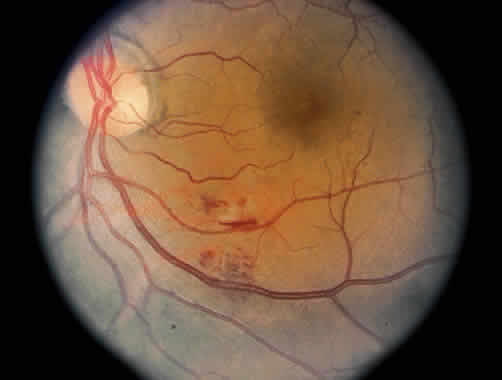
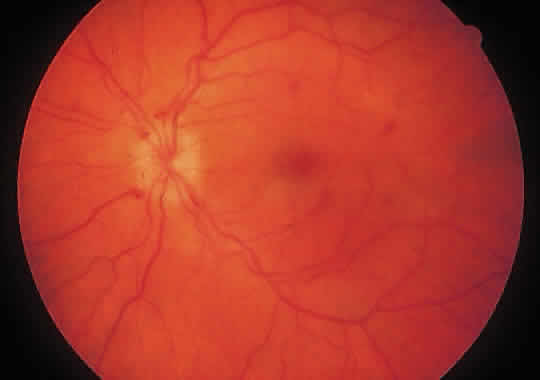
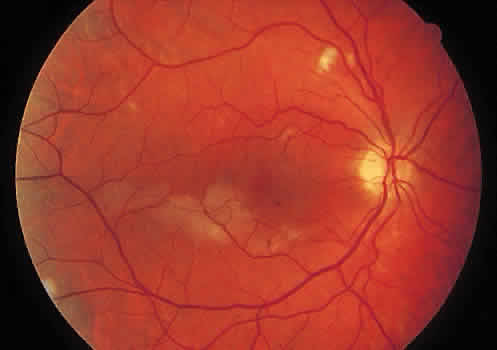
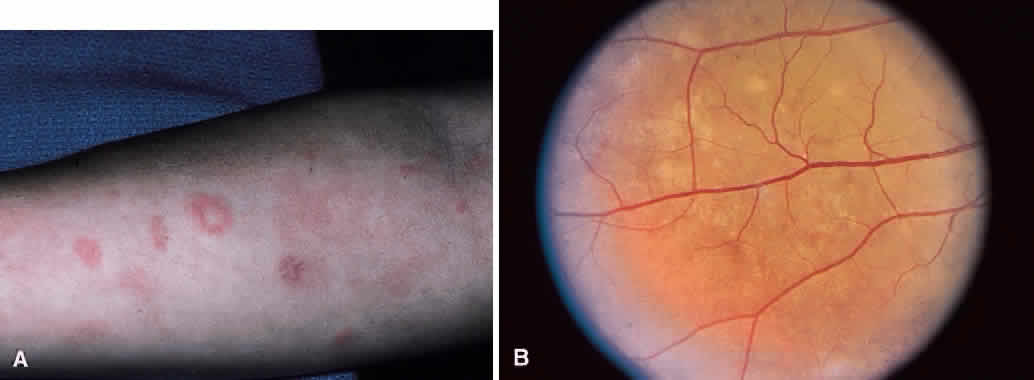
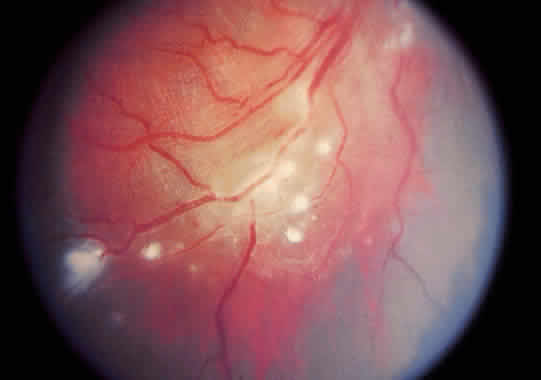
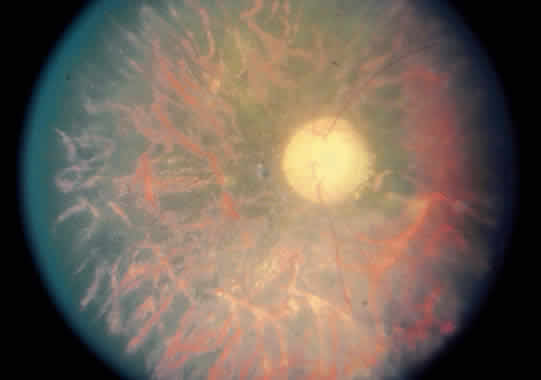
1. Graham E, Spalton DJ, Sanders MD. Immunological investigations in retinal
vasculitis. Trans Ophthalmol Soc UK 1981;101:12–16 2. Vinje O, Moller P, Mellbye J. Immunological variables and acute-phase reactants
in patients with ankylosing spondylitis (Bechterew's syndrome) and
their relatives. Clin Rheumatol 1984;3:501–513 3. Lehner T, Almeida JD, Levinsky RJ. Damaged membrane fragments and immune
complexes in the blood of patients with Behçet's syndrome. Clin
Exp Immunol 1978;34:206– 212 4. Oniki S, Inoue Y, Kawata K. Ocular Arthus reaction with reference to immunologic
retinal vasculitis: Its possible relation to Behçet's
disease. Jpn J Ophthalmol 1976;20:41 5. O'Connor GR. Factors related to the initiation and recurrence of uveitis. Am
J Ophthalmol 1983;96:577–599 6. Dumonde DC, Kasp-Grochowska E, Banga JP et al. Autoimmune mechanisms in
inflammatory eye disease. Trans Ophthalmol Soc UK 1985;104:232–238 7. Kasp E, Graham EM, Stanford MR et al. A point prevalence study of 150 patients
with idiopathic retinal vasculitis: 2. Clinical relevance of antiretinal
autoimmunity and circulating immune complexes. Br J Ophthalmol 1989;73:722– 730 8. Haynes B, Fauci A. Disorders of the immune system. In Isselbacher K, Braunwald
E, Wilson J et al (eds). Harrison's Principles of Internal
Medicine, p 1774. Vol 2. New York: McGraw-Hill, 1998 9. Thirkill C, Roth A, Keltner J. Cancer-associated retinopathy. Arch Ophthalmol 1987;105:372–375 10. Brasile L, Kremer JM, Clarke JL, Cerilli J. Identification of an autoantibody
to vascular endothelial cell-specific antigens in patients with
systemic vasculitis. Am J Med 1989; 87:74–80 11. Del Papa N, Conforti G, Gambini D et al. Characterization of the endothelial
surface proteins recognized by anti-endothelial antibodies in primary
and secondary autoimmune vasculitis. Clin Immunol Immunopathol 1994;70:211–216 12. Ferraro G, Meroni PL, Tincani A et al. Anti-endothelial cell antibodies
in patients with Wegener's granulomatosis and micropolyarteritis. Clin
Exp Immunol 1990;79:47–53 13. Savage CO, Pottinger BE, Gaskin G et al. Vascular damage in Wegener's
granulomatosis and microscopic polyarteritis: Presence of anti-endothelial
cell antibodies and their relation to anti-neutrophil cytoplasm
antibodies. Clin Exp Immunol 1991;85:14–19 14. Edelsten C, D'Cruz D, Hughes GR, Graham EM. Anti-endothelial cell antibodies
in retinal vasculitis. Curr Eye Res 1992;11:203–208 15. Kallenberg CG. Autoantibodies in vasculitis: Current perspectives [editorial]. Clin
Exp Rheumatol 1993;11:355–360 16. Sneller MC, Fauci AS. Pathogenesis of vasculitis syndromes. Med Clin North
Am 1997;81:221–242 17. Falk RJ, Terrell RS, Charles LA, Jennette JC. Anti-neutrophil cytoplasmic
autoantibodies induce neutrophils to degranulate and produce oxygen
radicals in vitro. Proc Natl Acad Sci USA 1990;87:4115–4119 18. Thomas PD, Hunninghake GW. Current concepts of the pathogenesis of sarcoidosis. Am
Rev Respir Dis 1987; 135:747–760 19. Nussenblatt RB, Gery I, Ballintine EJ, Wacker WB. Cellular immune responsiveness
of uveitis patients to retinal S-antigen. Am J Ophthalmol 1980;89:173–179 20. Youn J, Belehradek J, Quang T. Natural killer cell activity in patients
with uveitis. In O'Connor G, Chandler J (eds). Advances in Immunology
and Immunopathology of the Eye, p 246. Chicago: Year Book Medical Publishers, 1985 21. Abu El-Asrar A, Tabbara K. Retinal vasculitis. Curr Opinion Ophthalmol 1997;8:68–79 22. Pivetti-Pezzi P, Priori R, Catarinelli G et al. Markers of vascular injury
in Behçet's disease associated with retinal vasculitis. Ann
Ophthalmol 1992;24:411–414 23. Malinowski SM, Pulido JS, Goeken NE et al. The association of HLA-B8, B51, DR2, and
multiple sclerosis in pars planitis. Ophthalmology 1993;100:1199–1205 24. Bloch-Michel E, Frau E. Birdshot retinochoroidopathy and HLA-A29+ and
HLA-A29- idiopathic retinal vasculitis: Comparative study of 56 cases. Can
J Ophthalmol 1991; 26:361–366 25. Wise G, Dollery C, Henkind P. Clinical and microscopic features of retinal
lesions. In Wise G, Dollery C, Henkind P (eds). The Retinal Circulation, pp 203–210. New York: Harper & Row, 1971 26. Sanders MD. Retinal vasculitis: A review. J R Soc Med 1979;72:908–915 27. Karpik AG, Schwartz MM, Dickey LE et al. Ocular immune reactants in patients
dying with systemic lupus erythematosus. Clin Immunol Immunopathol 1985;35:295–312 28. Pepose JS, Holland GN, Nestor MS et al. Acquired immune deficiency syndrome. Pathogenic
mechanisms of ocular disease. Ophthalmology 1985;92:472–484 29. Kincaid J, Schatz H. Bilateral retinal arteritis with multiple aneurysmal
dilatations. Retina 1983;3:171–178 30. Klein B. Retinal lesions associated with uveal disease: I. Am J Ophthalmol 1956;42:831–847 31. Griffin A, Bodian M. Segmental retinal periarteritis. Am J Ophthalmol 1959;47:544–548 32. Orzalesi N, Ricciardi L. Segmental retinal periarteritis. Am J Ophthalmol 1971;72:55–59 33. Jampol LM, Isenberg SJ, Goldberg MF. Occlusive retinal arteriolitis with
neovascularization. Am J Ophthalmol 1976; 81:583–589 34. Eales H. Cases of retinal hemorrhage associated with epistaxis and constipation. Birmingham
Med Rev 1880;9: 262–273 35. Murphy R, Gieser S, Fine S. Retinal and vitreous findings in Eales' disease. Invest
Ophthalmol (Suppl) 1986;27:121 36. Murphy R, Renie W, Proctor L et al. A survey of patients with Eales' disease. In
Fine S, Owens S (eds). Management of Retinal Vascular and Macular
Disorders, pp 28–31. Baltimore: Williams & Wilkins, 1983 37. Das T, Biswas J, Kumar A et al. Eales' disease. Indian J Ophthalmol 1994;42:3–18 38. Bhooma V, Sulochana KN, Biswas J, Ramakrishnan S. Eales' disease: Accumulation
of reactive oxygen intermediates and lipid peroxides and decrease
of antioxidants causing inflammation, neovascularization and retinal
damage. Curr Eye Res 1997;16:91–95 39. Spitznas M, Meyer-Schwickerath G, Stephan B. The clinical picture of Eales' disease. Albrecht
Von Graefes Arch Klin Exp Ophthalmol 1975;194:73–85 40. Elliot AJ. 30-year observation of patients with Eales' disease. Am J Ophthalmol 1975;80:404–408 41. Theodossiadis G. Fluorescein angiography in Eales' disease. Am J Ophthalmol 1970;69:271–277 42. Loewenstein A, Michelson I, Hill J. Retinal vasculitis of the young. Trans
Ophthalmol Soc UK 1946;66:211–230 43. Elliot A. Recurrent intraocular hemorrhage in young adults (Eales' disease): A
report of thirty-one cases. Trans Am Ophthalmol Soc 1954;52:811–875 44. Verhoeff F, Simpson G. Tubercle within central retinal vein. Arch Ophthalmol 1940;24:645 45. Johnson G, Block K. Immunoglobulin levels in retinal vascular abnormalities
in pseudoxanthoma elasticum. Arch Ophthalmol 1969;81:322–324 46. Muthukkaruppan V, Rengarajan K, Chakkalath H. Immunological status of patients
with Eales' disease. Indian J Med Res 1989;90:351–359 47. Koliopoulos J, Perkins E, Seitanides B. Serum immunoglobulins in retinal
vasculitis. Br J Ophthalmol 1970;54:233–236 48. Andrews BS, McIntosh J, Petts V, Penny R. Circulating immune complexes
in retinal vasculitis. Clin Exp Immunol 1977;29:23–29 49. Palmer HE, Stanford MR, Sanders MD, Graham EM. Visual outcome of patients
with idiopathic ischaemic and non-ischaemic retinal vasculitis. Eye 1996;10:343–348 50. Gass A, Graham E, Moseley IF et al. Cranial MRI in idiopathic retinal vasculitis. J
Neurol 1995;242:174–177 51. Karma A, Seppala I, Mikkila H et al. Diagnosis and clinical characteristics
of ocular Lyme borreliosis. Am J Ophthalmol 1995;119:127–135 52. Leys AM, Schonherr U, Lang GE et al. Retinal vasculitis in Lyme borreliosis. Bull
Soc Belge Ophtalmol 1995; 259:205–214 53. Soheilian M, Markomichelakis N, Foster CS. Intermediate uveitis and retinal
vasculitis as manifestations of cat scratch disease. Am J Ophthalmol 1996;122:582–584 54. Hayreh SS. Optic disc vasculitis. Br J Ophthalmol 1972; 56:652–670 55. Bienfait MF, Baarsma GS, Wijngaarde R. Ten cases of optic vasculitis. Doc
Ophthalmol 1986;64:225–232 56. Fong AC, Schatz H. Central retinal vein occlusion in young adults [published
erratum appears in Surv Ophthalmol 1993 Jul-Aug;38(1):88]. Surv
Ophthalmol 1993;37:393–417 57. Tessler H. What is intermediate uveitis? In Ernest J (ed). Yearbook of
Ophthalmology, p 155. Chicago: Yearbook Medical Publishers, 1985 58. Gorman B, Mines J, Coles R. Vasculitis associated with intermediate uveitis. Acta
Ophthalmol (Suppl) 1984;163:59 59. Pruett RC, Brockhurst J, Letts NF. Fluorescein angiography of peripheral
uveitis. Am J Ophthalmol 1974;77:448–453 60. Chester GH, Blach RK, Cleary PE. Inflammation in the region of the vitreous
base. Pars planitis. Trans Ophthalmol Soc UK 1976;96:151–157 61. Brockhurst R, Schepens C, Okamura I. Peripheral uveitis: Clinical description, complications, and
differential diagnosis. Am J Ophthalmol 1960;49:1257–1266 62. Smith RE, Godfrey WA, Kimura SJ. Chronic cyclitis. I. Course and visual
prognosis. Trans Am Acad Ophthalmol Otolaryngol 1973;77:OP760–768 63. Pederson JE, Kenyon KR, Green WR, Maumenee AE. Pathology of pars planitis. Am
J Ophthalmol 1978;86: 762–774 64. Hayreh SS. So-called acute retinal necrosis syndrome—an acute ocular
panvasculitis syndrome. Dev Ophthalmol 1985;10:40–77 65. Gartry DS, Spalton DJ, Tilzey A, Hykin PG. Acute retinal necrosis syndrome. Br
J Ophthalmol 1991;75:292–297 66. Culbertson WW, Blumenkranz MS, Pepose JS et al. Varicella zoster virus
is a cause of the acute retinal necrosis syndrome. Ophthalmology 1986;93:559–569 67. Culbertson WW, Blumenkranz MS, Haines H et al. The acute retinal necrosis
syndrome. Part 2: Histopathology and etiology. Ophthalmology 1982;89:1317–1325 68. Rungger-Brandle E, Roux L, Leuenberger PM. Bilateral acute retinal necrosis (BARN). Identification of the presumed infectious agent. Ophthalmology 1984;91:1648–1658 69. Batisse D, Eliaszewicz M, Zazoun L et al. Acute retinal necrosis in the
course of AIDS: Study of 26 cases. AIDS 1996;10:55–60 70. Garweg J, Bohnke M. Varicella-zoster virus is strongly associated with
atypical necrotizing herpetic retinopathies. Clin Infect Dis 1997;24:603–608 71. Kleiner RC, Kaplan HJ, Shakin JL et al. Acute frosted retinal periphlebitis. Am
J Ophthalmol 1988;106:27–34 72. Sugin SL, Henderly DE, Friedman SM et al. Unilateral frosted branch angiitis. Am
J Ophthalmol 1991;111:682–685 73. Rabb MF, Jampol LM, Fish RH et al. Retinal periphlebitis in patients with
acquired immunodeficiency syndrome with cytomegalovirus retinitis mimics
acute frosted retinal periphlebitis. Arch Ophthalmol 1992;110:1257–1260 74. Chang TS, Aylward GW, Davis JL et al. Idiopathic retinal vasculitis, aneurysms, and
neuro-retinitis. Retinal Vasculitis Study. Ophthalmology 1995;102:1089–1097 75. Lam S, Tessler H. “New stuff” in uveitis. Ophthalmology Clin
North Am 1993;6:81–95 76. Bergink GJ, Ooyman FM, Maas S, Rademakers AJ. Three HLA-A29 positive patients
with uveitis. Acta Ophthalmol Scand 1996;74:81–83 77. Groothuis DR, Mikhael MA. Focal cerebral vasculitis associated with circulating
immune complexes and brain irradiation. Ann Neurol 1986;19:590–592 78. Hahn B. Systemic lupus erythematosus. In Isselbacher K, Braunwald E, Wilson
J et al (eds). Harrison's Principles of Internal Medicine, pp 1643–1648. New
York: McGraw-Hill, 1994 79. Gold D, Feiner L, Henkind P. Retinal arterial occlusive disease in systemic
lupus erythematosus. Arch Ophthalmol 1977;95:1580–1585 80. Coppeto J, Lessell S. Retinopathy in systemic lupus erythematosus. Arch
Ophthalmol 1977;95:794–797 81. Quillen DA, Stathopoulos NA, Blankenship GW, Ferriss JA. Lupus associated
frosted branch periphlebitis and exudative maculopathy. Retina 1997;17:449–451 82. Jabs D. The rheumatic diseases. In Ryan S, Schachat A, Murphy R, Patz A (eds). Retina, pp 457–480. St Louis: Mosby, 1989 83. Jabs DA, Fine SL, Hochberg MC et al. Severe retinal vaso-occlusive disease
in systemic lupus erythematous. Arch Ophthalmol 1986;104:558–563 84. Coppeto JR, Currie JN, Monteiro ML, Lessell S. A syndrome of arterial-occlusive
retinopathy and encephalopathy. Am J Ophthalmol 1984;98:189–202 85. Asherson RA, Merry P, Acheson JF et al. Antiphospholipid antibodies: A
risk factor for occlusive ocular vascular disease in systemic lupus erythematosus
and the “primary” antiphospholipid syndrome. Ann
Rheum Dis 1989;48: 358–361 86. Diddie KR, Aronson AJ, Ernest JT. Chorioretinopathy in a case of systemic
lupus erythematosus. Trans Am Ophthalmol Soc 1977;75:122–131 87. Matsuo T, Koyama T, Morimoto N et al. Retinal vasculitis as a complication
of rheumatoid arthritis. Ophthalmologica 1990;201:196–200 88. Giordano N, D'Ettorre M, Biasi G et al. Retinal vasculitis in rheumatoid
arthritis: An angiographic study. Clin Exp Rheumatol 1990;8:121–125 89. Cohen BH, Sedwick LA, Burde RM. Retinopathy of dermatomyositis. J Clin
Neuroophthalmol 1985;5:177–179 90. Isaak BL, Liesegang TJ, Michet CJ Jr. Ocular and systemic findings in relapsing
polychondritis. Ophthalmology 1986; 93:681–689 91. Backhouse O, Griffiths B, Henderson T, Emery P. Ophthalmic manifestations
of dermatomyositis. Ann Rheum Dis 1998;57:447–449 92. Zeuner M, Straub RH, Rauh G et al. Relapsing polychondritis: Clinical and
immunogenetic analysis of 62 patients. J Rheumatol 1997;24:96–101 93. Sahin O, Goldstein D, Tessler H. Findings typical of Susac's syndrome
in a patient with scleroderma. Retina 1999; 19:476–477 94. Garcia-Diaz M, Mira M, Nevado L et al. Retinal vasculitis associated with
Crohn's disease. Postgrad Med J 1995;71: 170–172 95. Kelly IM, Frith PA, Hyman NM, Jewell DP. Retinal periphlebitis in ulcerative
colitis. Postgrad Med J 1990;66: 565–567 96. Duker JS, Brown GC, Brooks L. Retinal vasculitis in Crohn's disease. Am
J Ophthalmol 1987;103:664–668 97. Li CG, Reynolds I, Ponting JM et al. Serum levels of vascular endothelial
growth factor (VEGF) are markedly elevated in patients with Wegener's
granulomatosis. Br J Rheumatol 1998;37:1303–1306 98. Robin JB, Schanzlin DJ, Meisler DM et al. Ocular involvement in the respiratory
vasculitides. Surv Ophthalmol 1985;30:127–140 99. Rao JK, Weinberger M, Oddone EZ et al. The role of antineutrophil cytoplasmic
antibody (c-ANCA) testing in the diagnosis of Wegener granulomatosis. A
literature review and meta-analysis. Ann Intern Med 1995;123:925–932 100. Cohen Tervaert JW, van der Woude FJ, Fauci AS et al. Association between
active Wegener's granulomatosis and anticytoplasmic antibodies. Arch
Intern Med 1989;149: 2461–2465 101. Mangouritsas G, Ulbig M. Cotton-wool spots as the initial ocular manifestation
in Wegener's granulomatosis. Ger J Ophthalmol 1994;3:68–70 102. Morgan CM, Foster CS, D'Amico DJ, Gragoudas ES. Retinal vasculitis in polyarteritis
nodosa. Retina 1986;6:205–209 103. Akova YA, Jabbur NS, Foster CS. Ocular presentation of polyarteritis nodosa. Clinical
course and management with steroid and cytotoxic therapy. Ophthalmology 1993;100: 1775–1781 104. Kozak M, Gill EA, Green LS. The Churg-Strauss syndrome. A case report with
angiographically documented coronary involvement and a review of the
literature. Chest 1995; 107:578–580 105. Hellemans S, Dens J, Knockaert D. Coronary involvement in the Churg-Strauss
syndrome. Heart 1997;77:576–578 106. Weinstein JM, Chui H, Lane S et al. Churg-Strauss syndrome (allergic granulomatous
angiitis). Neuro-ophthalmologic manifestations. Arch Ophthalmol 1983;101:1217–1220 107. Spencer W (ed). Ophthalmic Pathology: An Atlas and Textbook. Vol 4. Philadelphia: WB
Saunders, 1996 108. Vitali C, Genovesi-Ebert F, Romani A et al. Ophthalmological and neuro-ophthalmological
involvement in Churg-Strauss syndrome: A case report. Graefes
Arch Clin Exp Ophthalmol 1996;234:404–408 109. Laroche L, Saraux H, Pelosse B, Brissaud P. [Chorioretinal involvement
in Churg-Strauss angiitis]. Bull Soc Ophtalmol Fr 1986;86:757–760 110. Tse DT, Mandelbaum S, Chuck DA et al. Lymphomatoid granulomatosis with
ocular involvement. Retina 1985;5: 94–97 111. Forman S, Rosenbaum PS. Lymphomatoid granulomatosis presenting as an isolated
unilateral optic neuropathy. A clinicopathologic report. J Neuroophthalmol 1998;18: 150–152 112. Medeiros LJ, Jaffe ES, Chen YY, Weiss LM. Localization of Epstein-Barr
viral genomes in angiocentric immunoproliferative lesions. Am J Surg Pathol 1992;16:439–447 113. Hunder GG, Bloch DA, Michel BA et al. The American College of Rheumatology 1990 criteria
for the classification of giant cell arteritis. Arthritis
Rheum 1990;33:1122–1128 114. Fineman MS, Savino PJ, Federman JL, Eagle RC Jr. Branch retinal artery
occlusion as the initial sign of giant cell arteritis. Am J Ophthalmol 1996;122:428–430 115. Melberg NS, Grand MG, Dieckert JP et al. Cotton-wool spots and the early
diagnosis of giant cell arteritis. Ophthalmology 1995;102:1611–1614 116. Sagar S, Kar S, Gupta A, Sharma BK. Ocular changes in Takayasu's arteritis
in India. Jpn J Ophthalmol 1994;38: 97–102 117. Fauci A. The vasculitis syndrome. In Braunwald E, Wilson J, Martin J et
al (eds). Harrison's Principles of Internal Medicine, pp 1677–1678. New
York: Mosby, 1994 118. Bodker FS, Tessler HH, Shapiro MJ. Ocular complications of Takayasu's
disease in a Hispanic woman [letter]. Am J Ophthalmol 1993;115:676–677 119. Rock T, Dinar Y, Romem M. Retinal periphlebitis after hormonal treatment. Ann
Ophthalmol 1989;21:75–76 120. Shaw HE Jr, Lawson JG, Stulting RD. Amaurosis fugax and retinal vasculitis
associated with methamphetamine inhalation. J Clin Neuroophthalmol 1985;5:169–176 121. Citron BP, Halpern M, McCarron M et al. Necrotizing angiitis associated
with drug abuse. N Engl J Med 1970; 283:1003–1011 122. Nichols CJ, Mieler WF. Severe retinal vaso-occlusive disease secondary
to procainamide-induced lupus. Ophthalmology 1989;96:1535–1540 123. Tsai JC, Forster DJ, Ober RR, Rao NA. Panuveitis and multifocal retinitis
in a patient with leucocytoclastic vasculitis. Br J Ophthalmol 1993;77:318–320 124. Corwin JM, Baum J. Iridocyclitis in two patients with hypocomplementemic
cutaneous vasculitis. Am J Ophthalmol 1982;94:111–113 125. Ryan LM, Kozin F, Eiferman R. Immune complex uveitis: A case. Ann Intern
Med 1978;88:62–63 126. Doutre MS, Beylot C, Morel P et al. [Ophthalmologic manifestations
of leukocytoclastic vasculitis. Apropos of 3 case reports]. Ann
Dermatol Venereol 1986;113:419–425 127. Jabs DA, Johns CJ. Ocular involvement in chronic sarcoidosis. Am J Ophthalmol 1986;102:297–301 128. Thorne JE, Galetta SL. Disc edema and retinal periphlebitis as the initial
manifestation of sarcoidosis. Arch Neurol 1998;55:862–863 129. Karma A, Huhti E, Poukkula A. Course and outcome of ocular sarcoidosis. Am
J Ophthalmol 1988;106:467–472 130. Ohara K, Okubo A, Sasaki H, Kamata K. Intraocular manifestations of systemic
sarcoidosis. Jpn J Ophthalmol 1992;36:452–457 131. Nussenblatt R, Whitcup S, Palestine A. Uveitis: Fundamentals and Clinical
Practice. Bethesda: Mosby, 1996 132. Obenauf CD, Shaw HE, Sydnor CF, Klintworth GK. Sarcoidosis and its ophthalmic
manifestations. Am J Ophthalmol 1978;86:648–655 133. James DG, Neville E, Langley DA. Ocular sarcoidosis. Trans Ophthalmol Soc
UK 1976;96:133–139 134. Duker JS, Brown GC, McNamara JA. Proliferative sarcoid retinopathy. Ophthalmology 1988;95:1680–1686 135. Spalton DJ, Sanders MD. Fundus changes in histologically confirmed sarcoidosis. Br
J Ophthalmol 1981;65:348–358 136. Mizuno K, Takahashi J. Sarcoid cyclitis. Ophthalmology 1986;93:511–517 137. Ohara K, Okubo A, Sasaki H, Kamata K. Branch retinal vein occlusion in
a child with ocular sarcoidosis. Am J Ophthalmol 1995;119:806–807 138. Kimmel AS, McCarthy MJ, Blodi CF, Folk JC. Branch retinal vein occlusion
in sarcoidosis. Am J Ophthalmol 1989;107:561–562 139. Denis P, Nordmann JP, Laroche L, Saraux H. Branch retinal vein occlusion
associated with a sarcoid choroidal granuloma [letter]. Am
J Ophthalmol 1992;113:333–334 140. Palmer HE, Stanford MR, McCartney AC, Graham EM. Non-caseating granulomas
as a cause of ischaemic retinal vasculitis [letter]. Br J
Ophthalmol 1997;81:1018–1019 141. DeRosa AJ, Margo CE, Orlick ME. Hemorrhagic retinopathy as the presenting
manifestation of sarcoidosis. Retina 1995;15:422–427 142. Sanders MD, Shilling JS. Retinal, choroidal, and optic disc involvement
in sarcoidosis. Trans Ophthalmol Soc UK 1976;96:140–144 143. Gass JD, Olson CL. Sarcoidosis with optic nerve and retinal involvement. A
clinicopathologic case report. Trans Am Acad Ophthalmol Otolaryngol 1973;77:OP739–750 144. Michelson JB, Chisari FV. Behçet's disease. Surv Ophthalmol 1982;26:190–203 145. Mishima S, Masuda K, Izawa Y et al. Behçet's disease in Japan: Ophthalmologic
aspects. Trans Am Ophthalmol Soc 1979;77:225–279 146. Atmaca LS. Fundus changes associated with Behçet's disease. Graefes
Arch Clin Exp Ophthalmol 1989;227:340–344 147. Inoue T, Oniki S, Kajiyama K, Jimi S. Circulating immune complexes in Behçet's
disease. Jpn J Ophthalmol 1983; 27:35–39 148. Munke H, Stockmann F, Ramadori G. Possible association between Behçet's
syndrome and chronic hepatitis C virus infection [letter]. N
Engl J Med 1995;332:400–401 149. Porter R. Uveitis in association with multiple sclerosis. Br J Ophthalmol 1972;56:478–481 150. Younge BR. Fluorescein angiography and retinal venous sheathing in multiple
sclerosis. Can J Ophthalmol 1976; 11:31–36 151. Engell T, Hvidberg A, Uhrenholdt A. Multiple sclerosis: Periphlebitis retinalis
et cerebro-spinalis. A correlation between periphlebitis retinalis
and abnormal technetium brain scintigraphy. Acta Neurol Scand 1984;69:293–297 152. Vine AK. Severe periphlebitis, peripheral retinal ischemia, and preretinal
neovascularization in patients with multiple sclerosis. Am J Ophthalmol 1992;113:28–32 153. Birch MK, Barbosa S, Blumhardt LD et al. Retinal venous sheathing and the
blood-retinal barrier in multiple sclerosis. Arch Ophthalmol 1996;114:34–39 154. Ronzani M, Lang GE, Wagner P, Lang GK. Severe occlusive retinal periphlebitis
with vitreous hemorrhage in multiple sclerosis. Ger J Ophthalmol 1995;4:328–331 155. Soldan SS, Berti R, Salem N et al. Association of human herpes virus 6 (HHV-6) with
multiple sclerosis: Increased IgM response to HHV-6 early
antigen and detection of serum HHV-6 DNA. Nat Med 1997;3:1394–1397 156. Fillet AM, Lozeron P, Agut H et al. HHV-6 and multiple sclerosis. Nat Med 1998;4:537–538 157. Packer AJ, Weingeist TA, Abrams GW. Retinal periphlebitis as an early sign
of bacterial endophthalmitis. Am J Ophthalmol 1983;96:66–71 158. Lamb H. The retina in septic and chronic endophthalmitis of ectogenous
origin. Am J Ophthalmol 1939;22:258–266 159. King LP, Libby C, Coats DK, Lee WH. Presumed septic emboli following dental
extraction. Graefes Arch Clin Exp Ophthalmol 1993;231:667–668 160. Weissgold DJ, Decker PJ. Retinal hemorrhages from septic emboli in a patient
with a ventricular false chorda. Am J Ophthalmol 1996;122:117–119 161. Fountain JA, Werner RB. Tuberculous retinal vasculitis. Retina 1984;4:48–50 162. Rosen PH, Spalton DJ, Graham EM. Intraocular tuberculosis. Eye 1990;4:486–492 163. Reny JL, Challe G, Geisert P et al. Tuberculosis-related retinal vasculitis
in an immunocompetent patient. Clin Infect Dis 1996;22:873–874 164. Helm CJ, Holland GN. Ocular tuberculosis. Surv Ophthalmol 1993;38:229–256 165. Lobes LA Jr, Folk JC. Syphilitic phlebitis simulating branch vein occlusion. Ann
Ophthalmol 1981;13:825–827 166. Ross WH, Sutton HF. Acquired syphilitic uveitis. Arch Ophthalmol 1980;98:496–498 167. Crouch ER Jr, Goldberg MF. Retinal periarteritis secondary to syphilis. Arch
Ophthalmol 1975;93:384–387 168. Mendelsohn AD, Jampol LM. Syphilitic retinitis. A cause of necrotizing
retinitis. Retina 1984;4:221–224 169. Morgan CM, Webb RM, O'Connor GR. Atypical syphilitic chorioretinitis and
vasculitis. Retina 1984;4:225–231 170. Deutman AF, Klomp HJ. Rift Valley fever retinitis. Am J Ophthalmol 1981;92:38–42 171. Tabbara KF. Ocular toxoplasmosis: Toxoplasmic retinochoroiditis. Int Ophthalmol
Clin 1995;35:15–29 172. Hayashi S, Kim MK, Belfort R Jr. White-centered retinal hemorrhages in
ocular toxoplasmosis. Retina 1997;17: 351–352 173. Gentile RC, Berinstein DM, Oppenheim R, Walsh JB. Retinal vascular occlusions
complicating acute toxoplasmic retinochoroiditis. Can J Ophthalmol 1997;32:354–358 174. Schwartz PL. Segmental retinal periarteritis as a complication of toxoplasmosis. Ann
Ophthalmol 1977;9:157–162 175. Raab EL, Leopold IH, Hodes HL. Retinopathy in Rocky Mountain spotted fever. Am
J Ophthalmol 1969;68:42–46 176. Gorenflot A, Moubri K, Precigout E et al. Human babesiosis. Ann Trop Med
Parasitol 1998;92:489–501 177. Ortiz JM, Eagle RC Jr. Ocular findings in human babesiosis (Nantucket fever). Am
J Ophthalmol 1982;93:307–311 178. Semba RD, Donnelly JJ, Rockey JH et al. Experimental ocular onchocerciasis
in cynomolgus monkeys. II. Chorioretinitis elicited by intravitreal Onchocerca lienalis microfilariae. Invest Ophthalmol Vis Sci 1988;29:1642–1651 179. Umeh RE, Chijioke CP, Okonkwo PO. Eye disease in an onchocerciasis-endemic
area of the forest-savanna mosaic region of Nigeria. Bull World Health
Organ 1996;74:95–100 180. Knox DL, King J Jr. Retinal arteritis, iridocyclitis, and giardiasis. Ophthalmology 1982;89:1303–1308 181. Woods SL, Wakefield D, McCluskey P. The acquired immune deficiency syndrome: Ocular
findings and infection control guidelines. Aust NZ J Ophthalmol 1986;14:287–291 182. McCluskey PJ, Wakefield D. Posterior uveitis in the acquired immunodeficiency
syndrome. Int Ophthalmol Clin 1995;35:1–14 183. Geier SA, Rolinski B, Sadri I et al. [Ocular microangiopathy syndrome
in patients with AIDS is associated with increased plasma levels of
the vasoconstrictor endothelin-1]. Klin Monatsbl Augenheilkd 1995;207:353–360 184. Thierfelder S, Linnert D, Grehn F. Increased prevalence of HIV-related
retinal microangiopathy syndrome in patients with hepatitis C [letter]. Arch
Ophthalmol 1996;114:899 185. Kestelyn P, Van de Perre P, Rouvroy D et al. A prospective study of the
ophthalmologic findings in the acquired immune deficiency syndrome in
Africa. Am J Ophthalmol 1985;100:230–238 186. Kestelyn P, Lepage P, Van de Perre P. Perivasculitis of the retinal vessels
as an important sign in children with AIDS-related complex. Am J
Ophthalmol 1985;100:614–615 187. Kestelyn P, Van de Perre P, Sprecher-Goldberger S. Isolation of the human
T-cell leukemia/lymphotropic virus type III from aqueous humor in two
patients with perivasculitis of the retinal vessels. Int Ophthalmol 1986;9:247–251 188. Barr CC, Joondeph HC. Retinal periphlebitis as the initial clinical finding
in a patient with Hodgkin's disease. Retina 1983;3:253–257 189. Lewis RA, Clark RB. Infiltrative retinopathy in systemic lymphoma. Am J
Ophthalmol 1975;79:48–52 190. Char DH, Ljung BM, Miller T, Phillips T. Primary intraocular lymphoma (ocular
reticulum cell sarcoma) diagnosis and management. Ophthalmology 1988;95:625–630 191. Gass JD, Trattler HL. Retinal artery obstruction and atheromas associated
with non-Hodgkin's large cell lymphoma (reticulum cell sarcoma). Arch
Ophthalmol 1991;109: 1134–1139 192. Brown SM, Jampol LM, Cantrill HL. Intraocular lymphoma presenting as retinal
vasculitis. Surv Ophthalmol 1994; 39:133–140 193. Ormerod LD, Puklin JE. AIDS-associated intraocular lymphoma causing primary
retinal vasculitis. Ocul Immunol Inflamm 1997;5:271–278 194. Dev S, Pulido JS, Tessler HH et al. Progression of diabetic retinopathy
after endophthalmitis. Ophthalmology 1999; 106:774–781 195. Huamonte FU, Cyrlin MN, Tessler HT, Goldberg MF. Retinopathy in diabetes
associated with sarcoidosis. Ann Ophthalmol 1980;12:1290–1297 196. Forde AM, Feighery C, Jackson J. Anti-monocyte cytoplasmic antibodies in
granulomatous disease. Clin Immunol Immunopathol 1996;81:88–95 197. Cristea A, Badea T, Bodizs G, Olinic N. Clinical evaluation of antineutrophil
cytoplasmic autoantibodies in ANCA-associated diseases. J Clin
Lab Immunol 1995;46:85–94 198. Knox CM, Chandler D, Short GA, Margolis TP. Polymerase chain reaction-based
assays of vitreous samples for the diagnosis of viral retinitis. Use
in diagnostic dilemmas. Ophthalmology 1998;105:37–45 199. Abe T, Tsuchida K, Tamai M. A comparative study of the polymerase chain
reaction and local antibody production in acute retinal necrosis syndrome
and cytomegalovirus retinitis. Graefes Arch Clin Exp Ophthalmol 1996;234: 419–424 200. Chan CC, Whitcup SM, Solomon D, Nussenblatt RB. Interleukin-10 in the vitreous
of patients with primary intraocular lymphoma. Am J Ophthalmol 1995;120:671–673 201. Buggage RR, Velez G, Myers-Powell B et al. Primary intraocular lymphoma
with a low interleukin 10 to interleukin 6 ratio and heterogeneous IgH
gene rearrangement. Arch Ophthalmol 1999;117:1239–1242 202. Akpek EK, Maca SM, Christen WG, Foster CS. Elevated vitreous Interleukin-10 level
is not diagnostic of intraocular-central nervous system lymphoma. Ophthalmology 1999; 106:2291–2295 203. Davis JL, Viciana AL, Ruiz P. Diagnosis of intraocular lymphoma by flow
cytometry. Am J Ophthalmol 1997; 124:362–372 204. Matzkin DC, Slamovits TL, Sachs R, Burde RM. Visual recovery in two patients
after intravenous methylprednisolone treatment of central retinal
artery occlusion secondary to giant-cell arteritis. Ophthalmology 1992;99:68–71 205. Lynch JP, 3rd, Hoffman GS. Wegener's granulomatosis: Controversies
and current concepts. Compr Ther 1998; 24:421–440 206. Tessler HH, Jennings T. High-dose short-term chlorambucil for intractable
sympathetic ophthalmia and Behçet's disease. Br J Ophthalmol 1990;74:353–357 207. Nussenblatt RB, Palestine AG, Chan CC. Cyclosporin A therapy in the treatment
of intraocular inflammatory disease resistant to systemic corticosteroids
and cytotoxic agents. Am J Ophthalmol 1983;96:275–282 208. Suzuki N, Kaneko S, Ichino M et al. In vivo mechanisms for the inhibition
of T lymphocyte activation by long-term therapy with tacrolimus (FK-506): Experience
in patients with Behçet's disease. Arthritis
Rheum 1997;40:1157–1167 209. Okada AA. Cytokine therapy in eye disease [editorial]. Arch Ophthalmol 1998;116:1514–1516 210. Pivetti-Pezzi P, Accorinti M, Pirraglia MP et al. Interferon alpha for
ocular Behçet's disease. Acta Ophthalmol Scand 1997;75:720–722 211. Azizlerli G, Sarica R, Kose A et al. Interferon alfa-2a in the treatment
of Behçet's disease. Dermatology 1996;192: 239–241 |