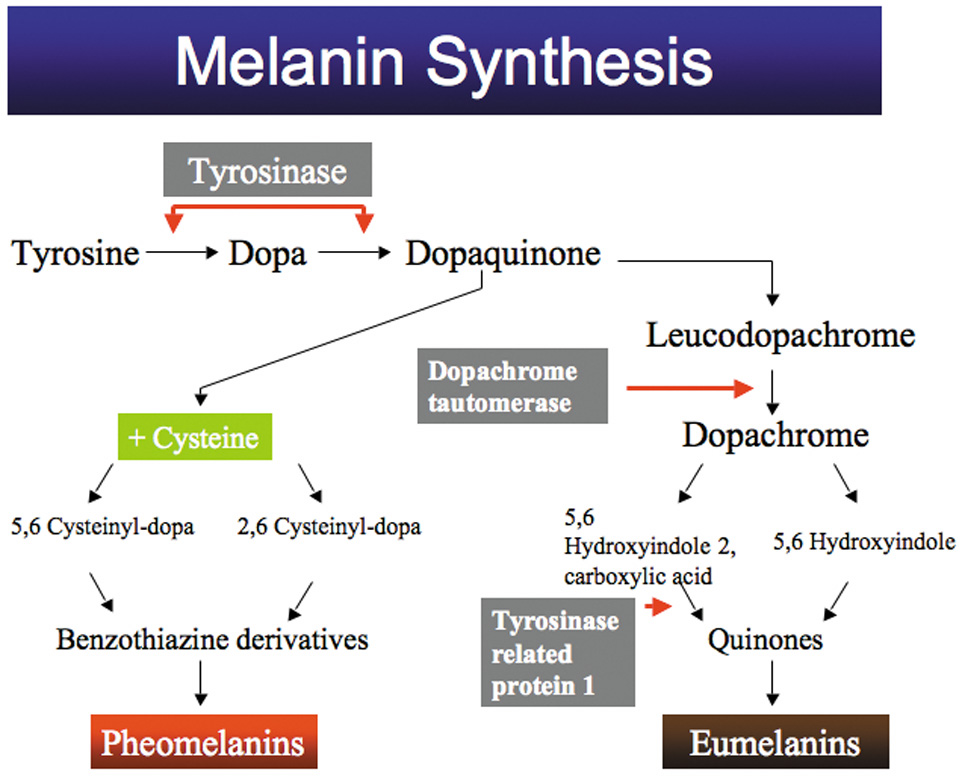
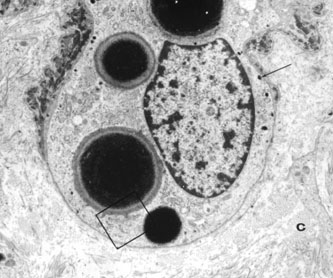
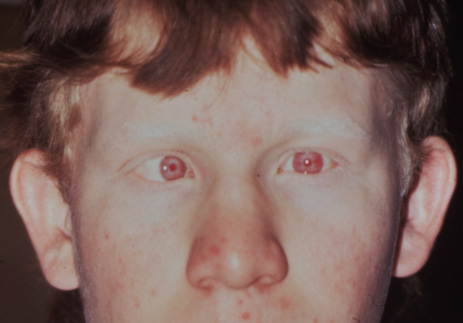
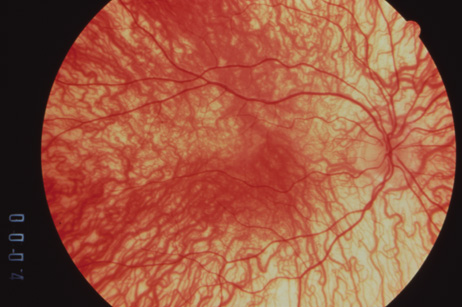
1. Abrams L, Traboulsi EI: Albinism. In: Traboulsi EI (ed). Genetic Diseases of the Eye. New York: Oxford University Press, 1999:697–721. 2. Oetting WS, Brilliant MH, King RA: The clinical spectrum of albinism in humans. Mol Med Today 2:330–335, 1996. 3. Spritz RA: Molecular genetics of oculocutaneous albinism. Hum Mol Genet 3(Spec No):1469–75, 1994. 4. Carden SM, Boissy RE, Schoettker PJ, Good WV: Albinism: modern molecular diagnosis. Br J Ophthalmol 82:189–195, 1998. 5. Oetting WS, King RA: Molecular basis of albinism: mutations and polymorphisms of pigmentation
genes associated with albinism. Hum Mutat 13:99–115, 1999. 6. Sorsby A: Noah—An albino. Br Med J 2:1587, 1958. 7. Froggatt P: Albinism in Northern Ireland. Ann Hum Genet 24:213, 1960. 8. Nance WE, Jackson CE, Witkop CJ Jr: Amish albinism: a distinctive autosomal recessive phenotype. Am J Hum Genet 22:579–586, 1970. 9. King RA, Lewis RA, Townsend D, et al: Brown oculocutaneous albinism. Ophthalmology 92:1496–1505, 1985. 10. Jay B, Witkop CJ, King RA: Albinism in England. Birth Defects 18:319, 1982. 11. Izquierdo NJ, Townsend W, Hussels IE: Ocular findings in the Hermansky-Pudlak syndrome. Trans Am Ophthalmol Soc 93:191–200, 1995. 12. Introne W, Boissy RE, Gahl WA. Clinical, molecular, and cell biological aspects of Chediak-Higashi syndrome. Mol Genet Metab 68:283–303, 1999. 13. Durham FM: On the presence of tyrosinase in the skin of some pigmented vertebrates. Proc R Soc Lond 74:310, 1904. 14. Kugelman TP, Van Scott EJ: Tyrosinase activity in melanocytes of human albinos. J Invest Dermatol 37:73, 1961. 15. Toyofuku K, Wada I, Spritz RA, Hearing VJ: The molecular basis of oculocutaneous albinism type 1 (OCA1): Sorting
failure and degradation of mutant tyrosinases results in a lack
of pigmentation. Biochem J 15;355(Pt 2):259–269, 2001. 16. Giebel LB, Tripathi RK, Strunk KM, et al: Tyrosinase gene mutations associated with type IB (“yellow”) oculocutaneous
albinism. Am J Hum Genet 48:1159–1167, 1991. Erratum in: Am J Hum Genet 49:696, 1991. 17. Giebel LB, Tripathi RK, King RA, Spritz RA: A tyrosinase gene missense mutation in temperature-sensitive type I oculocutaneous
albinism. A human homologue to the Siamese cat and the Himalayan
mouse. J Clin Invest 1991; 87:1119–1122. 18. Berson JF, Frank DW, Calvo PA, et al: A common temperature-sensitive allelic form of human tyrosinase is retained
in the endoplasmic reticulum at the nonpermissive temperature. J Biol Chem 275:12281–12289, 2000. 19. Rinchik EM, Bultman SJ, Horsthemke B, et al: A gene for the mouse pink-eyed dilution locus and for human type II oculocutaneous
albinism. Nature 36:72–76, 1993. 20. Toyofuku K, Valencia JC, Kushimoto T, et al: The etiology of oculocutaneous albinism (OCA) type II: The pink
protein modulates the processing and transport of tyrosinase. Pigment Cell Res 15:217–224, 2002. 21. Manga P, Kromberg J, Turner A, et al: In southern Africa, brown oculocutaneous albinism (BOCA) maps
to the OCA2 locus on chromosome 15q: P-gene mutations identified. Am J Hum Genet 68:782–787, 2001. 22. Manga P, Kromberg JGR, Box NF, et al: Rufous oculocutaneous albinism in southern African blacks is caused by
mutations in the TYRP1 gene. Am J Hum Genet 61:1095–1101, 1997. 23. Sarangarajan R, Boissy RE: Tyrp1 and oculocutaneous albinism type 3. Pigment Cell Res 14:437–444, 2001. 24. Newton JM, Cohen-Barak O, Hagiwara N, et al: Mutations in the human orthologue of the mouse underwhite gene (uw) underlie
a new form of oculocutaneous albinism, OCA4. Am J Hum Genet 69:981–988, 2001. 25. Costin GE, Valencia JC, Vieira WD, et al: Tyrosinase processing and intracellular trafficking is disrupted in mouse
primary melanocytes carrying the underwhite (uw) mutation. A
model for oculocutaneous albinism (OCA) type 4. J Cell Sci 116(Pt 15):3203–3212, 2003. 26. Rundshagen U, Zuhlke C, Opitz S, et al: Mutations in the MATP gene in five German patients affected by oculocutaneous
albinism type 4. Hum Mutat 23:106–110, 2004. 27. Bassi MT, Schiaffino MV, Renieri A, et al: Cloning of the gene for ocular albinism type I from the distal arm of the
X chromosome. Nat Genet 10:13–19, 1995. 28. Huizing M, Anikster Y, Gahl WA: Hermansky-Pudlak syndrome and related disorders of organelle formation. Traffic 1:823–835, 2000. 29. Schnur RE, Gao M, Wick PA, et al: OA1 mutations and deletions in X-linked ocular albinism. Am J Hum Genet 62:800–809, 1998. 30. d'Addio M, Pizzigoni A, Bassi MT, et al: Defective intracellular transport and processing of OCA1 is a major cause
of ocular albinism type 1. Hum Mol Genet 9:3011–3018, 2000. 31. van Dorp DB: Albinism, or the NOACH syndrome. Clin Genet 31:228, 1987. 32. Creel DJ, Summers CG, King RA: Visual anomalies associated with albinism. Ophthalmic Paediatr Genet 11:193–200, 1990. 33. Summers CG: Vision in albinism. Trans Am Ophthalmol Soc 94:1095–155, 1996. 34. Mietz H, Green R, Wolff SM, Abundo GP: Foveal hypoplasia in complete oculocutaneous albinism. Retina 12:254–260, 1992. 35. Meyer CH, Lapolice DJ, Freedman SF: Foveal hypoplasia in oculocutaneous albinism demonstrated by optical coherence
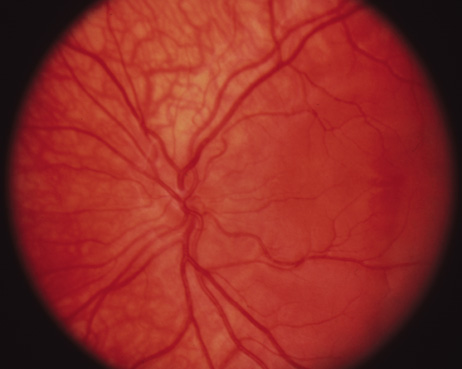
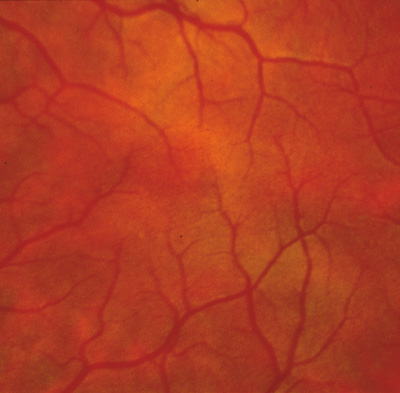
tomography. Am J Ophthalmol 133:409–410, 2002. 36. Gregor Z: The perifoveal vasculature in albinism. Br J Ophthalmol 62:554–557, 1978. 37. Spedick MJ, Beauchamp GR: Retinal vascular and optic nerve abnormalities in albinism. Pediatr Ophthalmol Strabismus 23:58–63, 1986. 38. Catalano RA, Nelson LB, Schaffer DB: Oculocutaneous albinism associated with congenital glaucoma. Ophthalmic Pediatr Genet 9:5–6, 1988. 39. Larkin DFP, O'Donoghue HN: Developmental glaucoma in oculocutaneous albinism. Ophthalmic Pediatr Genet 9:1–4, 1988. 40. Jeffery G: The albino retina: an abnormality that provides insight into normal retina
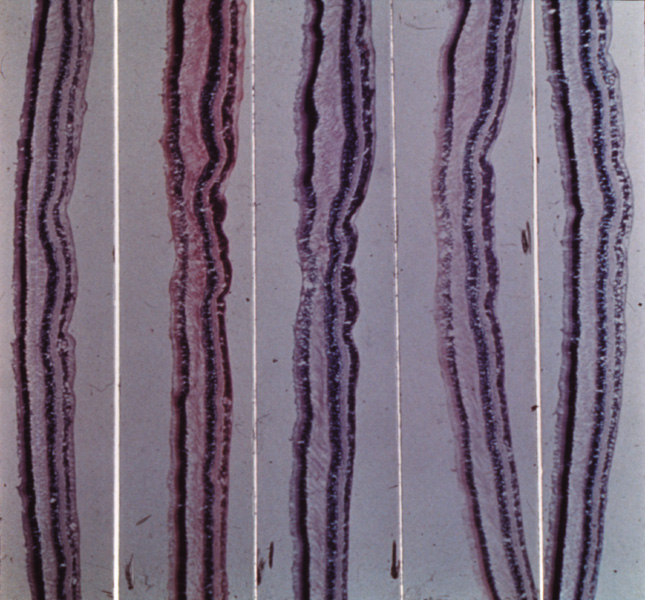
development. Trends Neurosci 20:165–169, 1997. 41. Beermann F, Schmid E, Schutz G: Expression of the mouse tyrosinase gene during embryonic development: Recapitulation
of the temporal regulation in transgenic mice. Proc Natl Acad Sci U S A 89:2809–2813, 1992. 42. Drager UC: Birth dates of retinal ganglion cells giving rise to the crossed and uncrossed
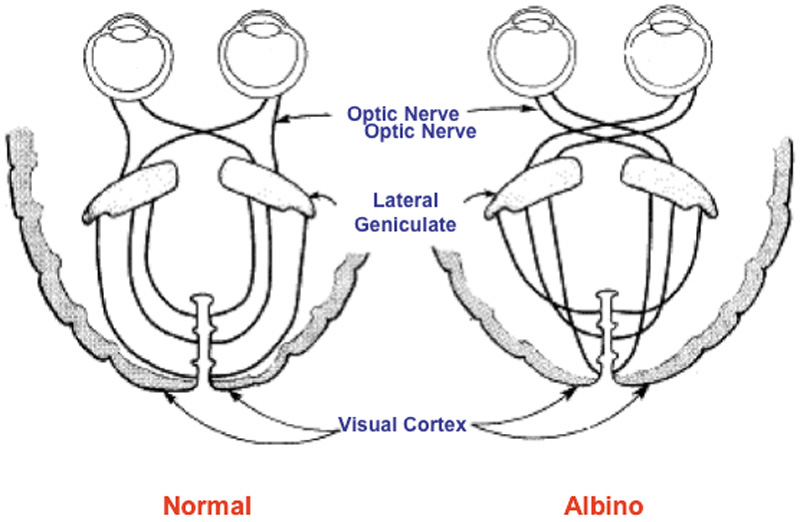
optic projections in the mouse. Proc R Soc Lond B Biol Sci 224:57–77, 1985. 43. Stellwagen D, Shatz CJ: An instructive role for retinal waves in the development of retinogeniculate
connectivity. Neuron 33:357–367, 2002. 44. Carroll WM, Jay BS, McDonald WI, et al: Two distinct patterns of visual evoked response asymmetry in human albinism. Nature 286:604, 1980. 45. Creel D, O'Donnell FE Jr, Witkop CJ: Visual system anomalies in human ocular albinos. Science 201:931, 1978. 46. Apkarian P, Reits D, Spekreijse H, et al: A decisive electrophysiological test for human albinism. Electroencephalogr Clin Neurophysiol 55:513, 1983. 47. Creel D, Garber SR, King RA, et al: Auditory brainstem anomalies in human albinos. Science 209:1253, 1980. 48. Fonda G: Characteristics and low-vision correction in albinism. Arch Ophthalmol 68:754, 1962. 49. Lanum J: The damaging effects of light on the retina: Empirical findings, theoretical
and practical implications. Surv Ophthalmol 22:221, 1978. 50. Feeney L, Berman ER: Oxygen toxicity: Membrane damage by free radicals. Invest Ophthalmol 15:789, 1976. 51. Yee RD, Wong EK, Baloh RW, et al: A study of congenital nystagmus: Waveforms. Neurology 26:326, 1976. 52. Naumann GOH, Lerche W, Schroeder W: Foveola-Aplasie bei tyrosinase-positivem oculocutanen albinismus. Graefes Arch Clin Exp Ophthalmol 200:39, 1976. 53. Fulton AB, Albert DM, Craft JL: Human albinism: Light and electron microscopy study. Arch Ophthalmol 96:305, 1978. 54. Mietz H, Green WR, Wolff SM, et al: Foveal hypoplasia in complete oculocutaneous albinism. Retina 12:254, 1992. 55. O'Donnell FE Jr, Hambrick GW, Green WR, et al: X-linked ocular albinism: An oculocutaneous macromelanosomal disorder. Arch Ophthalmol 94:1883, 1976. 56. Meyer CH, Lapolice DJ, Freedman SF: Foveal hypoplasia in oculocutaneous albinism demonstrated by optical coherence
tomography. Am J Ophthalmol 133:409–410, 2002. 57. Scott MJ Jr, Giacobetti R, Zugerman C: Malignant melanoma with oculocutaneous albinism. J Am Acad Dermatol 7:684, 1982. 58. Casswell AG, McCartney ACE, Hungerford JL: Choroidal malignant melanoma in an albino. Br J Ophthalmol 73:840, 1989. 59. Rinchik EM, Bultman SJ, Horsthemke B, et al: A gene for the mouse pink-eyed dilution locus and for human type II oculocutaneous
albinism. Nature 361:72, 1993. 60. Lee S-T, Nicholls RD, Bundey S, et al: Mutations of the P gene in oculocutaneous albinism, ocular albinism and
Prader-Willi syndrome plus albinism. N Engl J Med 330:529, 1994. 61. Ramsay M, Coleman M-A, Stevens G, et al: The tyrosinase-positive oculocutaneous albinism locus maps to chromosome 15q11.2–q12. Am J Hum Genet 51:879, 1992. 62. Gardner JM, Nakatsu Y, Gondo Y, et al: The mouse pink-eyed dilution gene: Association with human Prader-Willi
and Angelman syndromes. Science 257:1121, 1992. 63. Tripathi RK, Strunk KM, Giebel LB, et al: Tyrosinase gene mutations in type I (tyrosinase-deficient) oculocutaneous
albinism define two clusters of missense substitutions. Am J Med Genet 43:865, 1992. 64. Oetting WS, King RA: Molecular analysis of type I-A (tyrosine negative) oculocutaneous
albinism. Hum Genet 90:258, 1992. 65. Oetting WS, King RA: Molecular basis of type I (tyrosinase-related) oculocutaneous
albinism: Mutations and polymorphisms of the human tyrosinase gene. Hum Mutat 2:1, 1993. 66. Oetting WS, Mentink MM, Summers CG, et al: Three different frameshift mutations of the tyrosinase gene in type IA
oculocutaneous albinism. Am J Hum Genet 49:199, 1991. 67. Oetting WS, Witkop CJ Jr, Brown SA, et al: A frequent tyrosinase gene mutation associated with type I-A (tyrosinase-negative) oculocutaneous albinism in Puerto Rico. Am J Hum Genet 52:17, 1993. 68. Nance WE, Jackson CE, Witkop CJ Jr: Amish albinism: A distinctive autosomal recessive phenotype. Am J Hum Genet 22:579, 1970. 69. Hu F, Hanifin JM, Prescott GH, et al: Yellow mutant albinism: Cytochemical, ultrastructural, and genetic characterization
suggesting multiple allelism. Am J Hum Genet 32:387, 1980. 70. Spritz RA, Strunk K, King RA: Molecular analyses of the tyrosinase gene in patients with tyrosinase-deficient
oculocutaneous albinism [Abstract]. Am J Hum Genet 45(suppl):A221, 1989. 71. King RA, Townsend D, Oetting WS, et al: An unusual pigment pattern in type I oculocutaneous albinism (OCA) resulting
from a temperature-sensitive enzyme [Abstract]. Am J Hum Genet 45(suppl):A8, 1989. 72. King RA, Townsend D, Oetting W, et al: Temperature-sensitive tyrosinase associated with peripheral pigmentation
in oculocutaneous albinism. J Clin Invest 87:1046, 1991. 73. Giebel LB, Musarella MA, Spritz RA: A nonsense mutation in the tyrosinase gene of Afghan patients with tyrosinase
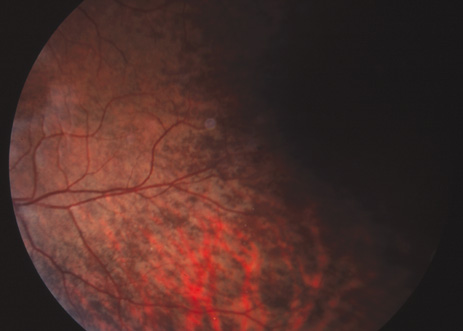
negative (type IA) oculocutaneous albinism. J Med Genet 28:464, 1991. 74. King RA, Creel D, Cervenka J, et al: Albinism in Nigeria with delineation of new recessive oculocutaneous type. Clin Genet 17:259, 1980. 75. King RA, Lewis RA, Townsend D, et al: Brown oculocutaneous albinism. Clinical, ophthalmological, and biochemical
characterization. Ophthalmology 92:1496, 1985. 76. King RA, Rich SS: Segregation analysis of brown oculocutaneous albinism. Clin Genet 29:496, 1986. 77. Silvers WK: The coat colors of mice. New York: Springer-Verlag, 1979. 78. Lyon M, Searle AG: Genetic variants and strains of the laboratory mouse. New York: Oxford University Press, 1989. 79. Jackson IJ: A cDNA encoding tyrosinase-related protein maps to the brown locus in mouse. Proc Natl Acad Sci U S A 85:4392, 1988; erratum in Proc Natl Acad Sci U S A 86:997, 1989. 80. Chintamaneni CD, Ramsay M, Colman MA, et al: Mapping the human CAS2 gene, the homologue of the mouse brown (b) locus
to human chromosome 9p22-pter. Biochem Biophys Res Commun 178:227, 1991. 81. Murty VV, Bouchard B, Mathew S, et al: Assignment of the human TYRP (brown) locus to chromosome region 9p23 by
nonradioactive in situ hybridization. Genomics 13:227, 1992. 82. Wagstaff J: A translocation-associated deletion defines a critical region for the 9p– syndrome [Abstract]. Am J Hum Genet 53(suppl):619, 1993. 83. Boissy RE, Zhao H, Austin LM, et al: Melanocytes from an individual with brown oculocutaneous albinism lack
expression of TRP-1, the product of the human homologue of the murine
brown locus [Abstract]. Am J Hum Genet 53(suppl):160, 1993. 84. Walsh RJ: A distinctive pigment of skin in New Guinea indigenes. Ann Hum Genet 34:379, 1971. 85. O'Donnell FE Jr, Green WR, Fleischman JA, et al: X-linked ocular albinism in blacks: Ocular albinism cum pigmento. Arch Ophthalmol 96:1189, 1978. 86. Falls HF: Sex-linked ocular albinism displaying typical fundus changes in the female
heterozygote. Am J Ophthalmol 34:41, 1951. 87. Gillespie FP, Covelli B: Carriers of ocular albinism with and without ocular changes. Arch Ophthalmol 70:209, 1963. 88. Lang GE, Rott H-D, Pfeiffer RA: X-linked ocular albinism: Characteristic pattern of affection in female
carriers. Ophthalmic Paediatr Genet 11:265, 1990. 89. Pearce WG, Johnson GL, Gillan JG: Nystagmus in a female carrier of ocular albinism. J Med Genet 9:126, 1972. 90. Maguire AM, Maumenee IH: Iris pigment mosaicism in carriers of X-linked ocular albinism cum pigmento. Am J Ophthalmol 107:298, 1989. 91. Charles SJ, Moore AT, Grant JW, et al: Genetic counseling in X-linked ocular albinism: Clinical features of the
carrier state. Eye 6:75, 1992. 92. Breathnach AS, Wyllie L: Ultrastructure of retinal pigment epithelium of the human fetus. J Ultrastruct Res 16:584, 1966. 93. Mund ML, Rodrigues MM, Fine BS: Light and electronmicroscopic observations on the pigmented layers of the
developing human eye. Am J Ophthalmol 73:167, 1972. 94. Wong L, O'Donnell FE Jr, Green WR: Giant pigment granules in the retinal pigment epithelium of a fetus with
X-linked ocular albinism. Ophthalmic Paediatr Genet 2:47, 1983. 95. Schnur RE, Wick PA, Bailey C, et al: Phenotypic variability in X-linked ocular albinism: Relationship to linkage
genotypes. Am J Hum Genet 55:484, 1994. 96. Benedict PH, Szabo G, Fitzpatrick TB, et al: Melanotic macules in Albright's syndrome and in neurofibromatosis. JAMA 205:618, 1968. 97. Jimbow K, Szabo G, Fitzpatrick TB: Ultrastructure of giant pigment granules (macromelanosomes) in
the cutaneous pigmented macules of neurofibromatosis. J Invest Dermatol 61:300, 1973. 98. Guerrier CJ, Lutzner MA, Devico V: An electronmicroscopical study of the skin in 18 cases of xeroderma pigmentosum. Dermatologica 146:211, 1973. 99. Nakagawa H, Hori Y, Sato S, et al: The nature and origin of the melanin macroglobule. J Invest Dermatol 83:134, 1984. 100. Cortin P, Tremblay M, Lemagne JM: X-linked ocular albinism: Relative value of skin biopsy, iris transillumination
and funduscopy in identifying affected males and carriers. Can J Ophthalmol 16:121, 1981. 101. Lyon MF: Sex chromatin and gene action in the mammalian X-chromosome. Am J Hum Genet 14:135, 1962. 102. O'Donnell FE, King RA, Green WR, et al: Autosomal recessively inherited ocular albinism: A new form of ocular albinism
affecting women as severely as men. Arch Ophthalmol 96:1621, 1978. 103. Morland AB, Hoffmann MB, Neveu M, Holder GE: Abnormal visual projection in a human albino studied with functional magnetic
resonance imaging and visual evoked potentials. J Neurol Neurosurg Psychiatry 72:523–526, 2002. 104. Wacks MA, Peachey NS, Fishman GA: Electroretinographic findings in human oculocutaneous albinism. Ophthalmology 96:1778–1785, 1989. 105. Hermansky F, Pudlak P: Albinism associated with hemorrhagic diathesis and unusual pigmented reticular
cells in the bone marrow: Report of two cases with histochemical
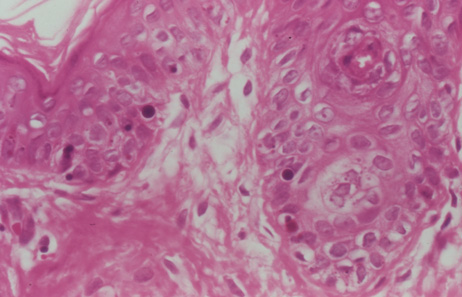
studies. Blood 14:162, 1959. 106. Witkop CJ, Almodavar C, Pineiro B, et al: Hermansky-Pudlak syndrome (HPS). An epidemiologic study. Ophthalmic Paediatr Genet 11:245, 1990. 107. Summers CG, Knobloch WH, Witkop CJ Jr, et al: Hermansky-Pudlak syndrome: Ophthalmic findings. Ophthalmology 95:545, 1988. 108. Scheinfeld NS: Syndromic albinism: a review of genetics and phenotypes. Dermatol Online J 9:5, 2003. 109. Beguez Cesar A: Neutropenia cronica maligna familar con granulaciones atipicas de los leucocitos. Biol Soc Cub Ped 15:900, 1943. 110. Tanaka T: Chédiak-Higashi syndrome: Abnormal lysosomal enzyme levels in granulocytes
of patients and family members. Pediatr Res 14:901, 1980. 111. De Beer HA, Anderson R, Findlay GH: Chédiak-Higashi syndrome in a “black” child: Clinical
features, immunological studies, and optics of the hair and skin. S Afr Med J 60:108, 1981. 112. Blume RS, Wolff SM: The Chédiak-Higashi syndrome: Studies in four patients and a review
of the literature. Medicine 51:247, 1972. 113. Spencer WH, Hogan MJ: Ocular manifestations of Chédiak-Higashi syndrome. Am J Ophthalmol 50:1197, 1960. 114. Johnson DL, Jacobson LW, Toyama R, et al: Histopathology of eyes in Chédiak-Higashi syndrome. Arch Ophthalmol 75:84, 1966. 115. Windhorst DB, Zelickson AS, Good RA: A human pigmentary dilution based on heritable subcellular structural defect: The
Chédiak-Higashi syndrome. J Invest Dermatol 50:9, 1966. 116. Bedoya V, Grimley PM, Duque O: Chédiak-Higashi syndrome. Arch Pathol 88:340, 1969. 117. Rausch PDG, Pryzwansky KB, Spitznagel JK: Immunohistochemical identification of azurophilic and specific granule
markers in the granules of Chédiak-Higashi neutrophils. N Engl J Med 298:693, 1978. 118. Windhorst DB, Zelickson AS, Good RA: Chédiak-Higashi syndrome: Hereditary gigantism of cytoplasmic organelles. Science 151:81, 1966. 119. Zhao H, Boissy YL, Abdel-Malek Z, et al: On the analysis of the pathophysiology of Chédiak-Higashi syndrome. Defects
expressed by cultured melanocytes. Lab Invest 71:25, 1994. 120. Boxer LA, Watanabe AM, Rister M, et al: Correction of leukocyte function in Chédiak-Higashi syndrome by
ascorbate. N Engl J Med 295:1041, 1976. 121. Introne W, Boissy RE, Gahl WA: Clinical, molecular, and cell biological aspects of Chediak-Higashi syndrome. Mol Genet Metab 68:283–303, 1999. 122. Karim MA, Suzuki K, Fukai K, et al: Apparent genotype-phenotype correlation in childhood, adolescent, and adult
Chediak-Higashi syndrome. Am J Med Genet 2002;108:16–22. 123. Cross HE, McKusick VA, Breen W: A new oculocerebral syndrome with hypopigmentation. J Pediatr 70:398, 1967. 124. Preus M, Fraser FC, Wiglesworth FW: An oculocerebral hypopigmentation syndrome. J Genet Hum 31:323–328, 1983. 125. Patton MA, Baraitser M, Heagerty AH, Eady RA: An oculocerebral hypopigmentation syndrome: a case report with clinical, histochemical, and
ultrastructural findings. J Med Genet 24:118–122, 1987. 126. Lerone M, Pessagno A, Taccone A, et al: Oculocerebral syndrome with hypopigmentation (Cross syndrome): Report
of a new case. Clin Genet 41:87–89, 1992. 127. Tezcan I, Demir E, Asan E, et al: A new case of oculocerebral hypopigmentation syndrome (Cross syndrome) with
additional findings. Clin Genet 51:118–121, 1997. 128. Ozkan H, Unsal E, Kose G: Oculocerebral hypopigmentation syndrome (Cross syndrome). Turk J Pediatr 33:247–252, 1991. 129. De Jong G, Fryns JP: Oculocerebral syndrome with hypopigmentation (Cross syndrome): The
mixed pattern of hair pigmentation as an important diagnostic sign. Genet Couns 2:151–155, 1991. 130. Fryns JP, Dereymaeker AM, Heremans G, et al: Oculocerebral syndrome with hypopigmentation (Cross syndrome). Report
of two siblings born to consanguineous parents. Clin Genet 34:81–84, 1988. 131. Courtens W, Broeckx W, Ledoux M, Vamosa E: Oculocerebral hypopigmentation syndrome (Cross syndrome) in a
gipsy child. Acta Paediatr Scand 78:806–810, 1989. 132. Castle DJ, Jenkins T, Shawinsky AA: The oculocerebral syndrome in association with generalised hypopigmentation. A
case report. S Afr Med J 1;76:35–36, 1989. 133. Alfadley A, Parkes RM: Two siblings born preterm with large ears and hypopigmented hair who developed
palmoplantar keratoderma and frontal skull bossing: A new syndrome? Pediatr Dermatol 19:224–228, 2002. 134. White CP, Waldron M, Jan JE, Carter JE. Oculocerebral hypopigmentation syndrome associated with Bartter syndrome. Am J Med Genet 46:592–596, 1993. 135. Winship IM, Babaya M, Ramesar RS: X-linked ocular albinism and sensorineural deafness: Linkage to Xp22.3. Genomics 18:444, 1993. 136. Lewis RA: Ocular albinism and deafness [Abstract]. Am J Hum Genet 30:57, 1978. 137. Bard LA: Heterogeneity in Waardenburg's syndrome. Arch Ophthalmol 96:1193, 1978. 138. Donaldson DD: Transillumination of the iris. Trans Am Ophthalmol Soc 72:89, 1974. 139. Bergsma DR, Kaiser-Kupfer M: A new form of albinism. Am J Ophthalmol 77:837, 1974. 140. Margolis S, Siegel IM, Choy A, et al: Oculocutaneous albinism associated with Apert's syndrome. Am J Ophthalmol 84:830, 1974. 141. Mosher DB, Fitzpatrick TB: Piebaldism. Arch Dermatol 124:364, 1988. 142. Hayashibe K, Mishima Y: Tyrosinase-positive melanocyte distribution and induction of pigmentation
in human piebald skin. Arch Dermatol 124:381, 1988. 143. Woolf CM, Dolowitz DA, Aldous HE: Congenital deafness associated with piebaldness. Arch Otolaryngol 82:244, 1965. 144. Telfer MA, Sugar M, Jaeger EA, et al: Dominant piebald trait (white forelock and leukoderma) with neurological
impairment. Am J Hum Genet 23:383, 1971. 145. Rozycki DL, Ruben RJ, Rapen I, et al: Autosomal recessive deafness associated with short stature, vitiligo, muscle
wasting and achalasia. Arch Otolaryngol 93:194, 1971. 146. Waardenburg PJ: New syndrome combining developmental anomalies of the eyelids, eyebrows, and
nose root with pigmentary defects of iris and head hair and with
congenital deafness. Am J Hum Genet 3:195, 1951. 147. Arias S: Genetic heterogeneity in the Waardenburg syndrome. Birth Defects 7:87, 1971. 148. Foy C, Newton VE, Wellesley D, et al: Assignment of WS1 locus to human 2q37 and possible homology between Waardenburg
syndrome and the Splotch mouse. Am J Hum Genet 46:1017, 1990. 149. Tassabehji M, Read AP, Newton VE, et al: Waardenburg syndrome patients have mutations in the human homologue of
the PAX-3 paired box gene. Nature 355:635, 1993. 150. Tassabehji M, Read AP, Newton VE, et al: Mutations in the PAX3 gene causing Waardenburg syndrome type 1 and type 2. Nat Genet 3:26, 1993. 151. Baldwin CT, Hoth CF, Amos JA, et al: An exonic mutation in the HuP2 paired domain gene causes Waardenburg's
syndrome. Nature 355:637, 1993. 152. Hoth CF, Milunsky A, Lipsky N, et al: Mutations in the paired domain of the human PAX3 gene cause Klein-Waardenburg
syndrome (WS-III) as well as Waardenburg syndrome type
I (WS-I). Am J Hum Genet 52:455, 1993. 153. Hughes AE, Newton VE, Liu XZ, et al: A gene for Waarburg syndrome type 2 maps close to the human homologue of
the microphthalmia gene at chromosome 3p12–p14.1. Nat Genet 7:509, 1994. 154. Tassabehji M, Newton VE, Read AP: Waardenburg syndrome type 2 caused by mutations in the human microphthalmia (MITF) gene. Nat Genet 8:251, 1994. 155. Griscelli C, Durandy A, Guy-Grand D, et al: A syndrome associating partial albinism and immunodeficiency. Am J Med 65:691, 1976. 156. Mancini AJ, Chan LS, Paller AS: Partial albinism with immunodeficiency: Griscelli syndrome: report of a
case and review of the literature. J Am Acad Dermatol 38:295–300, 1998. 157. Menasche G, Ho CH, Sanal O, et al: Griscelli syndrome restricted to hypopigmentation results from a melanophilin
defect (GS3) or a MYO5A F-exon deletion (GS1). J Clin Invest 112:450–456, 2003. 158. Chen Y, Samaraweera P, Sun TT, et al: Rab27b association with melanosomes: dominant negative mutants disrupt
melanosomal movement. J Invest Dermatol 118:933–940, 2002. 159. Sanal O, Ersoy F, Tezcan I, et al: Griscelli disease: genotype-phenotype correlation in an array of clinical
heterogeneity. J Clin Immunol 22:237–243, 2002. 160. Fukuda M, Kuroda TS, Mikoshiba K: Slac2-a/melanophilin, the missing link between Rab27 and myosin Va: implications
of a tripartite protein complex for melanosome transport. J Biol Chem 277:12432–12436, 2002. 161. Aksu G, Kutukculer N, Genel F, et al: Griscelli syndrome without hemophagocytosis in an eleven-year-old girl: Expanding
the phenotypic spectrum of Rab27A mutations in humans. Am J Med Genet 116A:329–333, 2003. 162. Duran-McKinster C, Rodriguez-Jurado R, Ridaura C, et al: Elejalde syndrome—A melanolysosomal neurocutaneous syndrome: clinical
and morphological findings in 7 patients. Arch Dermatol 135:182–186, 1999. 163. Bahadoran P, Ortonne JP, Ballotti R, De Saint-Basile G. Comment on Elejalde syndrome and relationship with Griscelli syndrome. Am J Med Genet 116A:408–409, 2003. 164. Perry PK, Silverberg NB: Cutaneous malignancy in albinism. Cutis 67:427–430, 2001. 165. Collins B, Silver J: Recent experiences in the management of visual impairment in albinism. Ophthalmic Pediatr Genet 11:225–228, 1990. 166. Scheinfield NS: Syndromic albinism: a review of genetics and phenotypes. Dermatol Online J 9:5, 2003. |