ophthalmol Vis Sci 19:203, 1980 74. Topper JE, Brubaker RF: Effects of timolol, epinephrine, and acetazolamide on aqueous flow during
sleep. Invest Ophthalmol Vis Sci 26:1315, 1985 75. Reiss GR, Lee DA, Topper JE et al: Aqueous humor flow during sleep. Invest Ophthalmol Vis Sci 25:776, 1984 76. LeDouarec JC et al: The effect of timolol, a beta-adrenergic blocking agent, on IOP in rabbits. Pharmacology 18:139, 1976 77. Katz IM, Hubbard WA, Getson AJ et al: Intraocular pressure decrease in normal volunteers following timolol ophthalmic
solution. Invest Ophthalmol 15:489, 1976 78. Zimmerman TJ, Kaufman HE: Timolol: Dose response and duration of action. Arch Ophthalmol 95:605, 1977 79. Radius RL et al: Timolol: A new drug for management of chronic simple glaucoma. Arch Ophthalmol 96:1003, 1978 80. Novack GD: Ophthalmic beta blockers since timolol. Surv Ophthalmol 31:301, 1987 81. Zimmerman TJ: Topical ophthalmic beta blockers: A comparative review. J Ocul Pharmacol 9:373, 1993 82. Katz IM et al: Intraocular pressure decrease in normal volunteers following timolol ophthalmic
solution. Invest Ophthalmol 15:489, 1976 83. Diamond GR et al: Extended clinical studies using timolol in patients with ocular hypertension
and chronic open-angle glaucoma. Glaucoma 1:63, 1979 84. Zimmerman TJ, Canale P: Timolol: Further observations. Ophthalmology 86:166, 1979 85. Zimmerman TJ et al: Timolol maleate: Efficacy and safety. Arch Ophthalmol 97:656, 1979 86. Boger WP et al: Long-term experience with timolol ophthalmic solution in patients with
open-angle glaucoma. Ophthalmology 85:259, 1979 87. Zimmerman TJ, Kaufman JE: Timolol: A ss-adrenergic blocking agent for the treatment of glaucoma. Arch Ophthalmol 95:601, 1977 88. Zimmerman TJ et al: Timolol plus maximum tolerated antiglaucoma therapy. Arch Ophthalmol 97:278, 1979 89. Gross FJ, Schuman JS: Reduced ocular hypotensive effect of topical ss-blockers in glaucoma patients
receiving oral ss-blockers. J Glaucoma 1:174, 1992 90. Krupin T et al: One hour intraocular pressure response to timolol: Lack of correlation
with long-term response. Arch Ophthalmol 99:840, 1981 91. Boger WP: Timolol: Short-term “escape” and long-term “drift.” Ann Ophthalmol 11:1239, 1979 92. Neufeld AH et al: Influences on the density of ss-adrenergic receptors in the cornea and
iris-ciliary body of the rabbit. Invest Ophthalmol Vis Sci 17:1069, 1978 93. Steinert RF, Thomas JV, Boger WP: Long-term drift and continued efficacy after multiyear timolol therapy. Arch Ophthalmol 99:100, 1981 94. Lin LL et al: Long-term timolol therapy. Surv Ophthalmol 23:377, 1979 95. Moss AP et al: A comparison of the effects of timolol and epinephrine on intraocular pressure. Am J Ophthalmol 86:489, 1978 96. Sonntag JR et al: Timolol and epinephrine: comparison of efficacy and side effects. Arch Ophthalmol 97:273, 1979 97. Boger WP et al: Clinical trial comparing timolol ophthalmic solution to pilocarpine in
open-angle glaucoma. Am J Ophthalmol 86:8, 1978 98. Strahlman ER et al: A controlled clinical trial of 2.0% dorzolamide (MK-507) compared to timolol
and betaxolol. Invest Ophthalmol Vis Sci 35(Suppl):2177, 1994 99. Nagasubramanian S et al: Comparison of apraclonidine and timolol in chronic open-angle glaucoma. Ophthalmology 100:1318, 1993 100. Alm AA, Stjernschantz J: The Scandinavian Latanoprost Study Group: Effects of intraocular pressure
and side effects of 0.005% latanoprost applied once daily, evening
or morning. Ophthalmology 102:1743, 1995 101. Watson PG, Stjernschantz J: U.K. Latanoprost Study Group: Intraocular pressure (IOP) reducing effect
and side-effects of latanoprost and timolol: A six-month double-masked
comparison. Ophthalmology 101(Suppl):80, 1994 102. Camras CB et al: Latanoprost, a potent ocular hypotensive prostaglandin analog, increases
pigmentation in peripherally hypopigmented irides. Ophthalmology 101(Suppl):128, 1994 103. Glaucoma Laser Trial Research Group T: The Glaucoma Laser Trial (GLT). IV. Contralateral
effects of timolol on the intraocular pressure of eyes
treated with ALT. Ophthalmic Surg 22:324, 1991 104. Laurence J et al: A double-masked, placebo-controlled evaluation of timolol in a gel vehicle. J Glaucoma 2:177, 1993 105. Letchinger SL et al: Can the concentration of timolol or the frequency of its administration
be reduced? Ophthalmology 100:1259, 1993 106. Mills KB: Blind randomized non-crossover long-term trial comparing topical timolol 0.25% with
timolol 0.5% in the treatment of simple chronic glaucoma. Br J Ophthalmol 67:216, 1983 107. Katz IM, Hubbard WA, Getson AJ et al: Intraocular pressure decrease in normal volunteers following timolol ophthalmic
solution. Invest Ophthalmol 15:489, 1976 108. Soll DB: Evaluation of timolol in chronic open angle glaucoma: once a day vs. twice
a day. Arch Ophthalmol 98:2178, 1980 109. Sheddan AH: Timolol maleate in gel-forming solution. Chibret Intl J Ophthalmol 10:2, 1994 110. Katz IM, Berger ET: Effects of iris pigmentation on response of ocular pressure to timolol. Surv Ophthalmol 23:395, 1979 111. Stewart WC, Leland TM, Cate EA: Efficacy and safety of timolol solution once daily versus timolol gel in
treating elevated intraocular pressure. J Glaucoma 7:402, 1998 112. Stewart WC, Cate EA, Stewart JA: Systemic beta-blockade with once daily Betimol or Timoptic-XE. J Ocular Pharm Ther 15:225, 1999 113. Schlecte LP, Brubaker RF: The effects of withdrawal of timolol in chronically treated glaucoma patients. Ophthalmology 95:1212, 1988 114. Calissendorff B et al: Timolol versus pilocarpine separately or combined with acetazolamide: Effects
on intraocular pressure. Acta Ophthalmol 58:624, 1980 115. Kass MA et al: Timolol and acetazolamide: A study of concurrent administration. Arch Ophthalmol 100:941, 1982 116. Airaksinen PJ et al: A double-masked study of timolol and pilocarpine combined. Am J Ophthalmol 104:587, 1987 117. Smith RJ et al: Addition of timolol maleate to routine medical therapy: A clinical trial. Br J Ophthalmol 64:779, 1980 118. Blasini M, Shields MB: Apraclonidine hydrochloride as an adjunct to timolol maleate therapy. J Glaucoma 1:148,1992 119. Morrison J, Robin A: Adjunctive glaucoma therapy: A comparison of apraclonidine to dipivefrin
when added to timolol maleate. Ophthalmology 96:3, 1989 120. Yaldo M et al: Additive effect of 1% apraclonidine hydrochloride to nonselective ss-blockers. Ophthalmology 98:1075, 1991 121. Rulo AH, Greve EL, Hoyng PF: Additive effect of latanoprost, a prostaglandin F3a analogue, and timolol in patients with elevated intraocular pressure. Br J Ophthalmol 78:899, 1994 122. Alm A et al: Latanoprost administered once daily caused a maintained reduction of intraocular
pressure in glaucoma patients treated concomitantly with timolol. Br J Ophthalmol 79:12, 1995 123. Thomas JV, Epstein DL: Timolol and epinephrine in primary open-angle glaucoma: Transient, additive
effect. Arch Ophthalmol 99:91, 1981 124. Goldberg I et al: Timolol and epinephrine: A clinical study of ocular interactions. Arch Ophthalmol 98:484, 1980 125. Keates EU, Stone RA: Safety and effectiveness of concomitant administration of dipivefrin and
timolol maleate. Am J Ophthalmol 91:243, 1981 126. Knupp JA et al: Combined timolol and epinephrine therapy for open-angle glaucoma. Surv Ophthalmol 28:280, 1983 127. Semla TP, Schwartz A, Koch H et al: Patterns of drug prescribing. In Van
Nostrand JF, Furner SE, Suzman R (eds): Health Data on Older Americans: United
States, 1992: National Center for Health Statistics. Vital
Health Stat 3:187, 1993 128. Atkins JM, Pugh BR, Timewell RM: Cardiovascular effects of topical ss-blockers during exercise. Am J Ophthalmol 99:173, 1985 129. Van Buskirk EM: Adverse reactions from timolol administration. Ophthalmology 87:447, 1980 130. The Norwegian Multicenter Study Group: Timolol-induced reduction in mortality
and reinfarction in patients surviving acute myocardial infarction. N
Engl J Med 304:801, 1981 131. Schoene RB, Abuan T, Ward RL et al: Effects of topical betaxolol, timolol, and placebo on pulmonary function
in asthmatic bronchitis. Am J Ophthalmol 97:86, 1984 132. Diggory P et al: Avoiding unsuspected respiratory side-effects of topical timolol with cardioselective
or sympathomimetic agents. Lancet 345:1604, 1995 133. Renwick DS, Connolly MJ: Prevalence and treatment of chronic airways obstruction (AO) in older adults. Age Ageing 23(Suppl 2):6, 1994 134. Duch S et al: Changes in depressive status associated with topical ss-blockers. Int Ophthalmol 16:331, 1992 135. McMahon CD et al: Adverse effects experienced by patients taking timolol. Am J Ophthalmol 88:736, 1979 136. Fraunfelder FT: Sexual dysfunction secondary to topical ophthalmic timolol. JAMA 253:3092, 1985 137. Caputo BJ, Katz LJ: The quality of life of a glaucoma patient in the light of treatment modalities. Curr Opin Ophthalmol 5:10, 1994 138. Bour J, Blanchard F, Segal A: Repercussion of glaucoma on a patient's life: Concerning 341 cases
in Marne, France. J Fr Ophtalmol 16:380, 1993 139. Carrier P, Gentile S, Fusco R et al: Mood disorders in patients with chronic simple glaucoma. Psychiatry Res 36:33, 1991 140. Glynn R, Seddon J, Krug J et al: Falls in elderly patients with glaucoma. Arch Ophthalmol 109:205, 1991 141. Gerber SL, Cantor LB, Brater DC: Systemic drug interactions with topical glaucoma medications. Surv Ophthalmol 35:205, 1990 142. Velde TM, Kaiser FE: Ophthalmic timolol treatment causing altered hypoglycemic response in a
diabetic patient. Arch Intern Med 143:1627, 1983 143. Coppeto JR: Timolol-associated myasthenia gravis. Am J Ophthalmol 98:244, 1984 144. Shaivitz SA: Timolol and myasthenia gravis. JAMA 252:1611, 1979 145. Kuppens EVMJ et al: Effect of timolol with and without preservative on the basal tear turnover
in glaucoma. Br J Ophthalmol 79:339, 1995 146. Van Buskirk EM: Corneal anesthesia after timolol maleate therapy. Am J Ophthalmol 88:739, 1979 147. Weissman SS, Asbell PA: Effects of topical timolol (0.5%) and betaxolol (0.5%) on corneal sensitivity. Am J Ophthalmol 74:409, 1990 148. Herraras JM et al: Ocular surface alteration after long-term treatment with an antiglaucomatous
drug. Ophthalmology 99:1082, 1992 149. Williams DE et al: Effects of timolol, betaxolol, and levobunolol on human Tenon's fibroblasts
in tissue culture. Invest Ophthalmol Vis Sci 33:2233, 1992 150. Baxter GM, Williamson TH, McKillop G et al: Color Doppler ultrasound of orbital and ocular nerve blood flow: Effects
of posture and timolol 0.5%. Invest Ophthalmol Vis Sci 33:604, 1992 151. Grunwald JE, Furubayashi C: Effect of topical timolol maleate on the ophthalmic artery blood pressure. Invest Ophthalmol Vis Sci 30:1095, 1989 152. Grunwald JE: Effect of topical timolol on the human retinal circulation. Invest Ophthalmol Vis Sci 27:1713, 1986 153. Grunwald JE: Effect of timolol maleate on the retinal circulation of human eyes with
ocular hypertension. Invest Ophthalmol Vis Sci 31:521, 1990 154. Grunwald JE: Effect of two weeks of timolol maleate treatment on the normal retinal
circulation. Invest Ophthalmol Vis Sci 32:39, 1991 155. Martin XD, Rabineau PA: Vasoconstriction effect of topical timolol on human retinal arteries. Graefes Arch Clin Exp Ophthalmol 227:526, 1989 156. Meuche C, Heidrich H, Blackman H: Raynaud syndrome following timolol containing eye drops. Fortschr Ophthalmol 87:45, 1990 157. Partamian LG, Kass MA, Gordon M: A dose-response study of the effect of levobunolol on ocular hypertension. Am J Ophthalmol 95:229, 1983 158. Bensinger RE et al: Levobunolol: A 3-month efficacy study in the treatment of glaucoma and
ocular hypertension. Arch Ophthalmol 103:375, 1985 159. Long DA et al: Levobunolol and betaxolol: A double-masked, controlled comparison of efficacy
and safety in patients with elevated intraocular pressure. Ophthalmology 95:735, 1988 160. Long D et al: Minimum concentration of levobunolol required to control intraocular pressure
in patients with primary open-angle glaucoma or ocular hypertension. Am J Ophthalmol 99:18, 1985 161. Levobunolol Study Group T: Levobunolol: A 4-year study of efficacy and
safety in glaucoma treatment. Ophthalmology 96:642, 1989 162. Berson FG et al: Levobunolol compared with timolol for the long-term control of elevated
intraocular pressure. Arch Ophthalmol 103:379, 1985 163. Boozeman FW et al: Long-term evaluation of 0.25% levobunolol and timolol for therapy for elevated
intraocular pressure. Arch Ophthalmol 106:614, 1988 164. Cinotti A et al: Levobunolol versus timolol for open-angle glaucoma and ocular hypertension. Am J Ophthalmol 99:11, 1985 165. Duzman E et al: A clinical evaluation of the effects of topically applied levobunolol and
timolol on increased intraocular pressure. Am J Ophthalmol 94:318, 1982 166. McMahon CD, Shaffer RN, Hoskins HD Jr et al: Adverse effects experienced by patients taking timolol. Am J Ophthalmol 88:736, 1979 167. Levobunolol Study Group T: Levobunolol: A β-adrenoreceptor antagonist
effective in the long-term treatment of glaucoma. Ophthalmology 92:1271, 1985 168. Duzman E et al: A clinical evaluation of the effects of topically applied levobunolol and
timolol on increased intraocular pressure. Am J Ophthalmol 94:318, 1982 169. Derick RJ et al: Once daily versus twice daily levobunolol (0.5%) therapy. Ophthalmology 99:424, 1992 170. Novack GD et al: Plasma levobunolol levels following topical administration with reference
to systemic side effects. Ophthalmologica 194:194, 1987 171. Rakofsky SI et al: A comparison of the ocular hypotensive effect of once daily and twice daily
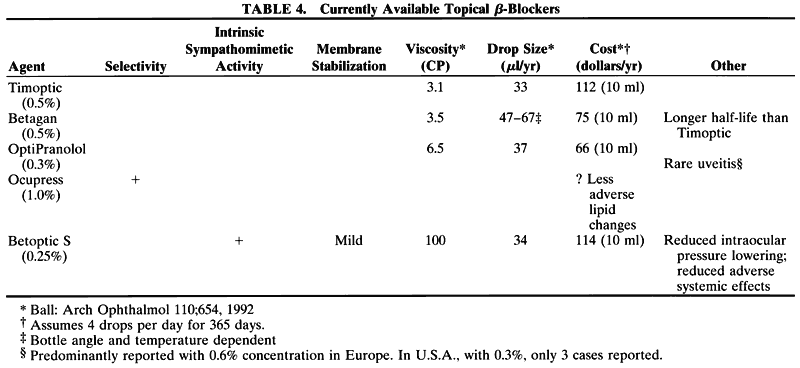
levobunolol treatment. Ophthalmology 96:8, 1989 172. Wandel T et al: Glaucoma treatment with once daily levobunolol. Am J Ophthalmol 101:298, 1986 173. Allen RC et al: A combination of levobunolol and dipivefrin for the treatment of glaucoma. Arch Ophthalmol 106:904, 1988 174. Rakofsky SI et al: Efficacy and safety of once daily levobunolol for glaucoma therapy. Can J Ophthalmol 24:2, 1989 175. Ball SF, Schneider E: Cost of β-adrenergic receptor blocking agents for ocular hypertension. Arch Ophthalmol 110:654, 1992 176. Serle JB, Lustgarten JS, Podos SM: A clinical trial of metipranolol, a noncardioselective β-adrenergic
antagonist, in ocular hypertension. Am J Ophthalmol 112:302, 1991 177. Hickey-Dwyer M, Campbell SH, Harding S: Double-masked, three-period crossover investigation of metipranolol in
control of raised intraocular pressure. J Ocul Pharmacol 7:277, 1991 178. Lyden M, Appelbaum HJ: Effect of ophthalmic metopranolol and timolol on exercise-induced tachycardia. J Glaucoma 4:124, 1995 179. Akingbehin T, Villada JR, Walley T: Metipranolol-induced adverse reactions. I. The rechallenge study. Eye 6:277, 1992 180. Melles RB, Wong IG: Metipranolol-associated granulomatous iritis. Am J Ophthalmol 118:712, 1994 181. Schultz JS, Hoenig JA, Charles H: Possible bilateral uveitis secondary to metipranolol (Optipranolol) therapy. Arch Ophthalmol 111:1607, 1993 182. Jasper JR, Michael MC, Insel PA: The ss-adrenoreceptor antagonist carteolol and its metabolite 8-hydroxycarteolol
have different intrinsic sympathomimetic activities. Br J Clin Pharmacol 30:109S, 1990 183. Stewart WC: Carteolol, an ophthalmic ss-adrenergic blocker with intrinsic sympathomimetic
activity. J Glaucoma 3:339, 1994 184. Kitazawa Y: Multicenter, double-blind comparison of carteolol and timolol in primary
open-angle glaucoma. Adv Ther 10:95, 1993 185. Scoville B et al: A double-masked comparison of carteolol and timolol in ocular hypertension. Arch Ophthalmol 107:1777, 1989 186. Duff GR: A double-masked crossover study comparing the effects of carteolol 1% and 2% on
intraocular pressure. Acta Ophthalmol 65:618, 1987 187. Duff GR, Graham PA: A double-crossover trial comparing the effects of topical carteolol and
placebo on intraocular pressure. Br J Ophthalmol 72:27, 1988 188. Duff GR, Newcombe RG: The 12-hour control of intraocular pressure on carteolol 2% twice daily. Br J Ophthalmol 72:890, 1988 189. Freedman SF et al: Effects of ocular carteolol and timolol on plasma high-density lipoprotein
cholesterol level. Am J Ophthalmol 116:600, 1993 190. Stewart WC et al: A 3-month comparison of 1% and 2% carteolol and 0.5% timolol in open-angle
glaucoma. Graefes Arch Clin Exp Ophthalmol 229:258, 1991 191. Nathanson JA: Human ciliary process adrenergic receptor. Invest Ophthalmol Vis Sci 21:798, 1981 192. Wax MB et al: Characterization of β-adrenergic receptors in cultured human trabecular
cells and in human trabecular meshwork. Invest Ophthalmol Vis Sci 30:51, 1989 193. Reiss GR, Brubaker RF: The mechanism of betaxolol, a new ocular hypotensive agent. Ophthalmology 90:1369, 1983 194. Berrospi AR, Leibowitz HM: Betaxolol: A new β-adrenergic blocking agent for treatment of glaucoma. Arch Ophthalmol 100:943, 1982 195. Caldwell DR, Salisbury CR, Guzek JP: Effects of topical betaxolol in ocular hypertensive patients. Arch Ophthalmol 102:539, 1984 196. Feghali JG, Kaufman PL: Decreased intraocular pressure in the hypertensive human eye with betaxolol, a β-adrenergic
antagonist. Am J Ophthalmol 100:777, 1985 197. Radius RL: Use of betaxolol in the reduction of elevated intraocular pressure. Arch Ophthalmol 101:898, 1983 198. Gaul GR, Will NJ, Brubaker RF: Comparison of a non-cardioselective ss-adrenergic blocker and a cardioselective
blocker in reducing aqueous flow in humans. Arch Ophthalmol 107:1308, 1989 199. Allen RC, Epstein DL: Additive effect of betaxolol and epinephrine in primary open-angle glaucoma. Arch Ophthalmol 104:1178, 1986 200. Collignon-Brach J: Long-term effect of ophthalmic ss-adrenoreceptor antagonists on intraocular
pressure and retinal sensitivity in primary open-angle glaucoma. Curr Eye Res 11:1, 1992 201. Stewart RH, Kimbrough RL, Ward RL: Betaxolol versus timolol: A 6-month double-blind comparison. Arch Ophthalmol 104:46, 1986 202. Albracht DC et al: A double-masked comparison of betaxolol and dipivefrin for the treatment
of increased intraocular pressure. Am J Ophthalmol 116:307, 1993 203. Kaiser HJ et al: Thirty-month visual field follow-up of glaucoma patients treated with ss-blockers. J Glaucoma 1:153, 1992 204. Harris A et al: Retrobulbar arterial hemodynamic effects of betaxolol and timolol in normal-tension
glaucoma. Am J Ophthalmol 120:168, 1995 205. Weinreb RN et al: A double-masked, 3-month comparison between 0.25% betaxolol suspension
and 0.5% betaxolol ophthalmic solution. Am J Ophthalmol 110:189, 1990 206. Vuori M et al: Plasma and aqueous humour concentrations and systemic effects of topical
betaxolol and timolol in man. Acta Ophthalmol 71:201, 1993 207. Ananthanarayan CR, Vaile SJ, Feldman F: Acute episode of asthma following topical administration of betaxolol eyedrops. Can J Ophthalmol 28:80, 1993 208. Harris LS, Greinstein SH, Bloom AF: Respiratory difficulties with betaxolol. Am J Ophthalmol 102:274, 1986 209. Brooks AMV, Burdon JGW, Gillies WE: The significance of reactions to betaxolol reported by patients. Aust NZ J Ophthalmol 17:353, 1989 210. Schoene RB et al: Effects of topical betaxolol, timolol, and placebo on pulmonary function
in asthmatic bronchitis. Am J Ophthalmol 97:86, 1984 211. Van Buskirk EM et al: Betaxolol in patients with glaucoma and asthma. Am J Ophthalmol 101:531, 1986 212. Weinreb RN et al: Long-term betaxolol therapy in glaucoma patients with pulmonary disease. Am J Ophthalmol 106:162, 1988 213. Zabel RW, MacDonald IM: Sinus arrest associated with betaxolol ophthalmic drops. Am J Ophthalmol 104:431, 1987 214. Ball S: Congestive heart failure from betaxolol. Arch Ophthalmol 10:320, 1987 215. Lynch MG et al: Topical ss-blocker therapy and central nervous system side effects: A preliminary
study comparing betaxolol and timolol. Arch Ophthalmol 106:908, 1988 216. Knapp A: The action of adrenalin on the glaucomatous eye. Arch Ophthalmol 50:556, 1921 217. Gifford SR: Some modern preparations used in the treatment of glaucoma. Arch Ophthalmol 57:612, 1928 218. Becker B: Topical epinephrine in the treatment of the glaucomas. In: Symposium
on Glaucoma, Transactions of the New Orleans Academy of Ophthalmology. St
Louis: CV Mosby, 1967:152 219. Schenker HI et al: Fluorophotometric study of epinephrine and timolol in human subjects. Arch Ophthalmol 99:1212, 1981 220. Townsend DJ, Brubaker RF: Immediate effect of epinephrine on aqueous formation in the normal human
eye as measured by fluorophotometry. Invest Ophthalmol Vis Sci 19:256, 1980 221. Becker B, Pettit TH, Gay AJ: Topical epinephrine therapy of open-angle glaucoma. Arch Ophthalmol 66:219, 1961 222. Robinson JC, Kaufman PL: Effects and interactions of epinephrine, norepinephrine, timolol, and betaxolol
on outflow facility in the cynomolgus monkey. Am J Ophthalmol 109:189, 1990 223. Bill A, Inomata H, Smelser GK: Unconventional routes of aqueous humor outflow in cynomolgus monkey (Macaca irus). Am J Ophthalmol 73:893, 1972 224. Neufeld AH, Jampol LM, Sears ML: Cyclic AMP in the aqueous humor: The effects of adrenergic agents. Exp Eye Res 14:242, 1972 225. Obstbaum SA, Kolker AE, Phelps CD: Low-dose epinephrine: Effect on intraocular pressure. Arch Ophthalmol 92:118, 1974 226. Alexander DW, Betson FG, Epstein DL: A clinical trial of timolol and epinephrine in the treatment of primary
open-angle glaucoma. Ophthalmology 95:247, 1988 227. Becker B, Ley AP: Epinephrine and acetazolamide in the therapy of the chronic glaucomas. Am J Ophthalmol 45:639, 1958 228. Cyrlin MN, Thomas JV, Epstein DL: Additive effect of epinephrine to timolol therapy in primary open-angle
glaucoma. Arch Ophthalmol 100:414, 1982 229. Higgins RG, Brubaker RF: Acute effect of epinephrine on aqueous humor formation in the timolol-treated
normal eye as measured by fluorophotometry. Invest Ophthalmol Vis Sci 19:420, 1980 230. Keates EU: Evaluation of timolol maleate combination therapy in chronic open-angle
glaucoma. Am J Ophthalmol 88:565, 1979 230a. Widengard I, Maepea O, Alm A: Effects of latanoprostand dipivefrin, alone or combined, on intraocular
pressure and on blood-aqueous barrier permeability. Br J Ophthalmol 82:404, 1988 231. Camras CB et al: Inhibition of the epinephrine-induced reduction of intraocular pressure
by systemic indomethacin in humans. Am J Ophthalmol 100:169, 1985 232. Becker B, Morton WR: Topical epinephrine in glaucoma suspects. Am J Ophthalmol 62:272, 1966 233. Kerr CR et al: Cardiovascular effects of epinephrine and dipivalyl epinephrine applied
topically to the eye in patients with glaucoma. Br J Ophthalmol 66:109, 1982 234. Becker B, Morton WR: Topical epinephrine in glaucoma suspects. Am J Ophthalmol 62:272, 1966 235. Kohn AN et al: Clinical comparison of dipivalyl epinephrine and epinephrine in the treatment
of glaucoma. Am J Ophthalmol 87:196, 1979 236. Keates EU, Stone RA: Safety and effectiveness of concomitant administration of dipivefrin and
timolol maleate. Am J Ophthalmol 91:243, 1981 237. Byron LT: Conjunctival reaction due to L-epinephrine bitartrate (Epitrate). Arch Ophthalmol 63:567, 1960 238. Corwin ME, Spencer WH: Conjunctival melanin depositions. Arch Ophthalmol 69:317, 1963 239. Miller D, Brooks SM, Mobilia E: Adrenochrome staining of soft contact lenses. Ann Ophthalmol 89:65, 1976 240. Spaeth GL: Nasolacrimal duct obstruction caused by topical epinephrine. Arch Ophthalmol 77:355, 1967 241. Waltman SR et al: Corneal endothelial changes with long-term topical epinephrine therapy. Arch Ophthalmol 95:1357, 1977 242. Miyake K, Miyake Y, Kuratomi R: Long-term effects of topically applied epinephrine on the blood-ocular
barrier in humans. Arch Ophthalmol 105:1360, 1987 243. Kolker AE, Becker B: Epinephrine maculopathy. Arch Ophthalmol 79:552, 1968 244. Mackool RJ et al: Epinephrine-induced cystoid macular edema in aphakic eyes. Arch Ophthalmol 95:791, 1977 245. Thomas JV et al: Correlation of epinephrine use and macular edema in aphakic glaucomatous
eyes. Arch Ophthalmol 96:625, 1978 246. Shell JW: Pharmacokinetics of topically applied ophthalmic drugs. Surv Ophthalmol 26:207, 1982 247. Mandell AI, Stentz F, Kitabchi AE: Dipivalyl epinephrine: A new pro-drug in the treatment of glaucoma. Ophthalmology 85:268, 1978 248. Anderson JA, David WL, Wei C: Site of ocular hydrolysis of a prodrug, dipivefrin, and a comparison of
its ocular metabolism with that of the parent compound, epinephrine. Invest Ophthalmol Vis Sci 19:817, 1980 249. Krieglstein GK, Leydhecker W: The dose-response relationships of dipivalyl epinephrine in open-angle
glaucoma. Graefes Arch Clin Exp Ophthalmol 205:141, 1978 250. Kaback MB et al: The effects of dipivalyl epinephrine on the eye. Am J Ophthalmol 81:768, 1976 251. Wandel T, Spinak M: Toxicity of dipivalyl epinephrine. Ophthalmology 88:259, 1981 252. Yablonski ME et al: Dipivefrin use in patients with intolerance to topically applied epinephrine. Arch Ophthalmol 95:2157, 1977 253. Van Zweiten PA: The central activity of antihypertensive drugs mediated via central α-receptors. J Pharm Belg 25:89, 1973 254. Hausler G: Cardiovascular regulation by central adrenergic mechanisms and its alteration
by hypotensive drugs. Circ Res 37:223, 1975 255. Makabe R: Ophthalmological studies with dichlorophenyl-aminoimidazoline. Dtsch Med Wochenschr 91:1686, 1966 256. Hasslinger R: Catapres: A new drug lowering intraocular pressure. Klin Monatsbl Augenheilkd 154:95, 1969 257. Harrison R, Kaufmann CS: Clonidine: Effects of a topically administered solution on intraocular
pressure and blood pressure in open-angle glaucoma. Arch Ophthalmol 95:1368, 1977 258. Heilmann K: Clonidine in glaucoma therapy: Interferences for therapy and obvious problems. Buch Augenarzt 3:56, 1974 259. Hodapp E et al: The effect of topical clonidine on intraocular pressure. Arch Ophthalmol 99:1208, 1981 260. Heilmann K: Studies on the effect of Catapres on intraocular pressure. Klin Monatsbl Augenheilkd 161:425, 1972 261. Krieglstein GK, Langham ME, Leydhecker W: The peripheral and central neural actions of clonidine in normal and glaucomatous
eyes. Invest Ophthalmol Vis Sci 17:149, 1982 262. Gharagozolo NZ, Relf SJ, Brubaker RF: Aqueous flow is reduced by the α3-adrenergic agonist, apraclonidine hydrochloride (ALO 2145). Ophthalmology 95:1217, 1988 263. Lee DA, Topper JE, Brubaker RF: Effect of clonidine on aqueous humor flow in normal human eyes. Exp Eye Res 38:239, 1984 264. Robin AL: Short-term effects of unilateral 1% apraclonidine therapy. Arch Ophthalmol 106:912, 1988 265. Mittag TW, Tormay A: Drug responses of adenylate cyclase in iris-ciliary body determined by
adenine labelling. Invest Ophthalmol Vis Sci 26:396, 1985 266. Thorig L, Bill A: Effects of BHG-920 in the eye and on regional blood flows in anesthetized
and conscious rabbits. Curr Eye Res 5:565, 1986 267. Abrams DA et al: The safety and efficacy of topical 1% ALO 2145 (p-aminoclonidine hydrochloride) in
normal volunteers. Arch Ophthalmol 105:1205, 1987 268. Jampel H et al: Apraclonidine: A 1-week dose response study. Arch Ophthalmol 106:1069, 1988 269. Chien DS et al: Corneal and conjunctival/scleral penetration of p-aminoclonidine, AGN 190342, and
clonidine in rabbit eyes. Curr Eye Res 9:1051, 1990 270. Mansberger S et al: Acute effect of topical apraclonidine on perimacular and orbital hemodynamics. Invest Ophthalmol Vis Sci 35:2176, 1994 271. Serdahl CL, Galustian J, Lewis RA: The effects of apraclonidine on conjunctival oxygen tension. Arch Ophthalmol 107:1777, 1989 272. Robin AL, Coleman AL: Apraclonidine hydrochloride: An evaluation of plasma concentrations and
a comparison of its intraocular pressure-lowering and cardiovascular
effects to those of timolol maleate. Trans Am Ophthalmol Soc 88:149, 1990 273. Abrams DA et al: A limited comparison of apraclonidine's dose response in subjects
with normal and increased intraocular pressure. Am J Ophthalmol 108:230, 1989 274. Robin AL et al: Effect of ALO-2145 on intraocular pressure following argon laser trabeculoplasty. Arch Ophthalmol 105:646, 1987 275. Robin AL: Argon laser trabeculoplasty: Medical therapy to prevent the intraocular
pressure rise associated with argon laser trabeculoplasty. Ophthalmic Surg 22:31, 1991 276. Pollack IP et al: Effectiveness of apraclonidine in preventing the rise in intraocular pressure
after neodymium:YAG posterior capsulotomy. Trans Am Ophthalmol Soc 86:461, 1988 277. Silverstone DE et al: Prophylactic use of apraclonidine for intraocular pressure increase after
Nd:YAG capsulotomies. Am J Ophthalmol 113:401, 1992 278. Kitazawa Y, Taniguchi T, Sugiyama K: Use of apraclonidine to reduce acute intraocular pressure rise following
Q-switched Nd:YAG laser iridotomy. Ophthalmic Surg 20:49, 1989 279. Wiles SB, MacKenzie D, Ide CH: Control of intraocular pressure with apraclonidine hydrochloride after
cataract extraction. Am J Ophthalmol 111:184, 1991 280. Fry LL: Comparison of the postoperative intraocular pressure with Betagan, Betoptic, Timoptic, Iopidine, Diamox, Pilopine Gel, and Miostat. J Cataract Refract Surg 18:14, 1992 281. Stewart RH et al: The efficacy of apraclonidine as an adjunct to timolol therapy. Arch Ophthalmol 113:287, 1995 282. Blasini M, Shields MB: Apraclonidine hydrochloride as an adjunct to timolol maleate therapy. J Glaucoma 1:148, 1992 283. Kosela T, Brubaker RF: Apraclonidine and timolol: Combined effects in previously untreated normal
subjects. Arch Ophthalmol 109:804, 1991 284. Robin AL, The Apraclonidine Maximum-Tolerated Medical Therapy Study Group: Short-term
efficacy of apraclonidine hydrochloride added to maximum-tolerated
medical therapy for glaucoma. Am J Ophthalmol 120:423, 1995 285. Robin AL, Ritch R, Shin DH: Short term efficacy of apraclonidine hydrochloride added to maximum-tolerated
medical therapy for glaucoma. Am J Ophthalmol 120:423, 1995 286. King MH, Richards DW: Near syncope and chest tightening after administration of apraclonidine
before argon laser iridotomy. Am J Ophthalmol 110:308,1990 287. Thompson CD, Vachaspati PR, Kolis SP et al: A proposed mechanism for p-aminoclonidine allergenicity based on its relative
oxidative lability. Chem Res Toxicol 10:1032, 1997 288. Thompson CD, Macdonald TL, Garst ME et al: Mechanism of adrenergic induced allergy bioactivation and antigen formation. Exp Eye Res 64:767, 1997 289. Pulido JS, Sneed SR, Blodi SF: Apraclonidine hydrochloride in vitreoretinal surgery. Arch Ophthalmol 107:316, 1989 290. Hill RA et al: Apraclonidine prophylaxis for postcycloplegic intraocular pressure spikes. Ophthalmology 98:1083, 1991 291. Gieser SC, Juzych M, Robin AL et al: Clinical pharmacology of adrenergic
drugs: In Ritch R, Shields MB, Krupin T (eds): The Glaucomas, Glaucoma
Therapy. St Louis, Mosby-Year Book, 1996:1425 292. Schadlu R, Maus TL, Nau CB et al: Comparison of the efficacy of apraclonidine and brimonidine as aqueous
suppressants in humans. Arch Ophthalmol 116:1441, 1998 293. Toris CB, Camras CB, Yablonski ME et al: Acute versus chronic effects of brimonidine on aqueous humor dynamics in
ocular hypertensive patients. Am J Ophthalmol 128:8, 1999 294. Barnes SD, Campagna JA, Dirks MS et al: Control of intraocular pressure elevations after argon laser trabeculoplasty: Comparison
of brimonidine 0.2% to apraclonidine 1.0%. Ophthalmology 106:2033, 1999 295. Chevrier RL, Assolian A, Dupeure J et al: Apraclonidine 0.5% versus brimonidine 0.2% for the control of intraocular
pressure elevations following anterior segment laser procedures. Ophthalmic Surg Lasers 30:199, 1999 296. Javitt JC, Schiffman RM: Clinical success and quality of life with brimonidine 0.2% or timolol 0.5% used
twice daily in glaucoma or ocular hypertension: A randomized
clinical trial. Brimonidine Outcomes Study Group I. J Glaucoma 9:224, 2000 297. Katz LJ: Brimonidine tartrate 0.2% twice daily vs timolol o.5% twice daily: 1-year
results in glaucoma patients. Brionidine Study Group. Am J Ophthalmol 127:20, 1999 298. Schuman JS, Horwitz B, Choplin NT et al: A 1-year study of brimonidine twice daily in glaucoma and ocular hypertension: A
controlled, randomized, multicenter clinical trial. Chronic
Brimonidine Study Group. Arch Ophthalmol 115:847, 1997 299. Hayreh SS, Zimmerman MB, Podhajsky P et al: Nocturnal arterial hypotension and its role in optic nerve head and ocular
ischemic disorders. Am J Ophthalmol 117:603,1994 300. Graham SL, Drance SM: Nocturnal hypotension: Role in glaucoma progression. Surv Ophthalmol 43:S10, 1999 301. Centofanti M, Manni G, Gregori D et al: Comparative acute effects of brimonidine 0.2% versus dorzolamide 2% combined
with beta-blockers in glaucoma. Graefes Archiv Clin Exp Ophthalmol 238:302, 2000 302. Theo Schwartzenberg OW, Buys YM: Efficacy of brimonidine 0.2% as adjunctive therapy for patients with glaucoma
inadequately controlled with otherwise maximal medical therapy. Ophthalmology 106:1616, 1999 303. Stewart WC, Sharpe ED, Day DG et al: Comparison of the efficacy and safety of latanoprost 0.005% compared to
brimonidine 0.2% or dorzolamide 2% when added to a topical beta-adrenergicblocker
in patients with primary open-angle glaucoma or ocular hypertension. J Ocul Pharmacol Ther 16:251, 2000 304. Kim DD: A case of suspected alphagan-induced psychosis. Arch Ophthalmol 118:1132, 2000 305. Walters G, Taylor RH: Severe systemic toxicity caused by brimonidine drops in an infant with
presumed juvenile xanthogranuloma. Eye 13:797, 1999 306. Carlsen JO, Zabriskie NA, Kwon YH et al: Apparent central nervous system depression in infants after the use of
topical brimonidine. Am J Ophthalmol 128:255, 1999 307. Korsch E, Grote A, Seybold M et al: Systemic adverse effects of topical treatment with brimonidine in an infant
with secondary glaucoma. Eur J Pediatr 158:685, 1999 308. Lark KK, Pasha AS, Yan X et al: The effect of latanoprost and brimonidine on rabbit subconjunctival fibroblasts.J Glaucoma 8:72, 1999 309. Serle JB et al: The effect of brimonidine tartrate in glaucoma patients on maximal medical
therapy. Invest Ophthalmol Vis Sci 34(Suppl):1137, 1993 310. Derick RJ et al: Brimonidine tartrate: A one-month dose response study. Invest Ophthalmol Vis Sci 34(Suppl):1138, 1993 311. Norlund JR et al: The cardiovascular, pulmonary, and ocular hypotensive effects of brimonidine
tartrate 0.2%. Arch Ophthalmol 113:77, 1995 312. Barnebey HS et al: The efficacy of brimonidine in decreasing elevation in intraocular pressure
after laser trabeculoplasty. Ophthalmology 100:1083, 1993 313. Laqueur L: Ueber eine neue therapeutische Verwendung des Physostigmins. Zentrabl Med Wissenschr 24:421, 1876 314. Velhagen K: Die Grundlager der Okularen: Pharmakologie und Toxikologie des Carbaminocholins. Arch Augenheilkd 107:319, 1933 315. Bonner TI: Domains of muscarinic acetylcholine receptors that confer specificity of
G protein coupling. Trends Pharmacol Sci 13:48, 1992 316. Kaufman PL: Aqueous humor dynamics following total iridectomy in the cynomolgus monkey. Invest Ophthalmol Vis Sci 18:870, 1979 317. Grierson I, Lee WR, Abraham S: Effect of pilocarpine on the morphology of the human outflow apparatus. Br J Ophthalmol 62:303, 1978 318. Lütjen-Drecoll E: Structural factors influencing outflow facility and its changeability under
drugs: A study in Macaca arctoides. Invest Ophthalmol 12:280, 1973 319. Kaufman PL, Bárány EH: Loss of acute pilocarpine effect on outflow facility following surgical
disinsertion and retrodisplacement of the ciliary muscle from the scleral
spur in the cynomolgus monkey. Invest Ophthalmol Vis Sci 15:793, 1976 320. Kaufman PL, Bárány EH: Residual pilocarpine effect on outflow facility after ciliary muscle disinsertion
in the cynomolgus monkey. Invest Ophthalmol 15:558, 1976 321. Bárány EH: The mode of action of miotics on outflow resistance: a study of pilocarpine
in the vervet monkey, Cercopithecus ethiops. Trans Ophthalmol Soc UK 86:539, 1966 322. Bárány EH: The mode of action of pilocarpine on outflow resistance in the eye of a
primate (Cercopithecus ethiops). Invest Ophthalmol 1:712, 1962 323. Shaffer RN: In Newell FW (ed): Glaucoma: Transactions of the Fifth Conference. New
York: Josiah Macy Jr Foundation, 1961 324. True-Gabelt B, Kaufman PL: Muscarinic receptor subtype antagonist inhibition of pilocarpine-stimulated
outflow facility, accommodation, and pupil constriction in rhesus
monkeys. Invest Ophthalmol Vis Sci 32(Suppl):1256, 1991 325. Lieberman TW, Leopold IH: The use of aceclidine in the treatment of glaucoma: Its effect on intraocular
pressure and facility of aqueous humor outflow as compared to that
of pilocarpine. Am J Ophthalmol 64:405, 1967 326. Drance SM, Fairclough M, Schulzer M: Dose response of human intraocular pressure to aceclidine. Arch Ophthalmol 88:394, 1972 327. Fechner PU, Teichmann KD, Weyrauch W: Accommodative effects of aceclidine in the treatment of glaucoma. Am J Ophthalmol 79:104, 1975 328. Pilza A, Lommatzsch P, Ulrich WD: Experimentelle und klinische Untersuchungen mit Aceclidin (Glaucostat). Ophthalmologica 168:37, 1974 329. McKinzie JW, Boggs MB Jr: Comparison of postoperative intraocular pressures after use of Miochol
and Miostat.J Cataract Refract Surg 15:185, 1989 330. Wedrich A, Menapace R: Intraocular pressure following small-incision cataract surgery and polyHEMA
posterior chamber lens implantation: A comparison between acetylcholine
and carbachol. J Cataract Refract Surg 18:500, 1992 331. West J et al: Prevention of acute postoperative pressure rises in glaucoma patients undergoing
cataract extraction with posterior chamber lens implantation. Br J Ophthalmol 76:534, 1992 332. Hollands RH, Drance SM, Schulzer M: The effect of acetylcholine on early postoperative intraocular pressure. Am J Ophthalmol 103:749, 1987 333. Grimmett MR et al: Corneal edema after Miochol. Am J Ophthalmol 115:236, 1993 (Letter) 334. Kronfeld PC: Eserine and pilocarpine: Our 100-year-old allies. Surv Ophthalmol 14:479, 1970 335. Weber A: Die Ursache des Glaukoms. Graefes Arch Ophthalmol 23:1, 1877 336. Goodman LS, Gilman A (eds): The Pharmacological Basis of Therapeutics. 5th
ed. New York: Macmillan, 1985 337. Harris LS, Galin MA: Dose response analysis of pilocarpine-induced ocular hypotension. Arch Ophthalmol 93:42, 1975 338. Drance SM, Nash PA: The dose response in human intra-ocular pressure to pilocarpine. Can J Ophthalmol 6:9, 1971 339. Drance SM, Bernsted M, Schulzer M: Pilocarpine and intraocular pressure: Duration of effectiveness of 4% and 8% pilocarpine
instillation. Arch Ophthalmol 91:104, 1974 340. March WF et al: Duration of effect of pilocarpine gel. Arch Ophthalmol 100:1270, 1982 341. Zimmerman TJ et al: Pilocarpine: A re-look at dose response and duration of action. Invest Ophthalmol Vis Sci 28(Suppl):377, 1987 342. Goldberg I et al: Efficacy and patient acceptance of pilocarpine gel. Am J Ophthalmol 88:843, 1979 343. Magder H, Boyaner D: The use of a longer-acting pilocarpine in the management of chronic simple
glaucoma. Can J Ophthalmol 9:285, 1974 344. Nagasubramanian S, Stewart RH, Hitchings RA: Long-term effects of glaucoma therapy with 4% pilocarpine gel on cornea
clarity and endothelial cell density. Int Ophthalmol 18:5, 1994 345. Johnson DH et al: A one-year multicenter clinical trial of pilocarpine gel. Am J Ophthalmol 97:723, 1984 346. Armaly MF, Rao KR: The effect of pilocarpine Ocusert with different release rates on ocular
pressure. Invest Ophthalmol 12:491, 1973 347. Lee P, Shen Y, Eberle M: The long-term Ocusert pilocarpine system in the management of glaucoma. Invest Ophthalmol 14:43, 1975 348. Quigley HA, Pollack IP, Harbin TSJ: Pilocarpine Ocuserts: Long-term clinical trials and selected pharmacodynamics. Arch Ophthalmol 93;771, 1975 349. Worthen DM, Zimmerman TJ, Wind CA: An evaluation of the pilocarpine Ocusert. Invest Ophthalmol 13:296, 1974 350. Place VA et al: Comparative pharmacologic effects of pilocarpine administered to normal
subjects by eye drops or by ocular therapeutic systems. Am J Ophthalmol 80:706, 1975 351. Hillman JS, Walker A, Davies EM: The management of chronic glaucoma with pilocarpine Ocusert. Trans Ophthalmol Soc UK 97:206, 1977 352. Brown HS et al: Visual effects of pilocarpine in glaucoma: Comparative study of administration
by eyedrops or by ocular therapeutic systems. Arch Ophthalmol 94:1716, 1977 353. Tymer G, Scheie H: Mechanism of the miotic-resistant pupil with increased intraocular pressure. Arch Ophthalmol 50:572, 1953 354. Francois J, Goes F: Ultrasonographic study of the effect of different miotics on the eye components. Ophthalmologica 175:328, 1977 355. Francois J, Goes F: Comparative ultrasonographic study of the effect of pilocarpine 2% and
Ocusert P 20 on the eye components. Am J Ophthalmol 86:233, 1978 356. Airaksinen PJ: The long-term hypotensive effect of timolol maleate compared with the effect
of pilocarpine in simple and capsular glaucoma. Acta Ophthalmol 57:425, 1979 357. Boger WP, Puliafito CA, Steinert RF et al: Long-term experience with timolol ophthalmic solution in patients with
open-angle glaucoma. Ophthalmology 85:259, 1978 358. Ashburn FS, Gillespie JE, Kass MA et al: Timolol plus maximum tolerated antiglaucoma therapy: A one-year follow-up
study. Surv Ophthalmol 23:389, 1979 359. Bill A, Walinder P: The effects of pilocarpine on the dynamics of aqueous humor in a primate (Macaca irus). Invest Ophthalmol 5:170, 1966 360. Friström B, Nilsson SE: Interaction with PhXA41, a new prostaglandin analogue, with pilocarpine. Arch Ophthalmol 111:662, 1993 361. Quaranta L, Ripandelli G, Manni GL et al: Hypotensive effect of pilocarpine after laser trabeculoplasty. J Glaucoma 1:233, 1992 362. Wilkie J, Drance SM, Schulzer M: The effects of miotics on anterior chamber depth. Am J Ophthalmol 68:78, 1969 363. Abramson DH, Coleman DJ, Forbes M et al: Pilocarpine: Effect on the anterior chamber and lens thickness. Arch Ophthalmol 87:615, 1972 364. Abramson DH, Franzen LA, Coleman DJ: Pilocarpine in the presbyope: Demonstration of an effect on the anterior
chamber and lens thickness. Arch Ophthalmol 89:100, 1973 365. Puustjari T: Retinal detachment during glaucoma therapy: A review of a case. Ophthalmologica 190:40, 1985 366. Weseley P, Liebmann J, Ritch R: Rhegmatogenous retinal detachment after initiation of Ocusert therapy. Am J Ophthalmol 112:458, 1991 367. Kalenak JW, Zavok ZN: Presumed sudden leakage of a pilocarpine Ocusert and rhegmatogenous retinal
detachment. J Glaucoma 3:152, 1994 368. Kraushar MF, Steinberg JA: Miotics and retinal detachment: Upgrading the community standard. Surv Ophthalmol 35:311, 1991 369. Pietch RL, Bobo CB, Vallotton WW: Lens opacities and organophosphate cholinesterase-inhibiting agents. Am J Ophthalmol 73:236, 1972 370. Shaffer RN, Hetherington I: Anticholinesterase drugs and cataracts. Am J Ophthalmol 62:613, 1966 371. Mori M, Araie M, Sakurai M et al: Effects of pilocarpine and tropicamide on blood-aqueous barrier permeability
in man. Invest Ophthalmol Vis Sci 33:416, 1992 372. Camras C, Campbell DG: Cases in controversy: Initial treatment of pigmentary glaucoma. J Glaucoma 2:44, 1993 373. Militor H: A comparative study of the effects of five choline compounds used in therapeutics: Acetylcholine
chloride, acetyl-beta-methylcholine chloride, carbaminoyl
beta-methylcholine chloride. J Pharmacol Exp Ther 58:337, 1936 374. O'Brien CS, Swan KC: Carbaminoylcholine chloride in the treatment of glaucoma simplex. Arch Ophthalmol 27:253, 1942 375. Smolen VF et al: Biophysical availability of ophthalmic carbachol. I. Mechanisms of cationic
polymer- and surfactant-promoted miotic activity. J Pharm Sci 62:958, 1973 376. McKinstry DN, Koelle GB: Effects of drugs on acetylcholine release from the cat superior cervical
ganglion by carbachol and by preganglionic stimulation. J Pharmacol Exp Ther 157:328, 1967 377. Hollands RH, Drance SM, Schulzer M: The effect of intracameral carbachol on intraocular pressure after cataract
extraction. Am J Ophthalmol 104:225, 1987 378. Stocker FW: Experimental studies on the blood-aqueous barrier. Arch Ophthalmol 37:583, 1947 379. McCombie H, Saunders BC: Alkyl fluorophosphonates: Preparation and physiological properties. Nature 157:287, 1946 380. Reichert RW, Shields MB: Intraocular pressure response to the replacement of pilocarpine or carbachol
with echo-thiophate. Graefes Arch Clin Exp Ophthalmol 229:252, 1991 381. Havener WH: Ocular Pharmacology. Vol 4. St Louis: CV Mosby, 1978 382. Barsam PC, Vogel HP: The effect of phospholine iodide on the diurnal variation of intraocular
pressure in glaucoma. Am J Ophthalmol 57:241, 1964 383. Drance SM, Carr F: Effects of Phospholine Iodide (217MI) on intraocular pressure in man. Am J Ophthalmol 49:470, 1960 384. Harris LS: Dose-response analysis of echothiophate iodide. Arch Ophthalmol 86:502, 1971 385. Lipson ML, Holmes JH, Ellis PP: Oral administration of pralidoxime chloride in echothiophate iodide toxicity. Arch Ophthalmol 82:830, 1969 386. de Roetth A Jr et al: Blood cholinesterase activity in glaucoma patients with phospholine iodide. Am J Ophthalmol 62:834, 1966 387. Kalow W: Hydrolysis of local anesthetics by human serum cholinesterase. J Pharmacol Exp Ther 104:122, 1952 388. Axelsson U, Holberg A: The frequency of cataract after miotic therapy. Acta Ophthalmol 44:421, 1966 389. Thoft RA: Incidence of lens changes in patients treated with echothiophate iodide. Arch Ophthalmol 80:317, 1968 390. Alpar JJ: Miotics and retinal detachment. Ann Ophthalmol 11:395, 1979 391. Pape LG, Forbes M: Retinal detachment and miotic therapy. Am J Ophthalmol 85:558, 1978 392. Patten JT, Cavanagh HD, Allansmith MR: Induced ocular pseudopemphigoid. Am J Ophthalmol 72:917, 1971 393. Drance SM, Carr F: Effect of demecarium bromide (BC 48) on intraocular pressure in man. Arch Ophthalmol 62:673, 1959 394. Friedenwald JS: Carbonic anhydrase inhibition and aqueous flow. Am J Ophthalmol 40:159, 1955 395. Friedenwald JS: The formation of intraocular fluid. Am J Ophthalmol 32:9, 1949 396. Wistrand PJ: Carbonic anhydrase in the anterior uvea of the rabbit. Acta Physiol Scand 24:144, 1951 397. Kinsey VE: Comparative chemistry of aqueous humor in posterior and anterior chambers
of rabbit eye. Arch Ophthalmol 50:401, 1953 398. Becker B: Decrease in intraocular pressure in man by a carbonic anhydrase inhibitor, Diamox. Am J Ophthalmol 37:13, 1954 399. Kupfer C, Lawrence C: Long-term administration of acetazolamide (Diamox) in the treatment of
glaucoma. Am J Ophthalmol 40:673, 1955 400. Dodgson SJ: The carbonic anhydrases: Overview of their importance in cellular
physiology and in molecular genetics. In Dodgson SJ et al (eds): The
Carbonic Anhydrases: Cellular Physiology and Molecular Genetics. New
York:Plenum Press, 1991 401. Wistrand PJ, Lindqvist A: Design of carbonic anhydrase inhibitors and the
relationship between the pharmacodynamics and pharmacokinetics of acetazolamide. In
Botré F, Gros G, Storey BT (eds): Carbonic Anhydrase
from Biochemistry and Genetics to Physiology and Clinical Medicine. New
York: VCH, 1991 402. Dobbs PC, Epstein DL, Anderson JP: Identification of isoenzyme C as the principal carbonic anhydrase in human
ciliary processes. Invest Ophthalmol Vis Sci 18:867, 1979 403. Wistrand PJ, Carg LC: Evidence of a high-activity C type of carbonic anhydrase inhibitor in human
ciliary processes. Invest Ophthalmol Vis Sci 18:802, 1979 404. Krupin T et al: Failure of acetazolamide to decrease intra-ocular pressure in patients
with carbonic anhydrase II deficiency. Am J Ophthalmol 99:396, 1985 405. Maren TH, Wynns GC, Wistrand PJ: Properties of carbonic anhydrase (CA) IV, the membrane-bound enzyme in
secretory tissues. Invest Ophthalmol Vis Sci 34(Suppl): 930, 1993 406. Murakami M et al: The loci of carbonic anhydrase activity in the ciliary epithelium of the
rabbit eye: Electrophysiological study with isolated ciliary epithelial
bilayer. Acta Histochem Cytochem 25:771, 1992 407. Schwam H et al: Identification and partial purification of a sodium dodecyl sulfate-resistant
carbonic anhydrase activity from the rabbit ciliary process. Invest Ophthalmol Vis Sci 34(Suppl):930, 1993 408. Wistrand PJ: The importance of carbonic anhydrase B and C for the unloading of CO3 by the human erythrocyte. Acta Physiol Scand 113:517, 1981 409. Dodgson SJ et al: Carbonic anhydrase activity of intact carbonic anhydrase II-eficient human
erythrocytes. J Appl Physiol 65:1472, 1988 410. Becker B: Use of methazolamide (Neptazane) in the therapy of glaucoma: Comparison
with acetazolamide (Diamox). Am J Ophthalmol 49:1307, 1960 411. Becker B: Carbonic anhydrase and the formation of aqueous humor. The Friedenwald
Memorial Lecture. Am J Ophthalmol 47:342, 1959 412. Becker B: The mechanism of the fall in intraocular pressure induced by the carbonic
anhydrase inhibitor, Diamox. Am J Ophthalmol 39:177, 1955 413. Becker B: The effects of carbonic anhydrase inhibitor acetazolamide on the composition
of the aqueous humor: Part II. Am J Ophthalmol 40:129, 1955 414. Holm O, Wiebert O: The effect of systemically given acetazolamide (Diamox) upon the formation
of aqueous humour in the human eye, measured with a new photogrammetric
method. Acta Ophthalmol 46:1243, 1968 415. Dailey RA, Brubaker RF, Bourne WM: The effect of timolol maleate and acetazolamide on the rate of aqueous
humor formation in normal human subjects. Am J Ophthalmol 93:232, 1982 416. Topper JE, Brubaker RF: Effects of timolol, epinephrine, and acetazolamide on aqueous flow during
sleep. Invest Ophthalmol Vis Sci 26:1315, 1985 417. Kupfer C, Gaasterland D, Ross K: Studies of aqueous humor dynamics in man. V. Effects of acetazolamide and
isoproterenol in young and old normal volunteers. Invest Ophthalmol 15:349, 1976 418. Kupfer C, Gaasterland D, Ross K: Studies of aqueous humor dynamics in man. II. Measurements in young normal
subjects using acetazolamide and L-epinephrine. Invest Ophthalmol 10:523, 1971 419. Maren TH: The relation between enzyme inhibition and physiological response in the
carbonic anhydrase system.J Pharmacol Exp Ther 139:140, 1963 420. Wistrand PJ: The use of carbonic anhydrase inhibitors in ophthalmology and clinical
medicine. Ann NY Acad Sci 429:609, 1984 421. McCannel CA, Heinrich SR, Brubaker RF: Acetazolamide but not timolol lowers aqueous humor flow in sleeping humans. Graefes Arch Clin Exp Ophthalmol 230:518, 1992 422. Bietti G et al: Acetazolamide, metabolic acidosis, and intraocular pressure. Am J Ophthalmol 80:360, 1975 423. Krupin T et al: Acidosis, alkalosis, and aqueous humor dynamics in rabbits. Invest Ophthalmol Vis Sci 16:997, 1977 424. Benedikt O, Zirm M, Harnocourt K: Die Beziehungen zwischen metabolischer Acidose und intraocularem Druck
nach Carboanhyudrasehemmung mit Acetazolamid. Graefes Arch Clin Exp Ophthalmol 190:247, 1974 425. Friedman Z, Krupin T, Becker B: Ocular and systemic effects of acetazolamide in nephrectomized rabbits. Invest Ophthalmol Vis Sci 23:209, 1982 426. Maren TH: The development of ideas concerning the role of carbonic anhydrase
in the secretion of aqueous humor: Relation to the treatment of
glaucoma. In Drance SM, Neufeld AH (eds): Glaucoma: Applied Pharmacology
in Medical Treatment. Orlando: Grune & Stratton, 1984:325 427. Maren TH: Carbonic anhydrase: Chemistry, physiology, and inhibition. Physiol Rev 47:595, 1967 428. Maren TH et al: The pharmacology of methazolamide in relation to the treatment of glaucoma. Invest Ophthalmol Vis Sci 16:730, 1977 429. Maren TH, Mayer E, Wadsworth BC: The pharmacology of Diamox: 2-acetylamino-1,3,4 thiadiazole-5-sulfonamide. Bull Johns Hopkins Hosp 95:199, 1954 430. Friedland BR, Mallonee J, Anderson DR: Short-term dose-response characteristics of acetazolamide in man. Arch Ophthalmol 95:1809, 1977 431. Lehmann B, Linnér E, Wistrand PJ: The pharmacokinetics of acetazolamide
in relation to its use in the treatment of glaucoma and its effects
as an inhibitor of carbonic anhydrases. In Rospe G (ed): Schering
Workshop in Pharmacokinetics. Vol 5. New York: Pergamon Press, 1970 432. Linnér E, Wistrand PJ: The initial drop of the intraocular pressure following intravenous administration
of acet-azolamide in man. Acta Ophthalmol 37:209, 1959 433. Berson FG et al: Acetazolamide dosage forms in the treatment of glaucoma. Arch Ophthalmol 98:1051, 1980 434. Berson FG, Epstein DL: Separate and combined effects of timolol maleate and acetazolamide in open-angle
glaucoma. Am J Ophthalmol 92:788, 1981 435. Kass M et al: Timolol and acetazolamide: A study of concurrent administration. Arch Ophthalmol 100:941, 1982 436. Smith JP et al: Betaxolol and acetazolamide: Combined ocular hypotensive effect. Arch Ophthalmol 102:1794, 1984 437. Lichter PR et al: Patient tolerance to carbonic anhydrase inhibitors. Am J Ophthalmol 85:495, 1978 438. Lichter PR: Reducing side effects of carbonic anhydrase inhibitors. Ophthalmology 88:266, 1981 439. Alward PD, Wilensky JT: Determination of acetazolamide compliance in patients with glaucoma. Arch Ophthalmol 99:1973, 1981 440. Epstein DL, Grant WM: Carbonic anhydrase inhibitor side effects: Serum chemical analysis. Arch Ophthalmol 95:1378, 1977 441. Epstein RJ, Allen RC, Lunde MW: Organic impotence associated with carbonic anhydrase inhibitor therapy
for glaucoma. Ann Ophthalmol 19:48, 1987 442. Kass MA et al: Acetazolamide and urolithiasis. Ophthalmology 88:261, 1981 443. Constant MA, Becker B: The effect of carbonic anhydrase inhibitors on urinary excretion of citrate
by humans. Am J Ophthalmol 49:929, 1960 444. Higashihara E et al: Calcium metabolism in acidotic patients induced by carbonic anhydrase inhibitors: Response
to citrate. J Urol 145:942, 1991 445. Parfitt AM: Acetazolamide and sodium bicarbonate-induced nephrocalcinosis and nephrolithiasis: Relationship
to citrate and calcium excretion. Arch Intern Med 124:736, 1969 446. Fraunfelder FT, Bagby GC: Possible hematologic reactions associated with carbonic anhydrase inhibitors. JAMA 261:2257, 1989 447. Fraunfelder FT et al: Hematologic reactions to carbonic anhydrase inhibitors. Am J Ophthalmol 100:79, 1985 448. Mogk L, Cyrlin MN: Blood dyscrasias and carbonic anhydrase inhibitors. Ophthalmology 95:768, 1988 449. Zimran A, Beutler E: Can the risk of acetazolamide-induced aplastic anemia be decreased by periodic
monitoring of blood cell counts? Am J Ophthalmol 104:654, 1987 450. Blocker ER, Rostand RA: Carbonic anhydrase inhibition in glaucoma: Hazard or benefit for the chronic
lunger? Surv Ophthalmol 23:169, 1978 451. Goodfield M, Davis J, Jeffcoate W: Acetazolamide and symptomatic metabolic acidosis in mild renal failure. BMJ 284:422, 1982 452. Ayvazian JH, Ayvazian LF: A study of the hyperuricemia induced by hydrochlorothiazide and acetazolamide. J Clin Invest 40:196, 1961 453. Wettrell K, Pandolfi M: Propanolol vs. acetazolamide: A long-term, double-masked study of the effect
on intraocular pressure and blood pressure. Arch Ophthalmol 97:280, 1979 454. Grant WM: Antiglaucoma drugs: Problems with carbonic anhydrase inhibitors. In
Leopold IH (ed): Symposium on Ocular Therapy. Vol 6. St Louis: CV
Mosby, 1973 455. Maren TH: Teratology and carbonic anhydrase inhibition. Arch Ophthalmol 85:1, 1971 456. Layton WM, Hallesy DW: Deformity of forelimb in rats: Association with high doses of acetazolamide. Science 149:306, 1965 457. Söderman P, Hartvig P, Fagerund C: Acetazolamide excretion into human breast milk. Br J Clin Pharmacol 17:599, 1984 458. Anderson CJ, Kaufman PL, Sturm RJ: Toxicity of combined therapy with carbonic anhydrase inhibitors and aspirin. Am J Ophthalmol 86:516, 1978 459. Coudon WL, Block AJ: Acute respiratory failure precipitated by a carbonic anhydrase inhibitor. Chest 69:112, 1976 460. Yablonski ME et al: Enhancement of the ocular hypotensive effect of acetazolamide by diflusinal. Am J Ophthalmol 106:332, 1988 461. Beasley FJ: Transient myopia and retinal edema during ethoxzolamide (Cardrase) therapy. Arch Ophthalmol 68:490, 1962 462. Galin MA, Baras I, Zweifash P: Diamox-induced myopia. Am J Ophthalmol 54:237, 1962 463. Vela A, Campbell DG: Hypotony and ciliochoroidal detachment following pharmacologic aqueous
suppressant therapy in previously filtered patients. Ophthalmology 92:50, 1985 464. Bayne WF et al: Time course and disposition of methazolamide in human plasma and red blood
cells. J Pharm Sci 70:78, 1981 465. Maren RH, Robinson B: The pharmacology of acetazol-amide as related to cerebrospinal fluid and
the treatment of hydrocephalus. Bull Johns Hopkins Hosp 106:1, 1960 466. Stone RA et al: Low-dose methazolamide and intraocular pressure. Am J Ophthalmol 83:674, 1977 467. Dahlen K et al: A repeated dose-response study of methazolamide in glaucoma. Arch Ophthalmol 96:2214, 1978 468. Merkle W: Untersuchungen über die Wirkung von Methazolamid auf den intraokularen
Druck. Klin Monatsbl Augenheilkd 176:181, 1980 469. Garrison L et al: A clinical comparison of three carbonic anhydrase inhibitors. Trans Pacific Coast Otolaryngol Ophthalmol Soc 48:137, 1967 470. Lichter PR et al: Patient tolerance to carbonic anhydrase inhibitors. Am J Ophthalmol 85:495, 1978 471. Sugrue MF et al: A study of the in vitro inhibition of human carbonic anhydrase isoenzymes
I, II, and IV. Invest Ophthalmol Vis Sci 34(Suppl):1143, 1993 472. Baldwin J et al: Thienothiopyran-2-sulfonamides: Novel typically active
carbonic anhydrase inhibitors for the treatment of glaucoma. J Med Chem 32:2510, 1989 (Letter) 473. Lippa EA: Topical carbonic anhydrase inhibitors. In Dodgson SJ et al (eds): The
Carbonic Anhydrases: Cellular Physiology and Molecular Genetics. New
York: Plenum Press, 1991 474. Wang RF et al: MK-507 (L-671, 152), a topically active carbonic anhydrase inhibitor, reduces
aqueous humor production in monkeys. Arch Ophthalmol 109:1297, 1991 475. Sugrue MF, Funk H, Klotzbuecher C: Comparison of the topical carbonic anhydrase inhibitor L-671,152 and timolol
in glaucoma monkeys. Invest Ophthalmol Vis Sci 31(Suppl):232, 1990 476. Wang RF et al: The ocular hypotensive effect of the topical carbonic anhydrase inhibitor
L-671,152 in glaucomatous monkeys. Arch Ophthalmol 108:511, 1990 477. Hoffman HM et al: MK-507 (L-671,152): Lokale Verträglichkeit und Wirksamkeit eines neuen
lokalen Karboanhydrasehemmers bei gesunden Probande. Fortschr Ophthalmol 88:513, 1991 478. Hoffman H et al: L-671,152: Local tolerability and activity of a new topical
carbonic anhydrase inhibitor in normal volunteers. Presented at
the Glaucoma Symposium of the XXVI International Congress of Ophthalmology, Singapore, March 17, 1990 479. Serle JB, Podos SM: Update on dorzolamide: A topical carbonic anhydrase
inhibitor. In Krieglstein GK (ed): Glaucoma Update. Vol V. Heidelberg: Kaden
Verlag, 1995:261 480. Bourgeois H et al: 2% L-671,152: Multiple-dose activity bid. Invest Ophthalmol Vis Sci 31(Suppl):233, 1990 481. Lippa E, Laibovitz R, Clineschmid C: L-671,152: A novel, active topical
carbonic anhydrase inhibitor in patients. Presented at the Glaucoma Symposium
of the XXVI International Congress of Ophthalmology, Singapore, 1990 482. Lippa EA et al: MK-507 vs. sezolamide: Comparative efficacy of two topically active carbonic
anhydrase inhibitors. Ophthalmology 98:308, 1991 483. Lippa EA et al: Dorzolamide hydrochloride: Six-week, dose-response study of an active, topical
carbonic anhydrase inhibitor. Invest Ophthalmol Vis Sci 34(Suppl): 931, 1993 484. Wilkerson M et al: Four-week safety and efficacy study of dorzolamide, a novel, active topical
carbonic anhydrase inhibitor. Arch Ophthalmol 111:1343, 1993 485. Strahlman ER et al: A controlled clinical trial of 2.0% dorzolamide (MK-507) compared to timolol
and betaxolol. Invest Ophthalmol Vis Sci 35(Suppl):2177, 1994 486. Nardin G et al: Activity of the topical CAI MK-507 bid when added to timolol bid. Invest Ophthalmol Vis Sci 32(Suppl):989, 1991 487. Laibovitz R et al: Comparison of quality of life and patient preference of dorzolamide and
pilocarpine as adjunctive therapy to timolol in the treatment of glaucoma. J Glaucoma 4:306, 1995 488. Kitazawa Y et al: Topical dorzolamide hydrochloride can be a substitute for oral carbonic
anhydrase inhibitors. Invest Ophthalmol Vis Sci 35(Suppl):2177, 1994 489. Serle JB et al: Compassionate case usage of dorzolamide (MK-507) in 16 glaucoma patients. Invest Ophthalmol Vis Sci 35(Suppl):2177, 1994 490. Kaplan BH et al: The use of MK-507 2% ophthalmic solution in patients with glaucoma or ocular
hypertension. Invest Ophthalmol Vis Sci 35(Suppl):2177, 1994 491. Gervasoni JP et al: Absence of metabolic effects of a methyl-thienothiopyran-2-sulfonamide, a
new carbonic anhydrase inhibitor (CAI), during a 2-week ocular administration. Clin Pharmacol Ther 49:192, 1991 492. Werner EB, Gerber DS, Yoder YI: Effect of a topical carbonic anhydrase inhibitor, 6-hydroxybenzo(b)thiophene-2-sulfonamide, on
intraocular pressure in normotensive subjects. Can J Ophthalmol 22:316, 1987 493. Kitazawa Y: Long-term treatment with MK-507, a topical carbonic anhydrase inhibitor. Ophthalmology 100(Suppl):81, 1993 494. Konowal A, Morrison JC, Brown SV et al: Irreversible corneal decompensation in patients treated with topical dorzolamide. Am J Ophthalmol 127:403, 1999 495. Giasson CJ, Nguyen TQ, Boisjoly HM et al: Dorzolamide and corneal recovery from edema in patients with glaucoma or
ocular hypertension. Am J Ophthalmol 129:144, 2000 496. Ingram CJ, Brubaker RE: Effect of brinzolamide and dorzolamide on aqueous humor flow in human eyes. Am J Ophthalmol 128:292, 1999 497. Sall K: The efficacy and safety of brinzolamide 1% ophthalmic suspension (Azopt) as
a primary therapy in patients with open-angle glaucoma or ocular
hypertension. Brinzolamide Primary Therapy Study Group. Surv Ophthalmol 44:S155, 2000 498. Silver LH: Ocular comfort of brinzolamide 1.0% ophthalmic suspension compared with
dorzolamide 2.0% ophthalmic solution: Results from two multicenter comfort
studies. Brinzolamide comfort study group. Surv Ophthalmol 44:S141, 2000 499. Ambache N: Irin, a smooth-muscle contracting substance present in rabbit iris. J Physiol 129:65P, 1955 500. Ambache N: Properties of irin, a physiological constituent of the rabbit iris. J Physiol 135:114, 1957 501. Bito LZ: Prostaglandins, old concepts and new perspectives. Arch Ophthalmol 105:1036, 1987 502. Beitch BR, Eakins KE: The effects of prostaglandins on the intraocular pressure of the rabbit. Br J Pharmacol 37:158, 1969 503. Waitzman MB, King CD: Prostaglandin influences on intraocular pressure and pupil size. Am J Physiol 212:329, 1967 504. Podos SM, Becker B, Kass MA: Indomethacin blocks arachidonic acid-induced elevation of intraocular pressure. Prostaglandins 3:7, 1973 505. Neufeld AH, Jampol LM, Sears ML: Aspirin prevents the disruption of the blood-aqueous barrier in the rabbit
eye. Nature 238:158, 1972 506. Cole DR, Unger WG: Prostaglandins as mediators of the responses of the eye to trauma. Exp Eye Res 17:357, 1973 507. Eakins KE et al: Prostaglandin-like activity in ocular inflammation. BMJ 3:452, 1972 508. Masuda K, Izawa Y, Mishima S: Prostaglandins and uveitis: A preliminary report. Jpn J Ophthalmol 17:166, 1973 509. Camras CB, Chacko DM, Schlossman A: Posner-Schlossman syndrome. In Pepose
JS, Holland GN, Wilhelmus KR (eds): Ocular Infection and Immunity. St
Louis: CV Mosby, 1996:529 510. Casey WJ: Prostaglandin E3 and aqueous humor dynamics in the rhesus monkey eye. Prostaglandins 8:327, 1974 511. Green K, Kim K: Pattern of ocular response to topical and systemic prostaglandin. Invest Ophthalmol 14:36, 1975 512. Kass MA et al: Prostaglandin E and aqueous humor dynamics. Invest Ophthalmol 11:1022, 1972 513. Masuda K, Mishima S: Effects of prostaglandins on inflow and outflow of the aqueous humor in
rabbits. Jpn J Ophthalmol 17:300, 1973 514. Camras CB, Bito LZ, Eakins KE: Reduction of intraocular pressure by prostaglandins applied topically to
the eyes of conscious rabbits. Invest Ophthalmol Vis Sci 16:1125, 1977 515. Coleman RA, Smith WL, Narumiya S: Classification of prostanoid receptors: Properties, distribution, and structure
of the receptors and their subtypes. Pharmacol Rev 46:205, 1994 516. Crawford KS et al: The DP-receptor agonist SQ27986 raises but does not lower intraocular pressure
in ocular normotensive monkeys. J Glaucoma 1:94, 1992 517. Woodward DF et al: Marked species differences in the pharmacology of prostanoid induced ocular
hypertension. Invest Ophthalmol Vis Sci 32(Suppl):1257, 1994 518. Woodward DF et al: Studies on the ocular hypotensive effects of prostaglandin F3a ester prodrugs and receptor selective prostaglandin analogs. J Ocul Pharmacol 10:177, 1994 519. Woodward DF et al: Intraocular pressure effects of selective prostanoid receptor agonists
involve different receptor subtypes according to radioligand binding studies. J Lipid Mediat 7:545, 1993 520. Woodward DF et al: Definition of prostanoid receptor subtypes by radioligand binding and the
effects of selective agonists on intraocular pressure. Exp Eye Res 55(Suppl): S91, 1992 521. Camras CB et al: Multiple dosing of prostaglandin F3a or epinephrine on cynomolgus monkey eyes: I. Aqueous humor dynamics. Invest Ophthalmol Vis Sci 28:463, 1987 522. Crawford K, Kaufman PL, Gabelt BT: Effects of topical PGF3a on aqueous humor dynamics in cynomolgus monkeys. Curr Eye Res 6:1035, 1987 523. Kaufman PL: Effects of intracamerally infused prostaglandins on outflow facility in
cynomolgus monkey eyes with intact or retrodisplaced ciliary muscle. Exp Eye Res 43:819, 1986 524. Lee PY, Podos SM, Severin C: Effect of prostaglandin F3a on aqueous humor dynamics of rabbit, cat, and monkey. Invest Ophthalmol Vis Sci 25:1087, 1984 525. Gabelt BT, Kaufman PL: Prostaglandin F3a increases uveo-scleral outflow in the cynomolgus monkey. Exp Eye Res 49:389, 1989 526. Nilsson SFE et al: Increased uveoscleral outflow as a possible mechanism of ocular hypotension
caused by prostaglandin F3a-1-isopropylester in the cynomolgus monkey. Exp Eye Res 48:707, 1989 527. Selen G et al: Effects of PhXA34 and PhDA100A, two phenyl substituted prostaglandin esters, on
aqueous humor dynamics and microcirculation in the monkey eye. Invest Ophthalmol Vis Sci 32(Suppl):869, 1991 528. Toris CB, Camras CB, Yablonski ME: Effects of PhXA41, a new prostaglandin F3a analog, on aqueous humor dynamics in human eyes. Ophthalmology 100:1297, 1993 529. Ziai N et al: The effects on aqueous humor dynamics of PhXA41, a new prostaglandin F3a analogue, after topical application in normal and ocular hypertensive
human eyes. Arch Ophthalmol 111:1351, 1993 530. Alm A, Villumsen J: PhXA34, a new potent ocular hypotensive drug: A study on dose response
relationship and on aqueous humor dynamics in healthy volunteers. Arch Ophthalmol 109:1564, 1991 531. Tamm E, Rittig M, Lutjen-Drecoll E: Electromikroskopische and immunohistochemische Untersuchungen zur augendrucksenkender
Wirkung von Prostaglandin F3a. Fortschr Ophthalmol 87:623, 1990 532. Lutchen-Drecoll E, Tamm E: Morphological study of the anterior segment of cynomolgus monkey eyes following
treatment with prostaglandin F3a. Exp Eye Res 47:761, 1988 533. Ocklind A: Effect of latanoprost on the extracellular matrix of the ciliary muscle. A
study on cultured cells and tissue sections. Exp Eye Res 67:179,1998 534. Lindsey JD, Kashiwagi K, Kashiwagi F et al: Prostaglandins alter extracellular matrix adjacent to human ciliary muscle
cells in vitro. Invest Ophthalmol Vis Sci 38:2214, 1997 535. Alm A et al: Intraocular pressure-reducing effect of PhXA41 in patients with increased
eye pressure: A one-month study. Ophthalmology 100:1312, 1993 536. Camras CB: Discussion of intraocular pressure-reducing effect of PhXA41 in patients
with increased eye pressure: A one-month study. Ophthalmology 100:1316, 1993 537. Hotehama Y, Mishimi HK: Clinical efficacy of PhXA34 and PhXA41, two novel prostaglandin F3a-isopropyl ester analogues for glaucoma treatment. Jpn J Ophthalmol 37:259, 1993 538. Hotehama Y et al: Ocular hypotensive effect of PhXA41 in patients with ocular hypertension
or primary open-angle glaucoma. Jpn J Ophthalmol 37:2790, 1993 539. Nagasubramanian S et al: Intraocular pressure-reducing effect on PhXA41 in ocular hypertension: Comparison
of dose regimens. Ophthalmology 100:1305, 1993 540. Racz P et al: Maintained intraocular pressure reduction with once-a-day application of
a new prostaglandin F3a analogue (PhXA41). Arch Ophthalmol 111:657, 1993 541. Camras CB, United States Latanoprost Study Group: Comparison of latanoprost
and timolol in patients with ocular hypertension and glaucoma. Ophthalmology 103:138, 1996 542. Watson P, Stjernschantz J, Latanoprost Study Group: A six-month, randomized, double-masked
study comparing latanoprost with timolol in open-angle
glaucoma and ocular hypertension. Ophthalmology 103:126, 1996 543. Alm A, Stjernschantz J, Scandinavian Latanoprost Study Group: Effects of
intraocular pressure and side effects of 0.005% latanoprost applied
once daily, evening or morning: A comparison with timolol. Ophthalmology 102:1743, 1995 544. Aung T, Wong HT, Yip CC et al: Comparison of the intraocular pressure-lowering effect of latanoprost and
timolol in patients with chronic angle closure glaucoma: A preliminary
study. Ophthalmology 107:1178, 2000 545. Scherrer WJ, Hauber FA: Effect of latanoprost on intraocular pressure in steroid-induced glaucoma. J Glaucoma 9:179, 2000 546. Mastropasqua L, Carpineto P, Ciancaglini M et al: A 12-month, randomized, double-masked study comparing latanoprost with
timolol in pigmentary glaucoma. Ophthalmology 106:550,1999 547. Enyedi LB, Freedman SF, Buckley EG: The effectiveness of latanoprost for the treatment of pediatric-glaucoma.J AAPOS: J Am Assoc Pediatr Ophthalmol Strabismus 3:33, 1999 548. Altuna JC, Greenfield DS, Wand M et al: Latanoprost in glaucoma associated with Sturge-Weber syndrome: Benefits
and side-effects. J Glaucoma 8:199, 1999 549. Yang CB, Freedman SF, Myers JS et al: Use of latanoprost in the treatment of glaucoma associated with Sturge-Weber
syndrome. Am J Ophthalmol 126:600, 1998 550. Alm A et al: Latanoprost administered once daily caused a maintained reduction of intraocular
pressure in glaucoma patients treated concomitantly with timolol. Br J Ophthalmol 79:12, 1995 551. Rulo AH, Greve EL, Hoyng PF: Additive effect of latanoprost, a prostaglandin F3a analogue, and timolol in patients with elevated intraocular pressure. Br J Ophthalmol 78:899, 1994 552. Nordmann JP, Soderstrom M, Rouland JF et al: Comparison of the intraocular pressure lowering effect of latanoprost and
a fixed combination of timolol-pilocarpine eye drops in patients insufficiently
controlled with beta adrenergic antagonists. French latanoprost
study group, and the Swedish latanoprost study group. Br J Ophthalmol 84:181, 2000 553. Emmerich KH: Comparison of latanoprost monotherapy to dorzolamide combined with timolol
in patients with glaucoma or ocular hypertension. A 3-month randomised
study. Graefes Arch Clin Exp Ophthalmol 238:19, 2000 554. Linden C, Alm A: Latanoprost and physostigmine have mostly additive ocular hypotensive effects
in human eyes. Arch Ophthalmol 115:857, 1997 555. Shin DH, McCracken MS, Bendel RE et al: The additive effect of latanoprost to maximally-tolerated medications with
low-dose, high-dose or no pilocarpine therapy. Ophthalmol 106:386, 1999 556. Patelska B, Greenfield DS, Liebmann JM et al: Latanoprost for uncontrolled glaucoma in a compassionate case protocol. Am J Ophthalmol 124:279, 1997 557. Wang RF, Podos SM, Serle JB et al: Effect of latanoprost or 8-iso prostaglandin E2 alone and in combination
on intraocular pressure in glaucomatous monkey eyes. Arch Ophthalmol 118:74, 2000 558. Sjoquist B, Tajallaei S, Stjernschantz J: Pharmacokinetics of latanoprost in the cynomolgus monkey. 1st communication. Single
intravenous, oral or topical administration on the eye. Arzneimittel-Forschung 49:225, 1999 559. Sjoquist B, Uhlin A Byding P et al: Pharmacokinetics of latanoprost in the cynomolgus monkey. 2nd communication. Repeated
topical administration on the eye. Arz Forschung 49:234, 1999 560. Moroi SE, Gottfredsdottir MS, Schteingart MT et al: Cystoid macular edema associated with latanoprost therapy in a case series
of patients with glaucoma and ocular hypertension. Ophthalmol 106:1024, 1999 561. Ayyala RS, Cruz DA, Margo CE et al: Cystoid macular edema associated with lataonoprost in aphakic and pseudophakic
eyes. Am J Ophthalmol 126:602, 1998 562. Callanan D, Fellamn RL, Savage JA: Latanoprost-associated cystoid macular edema. Am J Ophthalmol 126:134, 1998 563. Warwar RE, Bullock JD, Ballal D: Cystoid macular edema and anterior uveitis associated with latanoprost
use. Experience and incidence in a retrospective review of 94 patients. Ophthalmology 105:263, 1998 564. Miyake K, Ota I, Maekubo K et al: Latanoprost accelerates disruption of the blood-aqueous barrier and the
incidence of angiographic cystoid macular edema in early postoperative-pseudophakia. Arch Ophthalmol 117:34, 1999 565. Dutkiewicz R, Albert DM, Levin LA: Effects of latanoprost on tyrosinase activity and mitotic index of cultured
melanoma lines. Exp Eye Res 70:563, 2000 566. Camras CB, Neely DG, Weiss EL: Latanoprost-induced iris color darkening: a case report with long-term
follow-up. J Glaucoma 9:95, 2000 567. Johnstone MA: Hypertrichosis and increased pigmentation of eyelashes and adjacent hair
in the region of the ipsilateral eyelids of patients treated with unilateral
topical latanoprost. Am J Ophthalmol 124:544, 1997 568. Kook MS, Lee K: Increased eyelid pigmentation associated with use of latanoprost. Am J Ophthalmol 129:804, 2000 569. Wand M, Gilbert CM, Liesegang TJ: Latanoprost and herpes simplex keratitis. Am J Ophthalmol 127:602, 1999 570. Kaufman HE, Yarnell ED, Thompson HW: Latanoprost increases the severity and recurrence of herpetic keratitis
in the rabbit. Am J Ophthalmol 127:531, 1999 571. Linden C, Alm A: Prostaglandin analogues in the treatment of glaucoma. Drugs Aging 14:387, 1999 572. Nordmann JP, Rouland JF, Mertz BP: A comparison of the intraocular pressure-lowering effect of 0.5% timolol
maleate and the docosanoid derivative of a prostaglandin F2 alpha metabolite, 0.12% unoprostone, in subjects with chronic open-angle glaucoma
or ocular hypertension. Curr Med Res Opin 15:87, 1999 573. Stewart WC, Stewart JA, Kapik BM: The effects of unoprostone isopropyl 0.12% and timolol maleate 0.5% on
diurnal intraocular pressure. J Glaucoma 7:388, 1998 574. Serle JB, Podos SM, Kitizawa Y et al: A comparative study of latanoprost (Xalatan) and isopropyl unoprostone (Rescula) in
normal and glaucomatous monkey eyes. Jpn J Ophthalmol 42:95, 1998 575. Yamamoto T, Kitizawa Y: Iris-color change developed after topical isopropyl unoprostone treatment. J Glaucoma 6:430, 1997 576. Robbins R, Galin MA: Effect of osmotic agents on the vitreous body. Arch Ophthalmol 82:694, 1969 577. Cantonnet A: Essai de traitement du glaucome par les substances osmotiques. Arch Ophthalmol 24:1, 1904 578. Hertel E: Experimentelle Untersuchungen über die Abhangigkeit des Augendrucks
von der Blutbeschaffenheit. Graefes Arch Clin Exp Ophthalmol 88:197, 1914 579. Javid M: Urea: New use of an old agent. Reduction of intracranial and intraocular
pressure. Surg Clin North Am 38:907, 1958 580. Javid M, Settlage P: Effect of urea on cerebrospinal fluid pressure in human subjects: Preliminary
report. JAMA 160:943, 1945 581. Weiss DI, Shaffer RN, Wise BL: Mannitol infusion to reduce intraocular pressure. Arch Ophthalmol 68:341, 1962 582. Virno M et al: Oral glycerol in ophthalmology: A valuable new method for the reduction
of intraocular pressure. Am J Ophthalmol 55:1133, 1963 583. Becker B, Kolker AE, Krupin T: Isosorbide, an oral hyperosmotic agent. Arch Ophthalmol 78:147, 1967 584. Krupin T, Podos SM, Becker B: Effect of optic nerve transection on osmotic alterations of intraocular
pressure. Am J Ophthalmol 70:214, 1970 585. Podos SM, Krupin T, Becker B: Effect of small dose hyperosmotic injection on intraocular pressure of
small animals and man when optic nerves are transected and intact. Am J Ophthalmol 71:898, 1971 586. Galin MA, Davidson R, Shachter N: Ophthalmological use of osmotic therapy. Am J Ophthalmol 62:629, 1966 587. Smith EW, Drance SM: Reduction of human intraocular pressure with intravenous mannitol. Arch Ophthalmol 68:734, 1962 588. Brogliotti B, Marello M, Cembrano S: Azlione ioptonizzante del mannitolo in somministrazione endovenosa a bolo. Boll Ocul 57:547, 1978 589. O'Keefe M, Nabil M: The use of mannitol in intraocular surgery. Ophthalmic Surg 14:55, 1983 590. Adams RE, Kirschner RJ, Leopold IH: Ocular hypotensive effect of intravenously administered mannitol: A preliminary
report. Arch Ophthalmol 69:55, 1963 591. Barry KG, Khoury AH, Brooks MH: Mannitol and isosorbide: Sequential effects on intraocular pressure, serum
osmolality, sodium, and solids in normal subjects. Arch Ophthalmol 81:695, 1969 592. Grabie MT et al: Contraindications for mannitol in aphakic glaucoma. Am J Ophthalmol 91:265, 1981 593. D'Alena P, Ferguson W: Adverse effects after glycerol orally and mannitol parenterally. Arch Ophthalmol 75:201, 1966 594. Almog Y, Eyer O, Laser M: Pulmonary edema as a complication of oral glycerol administration. Ann Ophthalmol 18:38, 1986 595. Borges MF, Mochs J, Kjellstrand CM: Mannitol intoxication in patients with renal failure. Arch Intern Med 142:63, 1982 596. Whelan T et al: Acute renal failure associated with mannitol intoxication. Arch Intern Med 144:2053, 1984 597. McNeill IJ: Hypersensitivity reaction to mannitol. Drug Intell Clin Pharm 19:552, 1985 598. Drance SM: Effect of oral glycerol on intraocular pressure in normal and glaucomatous
eyes. Arch Ophthalmol 72:491, 1964 599. Thomas RC: Glycerin: An orally effective osmotic agent. Arch Ophthalmol 70:625, 1963 600. Oakley DE, Ellis PP: Glycerol and hyperosmolar nonketotic coma. Am J Ophthalmol 81:469, 1976 601. Krupin T, Kolker AE, Becker B: A comparison of isosorbide and glycerol for cataract surgery. Am J Ophthalmol 69:737, 1970 602. Wisznia KI, Lazar M, Leopold IM: Oral isosorbide and intraocular pressure. Am J Ophthalmol 70:630, 1970 603. Mehra K, Singh R: Lowering of intraocular pressure by isosorbide: Effects of different doses
of drug. Arch Ophthalmol 86:623, 1971 604. Mehra K et al: Lowering of intraocular tension: Effects of isosorbide and glycerin. Arch Ophthalmol 85:167, 1971 605. Mao LK, Stewart WC, Shields MB: Correlation between intraocular pressure control and progressive glaucomatous
damage in primary open-angle glaucoma. Am J Ophthalmol 111:51, 1991 606. Araujo SV, Spaeth GL, Roth SM et al: A ten-year follow-up on a prospective, randomized trial of postoperative
corticosteroids after trabeculectomy. Ophthalmology 102:1753, 1995 607. Nouri-Mahdavi K, Brigatti L, Weitzman M et al: Outcomes of trabeculectomy for primary open-angle glaucoma. Ophthalmology 102:1760, 1995 608. Jay JL, Alan D: The benefit of early trabeculectomy versus conventional management in primary
open angle glaucoma relative to severity of disease. Eye 3:528, 1989 609. Sommer A: Intraocular pressure and glaucoma. Am J Ophthalmol 107:186, 1989 610. Cartwright MJ, Anderson DR: Correlation of asymmetric damage with asymmetric intraocular pressure in
normal-tension glaucoma. Arch Ophthalmol 106:898, 1988 611. Chauhan BC, Drance SM: The influence of intraocular pressure on visual field damage in patients
with normal-tension and high-tension glaucoma. Invest Ophthalmol Vis Sci 31:2367, 1990 612. Hitchings RA, Wu J, Poinoosawmy D et al: Surgery for normal tension glaucoma. Br J Ophthalmol 79:402, 1995 613. Schulzer M et al: Biostatistical evidence for two distinct chronic open angle glaucoma populations. Br J Ophthalmol 74:196, 1990 614. Caprioli J, Spaeth GL: Comparison of the optic nerve head in high- and low-tension glaucoma. Arch Ophthalmol 103:1145, 1985 615. Drance SM et al: Diffuse visual field loss in chronic open-angle and low-tension glaucoma. Am J Ophthalmol 104:577, 1987 616. Caprioli J, Sears M, Miller J: Patterns of early visual field loss in open-angle glaucoma. Am J Ophthalmol 103:512, 1987 617. Schumer RA, Podos SM: The nerve of glaucoma! Arch Ophthalmol 112:37, 1994 618. Flammer J, Guthauser U, Mahler F: Do ocular vasospasms help cause low tension
glaucoma? In Greve EL, Heijl A (eds): Seventh International Visual
Field Symposium, Amsterdam, September 1986. Dordrecht, The Netherlands: Martinus
Nijhoff/Dr W. Junk Publishers, 1987:397 619. Rojanapongpun P, Drance SM: The response of blood flow velocity in the ophthalmic artery and blood
flow of the finger to warm and cold stimuli in glaucomatous patients. Graefes Arch Clin Exp Ophthalmol 231:375, 1993 620. Netland PA et al: Calcium channel blockers in the management of low-tension and open-angle
glaucoma. Am J Ophthalmol 115:608, 1993 621. Kitazawa Y et al: The effect of Ca2(+ )-antagonist on visual field in low-tension glaucoma. Graefes Arch Clin Exp Ophthalmol 227:408, 1989 622. Dugan LL, Choi DW: Excitotoxicity, free radicals, and cell membrane changes. Ann Neurol 35:S17, 1994 623. Cummins D, Takahashi N, Caprioli J: Electrophysiology of cultured retinal
ganglion cells to investigate basic mechanisms of damage. In Krieglstein
GK (ed): Glaucoma Update IV. Proceedings of the Glaucoma Society
of the International Congress of Ophthalmology, Bali, March 1990. Berlin: Springer-Verlag, 1991:59 624. Olney J: Neurotoxicity of excitatory amino acids. In McGreer E, Olney J, McGreer
P (eds): Kainic Acid as a Tool in Neurobiology. New York: Raven
Press, 1978:95 625. Lucas D, Newhouse J: The toxic effects of sodium l-glutamate on the inner layers of the retina. Arch Ophthalmol 58:193, 1957 626. Choi D: Glutamate neurotoxicity and diseases of the nervous system. Neuron 1:623, 1988 627. Ehinger BZ: Glutamate as a retinal neurotransmitter. In Weiler R, Osbourne
N (eds): Neurobiology of the inner retina. Berlin: Springer-Verlag, 1989:1 628. David P, Lusky M, Teichberg V: Involvement of excitatory neurotransmitters in the damage produced in chick
embryo retinae by anoxia and extracellular high potassium. Exp Eye Res 46:657, 1988 629. Zeevalk G, Hyndman A, Nicklas W: Excitatory amino acid induced toxicity in chick retina: Amino acid release, histology
and effects of chloride channel blockers. J Neurochem 53:1610, 1989 630. Dreyer EB, Lipton SA: Excitatory amino acids in glaucoma: A potentially novel etiology of neuronal
loss. Invest Ophthalmol Vis Sci 33(Suppl):1093, 1992 631. Schumer RA, Podos SM, Lipton SA et al: Increased glutamate in the vitreous of monkeys with induced glaucoma. Invest Ophthalmol Vis Sci 35(Suppl):1484, 1994 632. Cummins D, Kitano S, Caprioli J: The effects of excitatory amino acids and ischemia on cultured rat retinal
ganglion cells. Invest Ophthalmol Vis Sci 33(Suppl):1031, 1992 633. Kittigawa K: Hyperthermia-induced neuronal protection against ischemic injury in gerbils. J Cereb Blood Flow Metab 11:449, 1991 634. Barbe M, Tytell M, Gower D et al: Hyperthermia protects against light damage in the rat retina. Science 241:1817, 1988 635. Raibowol K, Mizzen L, Welch W: Heat shock is lethal to fibroblasts microinjected with antibodies against
hsp70. Science 242:433, 1988 636. Kitano S, Caprioli J: Expression of 72kDa heat shock protein in cultured rat retinal ganglion
cells: Ischemic tolerance and protective effect of hyperthermia. Invest Ophthalmol Vis Sci 34(Suppl):1430, 1993 637. Kitano S, Hori S, Koseki Y et al: The heat shock protein synthesis inhibitor, quercetin, blocks the protection
effect of hyperthermia on anoxic rat retinal ganglion cells in culture. Invest Ophthalmol Vis Sci 35(Suppl):1868, 1994 638. Moncada S et al: Nitric oxide: Physiology, pathophysiology, and pharmacology. Pharmacol Rev 43:109, 1991 639. Koseki Y, Kitano S, Podos SM et al: A nitric oxide synthase inhibitor protects against anoxia in cultured rat
retinal ganglion cells. Invest Ophthalmol Vis Sci 35(Suppl): 1968, 1994 640. Pellegrini-Giampietro D et al: Excitatory amino acid release and free radical formation may cooperate
in the genesis of ischemia-induced neuronal damage. J Neurosci 10:1035, 1990 641. McNamara J, Fridovich I: Did radicals strike Lou Gehrig? Nature 362:20, 1993 642. Aguayo AJ et al: Synaptic connections made by axons regenerating in the central nervous
system of adult mammals. J Exp Biol 153:199, 1990 643. Villegas-Perez MP, Vidal-Sanz M, Bray GM et al: Influences of peripheral nerve grafts on the survival and regrowth of axotomized
retinal ganglion cells in adult rats.J Neurosci 8:265, 1988 |