TABLE 4. Diagnostic Criteria of Ankylosing Spondylitis*
Low back pain of over 3 months' duration, unrelieved by rest
Pain and stiffness in the thoracic cage
Limited chest expansion
Limited motion in the lumbar spine
Past or present evidence of iridocyclitis
Bilateral radiographic sacroiliitis
Radiographic syndesmophytosis
*Diagnosis requires four of the five clinical criteria or bilateral radiographic sacroiliitis and one other criterion.
(Bluestone R: Ankylosing spondylitis. In McCarty DJ (ed): Arthritis and Allied Conditions, p 819. Philadelphia, Lea & Febiger, 1985)
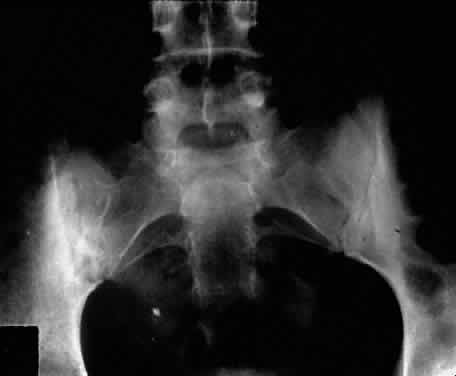
Ankylosing spondylitis is distinctly different from rheumatoid arthritis, although earlier literature did not make this distinction. The sacroiliac joint is usually the most easily visible area of involvement and is best seen in oblique views on radiography of the sacroiliac joint as periarticular sclerosis and irregular loss of the joint space. These patients are seronegative for rheumatoid factor and may have an elevated erythrocyte sedimentation rate.3,6
The presence of HLA-B27 antigen in 90% to 95% of white patients and 50% of black patients with ankylosing spondylitis is well documented, and the genetic predisposition to this disease has been recognized for many years. The relationship, however, is still incompletely understood at this point. HLA-B27 antigen is present in 6% to 14% of the white American population and 3% to 4% of the black American population. It is clear that people with this HLA type are at greater risk of developing one of the spondyloarthropathies, particularly ankylosing spondylitis, but as yet poorly defined environmental factors must be involved in triggering the development of the disease process.3,8 The development of acute iridocyclitis is even more strongly correlated with HLA-B27 antigen than is its association with joint changes. Of patients with HLA-B27 antigen, 20% to 25% will develop symptoms or radiographic evidence of spondylitis at some time in their life, and milder forms of the disease are commonly not diagnosed but believed to be due to back “strain” or injury. The incidence of clinical spondylitis is about equal between men and women, but the disease tends to be milder and more difficult to diagnose in women; its prevalence appears to approach 2% in those in the North American population who are HLA-B27 positive and who have been studied.
The mechanism of how HLA-B27 antigen is involved in this disease process is unclear. HLA determinants are located on chromosome 6. These determine cell surface markers that enable the immune system to recognize each cell as self. One theory that has been advanced suggests that the HLA-B27 antigen may be similar to antigens of the cell wall of certain exogenous agents or may be changed by the agents in a manner whereby the immune system has difficulty recognizing or responding appropriately to the agent or the altered HLA-B27 antigen. In one study, patients with ankylosing spondylitis had lower in vitro responsiveness of lymphocytes to Klebsiella antigens than HLA-B27-positive and HLA-B27-negative controls.9 Antisera to certain isolates of Klebsiella lysed the lymphocytes of HLA-B27-positive patients with ankylosing spondylitis, but not the lymphocytes of HLA-B27-positive or HLA-B27-negative controls. This suggests that perhaps some Klebsiella antigens cross-react with a gene product closely associated with HLA-B27 or a Klebsiella-modified B27 antigen in patients with ankylosing spondylitis. Cross-reacting antigens have been identified in HLA-B27 and Klebsiella, Shigella, and Yersinia.10
The role of Klebsiella has not been clarified, and no other agent has been substantiated with respect to ankylosing spondylitis. Chlamydia has been suggested in some cases of Reiter's syndrome, especially in instances of nonspecific urethritis.11–13 Shigella, Salmonella, and Yersinia have been involved in clinical epidemics of postinfectious arthropathies.14 Additional theories involve HLA-B27 linkage with a specific immune response gene that predisposes patients to the disease, perhaps making them more susceptible to infection. The role of these factors remains to be clarified.
The acute iridocyclitis associated with ankylosing spondylitis is characterized by rapid onset of pain, photophobia, and blurred vision. Conjunctival, episcleral, and scleral injection and edema are seen. Poorly defined keratic precipitates are seen in the lower half of the corneal endothelium, and the anterior chamber has heavy flare that may be uneven. If the process is severe, there may be clot formation in the pupil space.15,16 Cells in the anterior chamber may be so numerous that hypopyon will occur. Glaucoma can result from anterior chamber reaction blockage of the angle in the acute phase of inflammation and from pupil block from synechiae. Synechiae form early and, if not broken, will form lasting adhesions. Mydriatic and cycloplegic therapy is needed early in treatment. Spillover of inflammatory cells and inflammatory debris into the vitreous may occur, and the presence of disc blurring and macular edema is sometimes observed. This is sometimes also associated with hypotony. Posterior subcapsular cataracts and diffuse lens clouding are seen with severe prolonged episodes and repeated acute recurrences.15,16
The typical episode lasts from 2 to 6 weeks. Aggressive suppression of the inflammatory reaction with topical corticosteroids is usually sufficient and reduces tissue damage, if an early intensive schedule is used, rather than increasing the drop schedule as the reaction increases. These patients may need to be seen daily when they are acutely active and may need to be seen every 2 to 3 days until the process is stable or clearly resolving. Treatment must be continued for several weeks as the process is resolving or reactivation will occur. Oral corticosteroids can be given for short periods of time. Some patients experience elevation of intraocular pressure with corticosteroid therapy, particularly as the eye improves and the ciliary body is again more able to produce aqueous humor. The long-acting effects of periocluar injection of corticosteroids may become a more serious problem with persistent corticosteroid-induced glaucoma than the episode of acute iridocyclitis that was being treated.
Frequent episodes of recurrent iridocyclitis may cause significant disability that results in loss of work, discomfort, and structural damage to the eye. These patients may benefit from longer term treatment with oral nonsteroidal anti-inflammatory agents, such as indomethacin or naproxen. These medications may help to reduce the severity and frequency of recurrences, but the pain-decreasing effects of these medications may make it more difficult for the patient to recognize an acute recurrence of the iridocyclitis. Patients should be examined for exacerbations of the inflammation if they note any change in vision, even minor symptoms.
REITER'S SYNDROME OR POSTINFECTIOUS REACTIVE ARTHRITIS
Reiter's syndrome is a clinical syndrome usually described as arthritis, conjunctivitis, or iridocyclitis and nonbacterial urethritis or cervicitis. A better definition may be needed, because not only may these not all be present, but also dysentery and mucocutaneous disease with balanitis, oral ulceration, or keratoderma blennorrhagicum may be part of the clinical picture. Enthesopathy of the plantar fascia or Achilles tendon is also suggestive of Reiter's syndrome. It is more commonly identified in males, but may be more frequent in females than previously thought.14 The incidence reported by Noer17 in US Navy personnel over a 10-year period was 4 in 100,000 men per year. HLA-B27-positive persons have approximately a 25% risk for Reiter's syndrome development after Shigella infection.18 Although the cause is unknown, the high correlation with the presence of HLA-B27 (75%)19 is clearly recognized. The previous discussion in the section on spondyloarthropathies concerning this HLA-B27 association demonstrates a genetic predisposition in a high percentage of these patients. In Reiter's syndrome, infectious agents are suggested by reports of clinically indistinguishable acute disease after epidemic dysentery and sexually transmitted nongonococcal urethritis thought to be due to Chlamydia20 or possibly Mycoplasma (Ureaplasma urealyticum).21,22 Large epidemics of dysentery have been linked to multiple occurrences of arthritis, urethritis, and iridocyclitis.17,23 Shigella, Campylobacter, Salmonella, and Yersinia have all been implicated.24 Microbial antigens have been identified in the synovium after infectious with Chlamydia, Yersinia, and Salmonella.25–27 Salmonella typhimurium is a frequently associated Salmonella pathogen causing reactive arthritis.
This seronegative arthritis usually involves larger joints and the weight-bearing joints of the lower extremities. The knees and ankles are most frequently involved, with redness and diffuse swelling. Multiple joint involvement is usual. Periostitis and tendinitis may occur, especially involving the Achilles tendon, producing heel pain. Sacroiliac radiographic changes are present in up to 32% of patients.14 Children may be affected rarely.28 Reiter's syndrome tends to affect young adults in the range of 16 to 40 years of age. The diagnosis is sometimes hard to establish because the urethritis or cervicitis may be forgotten or suppressed and the enteritis and other symptoms may have been mild or not identified as abnormal. If a urethritis or cervicitis is present, cultures should be considered to make sure no treatable organism is present, such as gonococcus. Serologic testing for syphilis will help to rule out this sometimes-associated venereal problem.
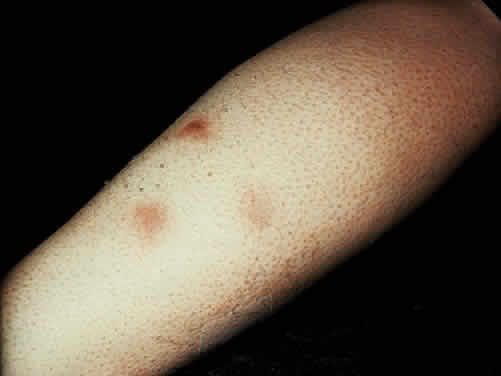
Low back pain due to insertional tendinitis and sacroiliitis is common. The dermal lesions typified by keratoderma blennorrhagicum, described as a hyperkeratotic erythematous dermatitis resembling pustular psoriasis, may not be present until later in the disease. It usually involves the hands and feet but may involve other areas. Superficial ulcers of the mucous membranes are frequent.29 Enteritis is usually a prolonged diarrheal episode with frequent passage of bloody, loose stools, but it may be a 24-hour episode of increased bowel activity.29
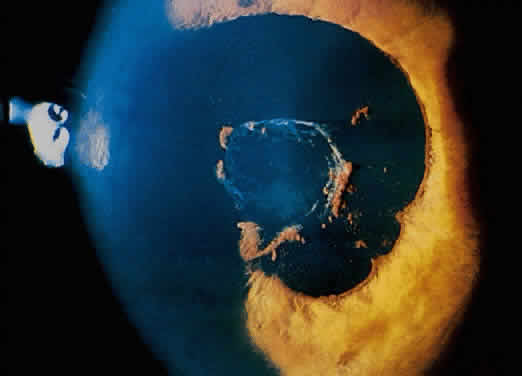
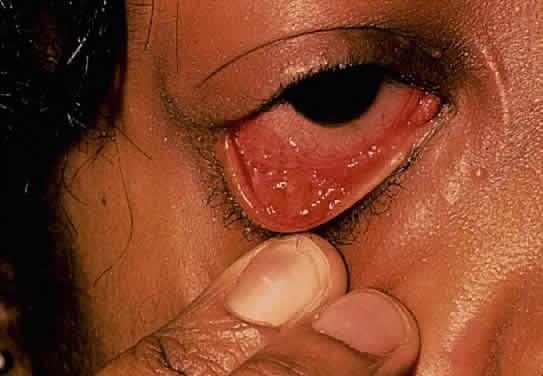
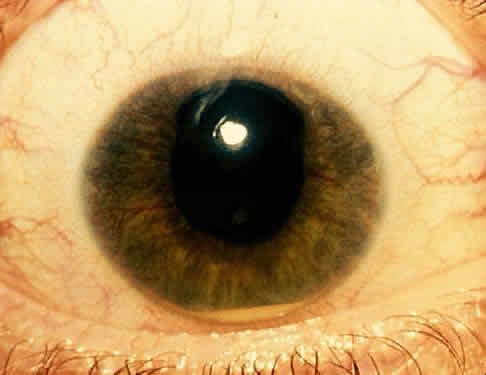
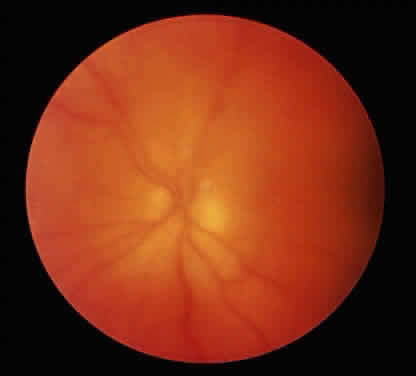
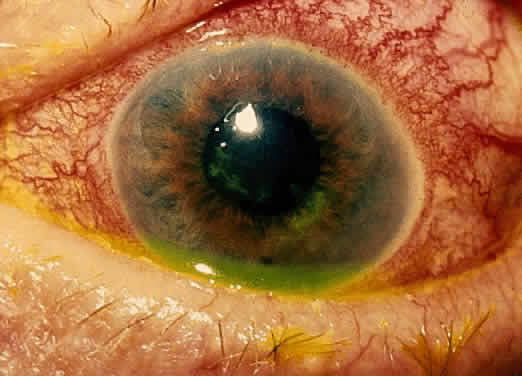
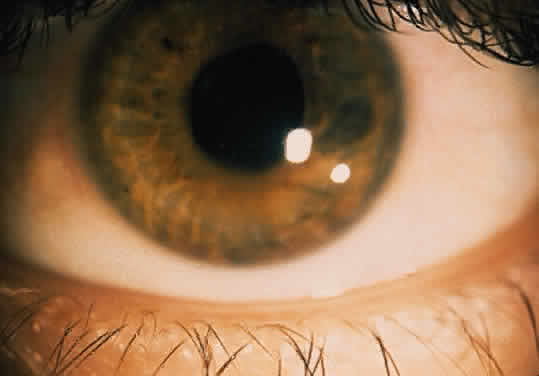
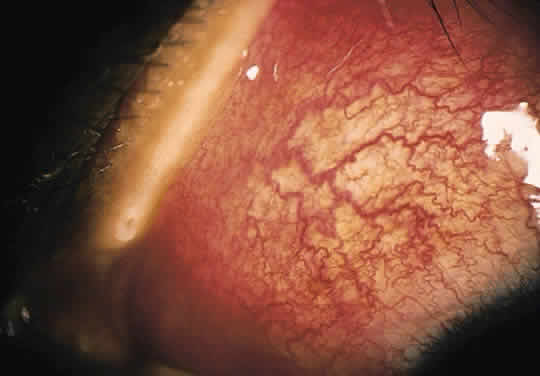
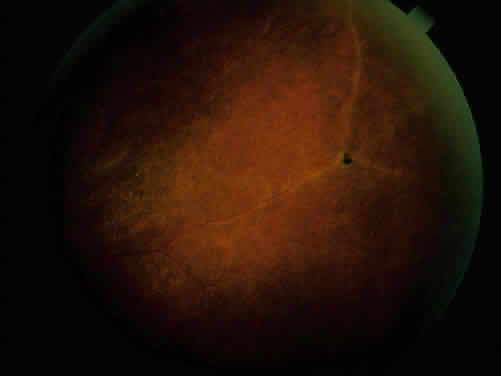
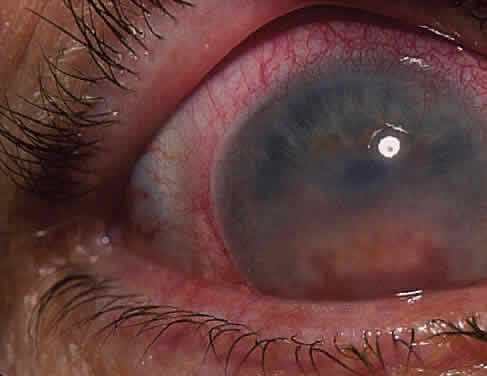
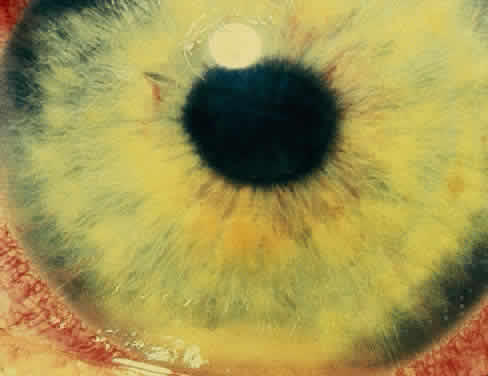
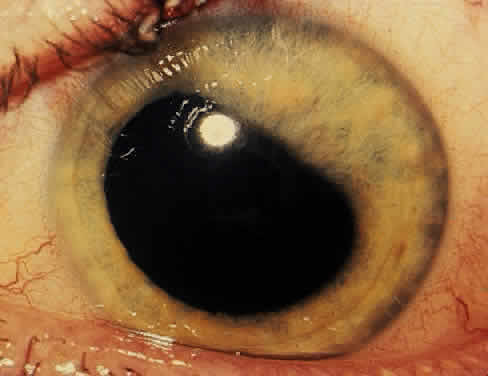
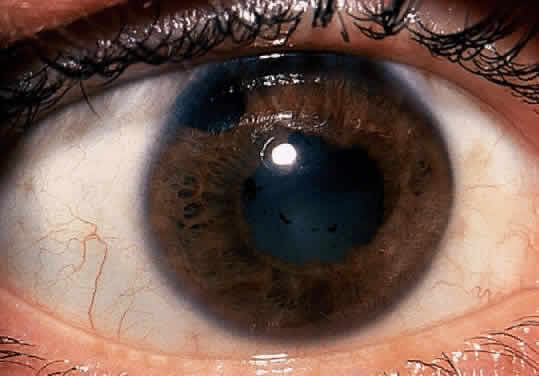
The conjunctivitis is usually described as mucopurulent and may be sterile. It may be accompanied by an iridocyclitis, episcleritis, or scleritis. Punctate and subepithelial corneal involvement has been reported. The iridocyclitis may be the presenting ocular manifestation. Recurrences are usually associated with an acute iridocyclitis, which is of rapid onset with conjunctival and episcleral edema and injection. The corneal endothelium has cellular debris and poorly defined, small-sized to medium-sized keratic precipitates.30 Heavy flare and cells and a very early tendency toward formation of posterior synechiae is characteristic, more so than in most other forms of acute iridocyclitis (Figs. 1, 2, and 3). Even the most aggressive pupil dilation management is sometimes inadequate for preventing synechiae. A peripheral iridectomy may be necessary to prevent iris bombé and angle closure if the synechiae cannot be broken enough to establish an opening for aqueous through the pupil (Fig. 4). The heavy flare is sometimes so plasmoid that cells are immobile and a fibrinlike clot may be seen in the pupil opening as the inflammation resolves. Cells and inflammatory debris may be seen in the vitreous, and blurring of the disc margins and macular edema may occur with severe or prolonged episodes. Lens clouding and posterior subcapsular cataracts occur with prolonged or repeated episodes. Hypotony can occur after a severe or prolonged course and may persist after resolution. Occasionally, secondary glaucoma may occur, owing to the anterior chamber reaction, in which case it will resolve as the inflammation resolves. With repeated recurrences, damage to the trabecular meshwork may occur, and prolonged recalcitrant glaucoma may result that may be poorly responsive to any medical or surgical management. This can be a serious factor in permanent visual loss with this type of iridocyclitis.15,16,30,31
|
|
|

|
The usual course is 2 to 6 weeks for acute iridocyclitis. Topical corticosteroids and mydriatics should be used early and aggressively to reduce tissue damage. Frequent follow-up visits are necessary until the process is clearly resolved. The use of systemic corticosteroids may be necessary and should be used with the same indications and precautions as in other inflammatory disease and for a short period of time. Prolonged topical treatment is necessary for several weeks after the inflammation has cleared, because early withdrawal of topical corticosteroids will frequently result in return of inflammatory changes.
Occasionally, this process will become chronically active or recur at frequent enough intervals to require long-term management. Oral nonsteroidal anti-inflammatory agents, such as indomethacin or naproxen, may be helpful and may be needed for many months for two reasons: (1) to help reduce the level of activity and the frequency of recurrences in the ocular inflammation; and (2) to allow decreasing the corticosteroid dosage to decrease the rate of cataract formation and other associated side effects of corticosteroids. The decreased awareness of pain sometimes seen with these medications may alter the patient's recognition of recurrences. If they have any change in symptoms, they should be examined to evaluate for recurrence of an acute episode of inflammation.