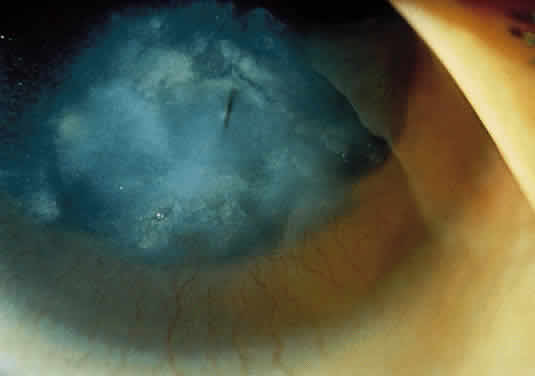
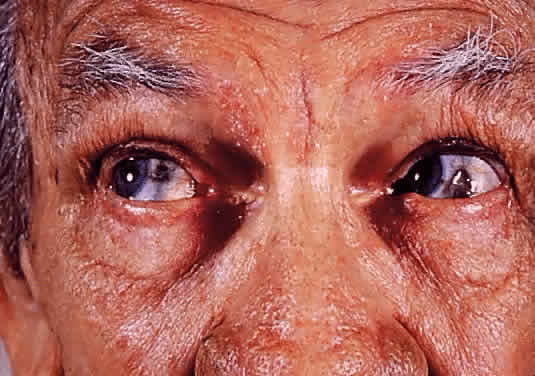
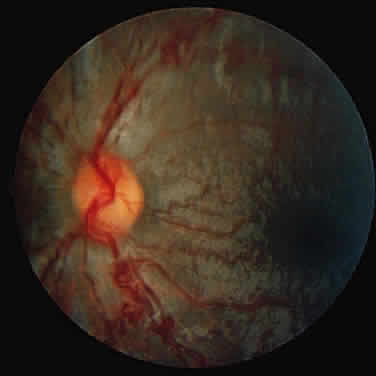
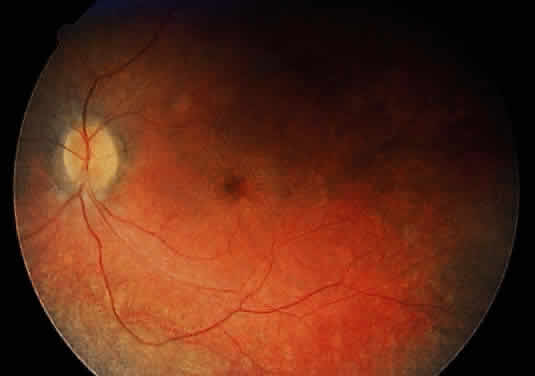
1. Garrod A: The Croonian lectures on inborn errors of metabolism: Lectures
I, II, III, IV. Lancet 2:1–7, 73–79, 142–148, 214–220, 1908 2. Garrod A: Inborn Errors of Metabolism. London: Oxford University Press, 1923 3. Bearn A: Archibald Garrod and the Individuality of Man. New York: Clarendon
Press, 1993 4. Scriver C, Beaudet A, Sly W et al: The Metabolic and Molecular Bases of
Inherited Disease. 7th ed. New York: McGraw-Hill, 1995 5. Endres W, Shin Y: Cataract and metabolic diseases. J Inherit Metab Dis 13:509, 1990 6. Sly W: The mucopolysaccharidoses in metabolic control and disease. In Bondy
P, Rosenberg L (eds): Metabolic Control and Disease, p 545. Philadelphia: WB
Saunders, 1980 7. McKusick V, Neufeld E: The mucopolysaccharide storage diseases. In Stanbury
J, Wyngaarden J, Fredrickson D et al (eds): The Metabolic Basis of
Inherited Disease, p 751. New York: McGraw-Hill, 1983 8. McKusick V: The mucopolysaccharidoses in heritable disorders of connective
tissue. In McKusick V (ed): Hentable Disorders of Corrective Tissue, pp 521, 641, 646. St. Louis, Mosby, 1972 9. Spranger J: The systemic mucopolysaccharidoses. Ergeb Inn Med Kinderheilkd 32: 165, 1972 10. Collins M, Traboulsi E, Maumenee I: Optic nerve head swelling and optic atrophy in the mucopolysaccharidoses. Ophthalmology 97:1445, 1990 11. Kenyon K, Quigley H, Hussels IE: The systemic mucopolysaccharidosis: Ultrastructural and histochemical studies
of conjunctiva and skin. Am J Ophthalmol 73:811, 1972 12. Berman E, Livni N, Shapira E et al: Congenital corneal clouding with abnormal systemic storage bodies: A new
variant of mucolipidosis. J Pediatr 84:519, 1974 13. Donaldson M, Pennock C, Berry P et al: Syndrome with cardiomyopathy in infancy. J Pediatr 114:430, 1989 14. Rosen D, Haust M, Yamashita T et al: Keratoplasty and electron microscopy of the cornea in systemic mucopolysaccharidosis (Hurler's disease). Can J Ophthalmol 3:218, 1968 15. Kenyon K: Ocular ultrastructure of inherited metabolic disease. In Goldberg
M (ed): Genetic and Metabolic Eye Disease, p 139. Boston: Little
Brown, 1974 16. Gills P, Hobson R, Hanley B et al: Electroretinography and fundus oculi findings in Hurler's disease
and allied mucopolysaccharidosis. Arch Ophthalmol 74:596, 1965 17. Nowaczyk M, Clarke J, Morin J: Glaucoma as an early complication of Hurler's disease. Arch Dis Child 63:1091, 1988 18. Spellacy E, Bankes J, Crow S et al: Glaucoma in a case of Hurler disease. Br J Ophthalmol 64:773, 1980 19. Duke-Elder S: Diseases of the Lens and Vitreous: System of Ophthalmology, p 172. London: Kimpton, 1969 20. Bach G, Friedman R, Weissmann B et al: The defect in the Hurler and Scheie syndromes: Deficiency of alpha-L-iduronidase. Proc Natl Acad Sci USA 69:2048, 1972 21. Scott H, Ashton L, Eyre H et al: Chromosomal localization of the human alpha-L-iduronidase gene (IDUA) to 4p16.3.Am J Hum Genet 47:802, 1990 22. Hopwood J, Vellodi A, Scott H et al: Long-term clinical progress in bone marrow transplanted mucopolysaccharidosis
type I patients with a defined genotype. J Inherit Metab Dis 16:1024, 1993 23. Fairbairn L, Lashford L, Spooncer E et al: Long-term in vitro correction of alpha-L-iduronidase deficiency (Hurler
syndrome) in human bone marrow. Proc Natl Acad Sci USA 93:2025, 1996 24. Scheie H, Hambrick GJ, Braner L: A newly recognized forme fruste of Hurler's disease (gargoylism): The
Sanford R. Gifford lecture. Am J Ophthalmol 53:753, 1952 25. Quigley H, Goldberg M: Scheie syndrome and macular corneal dystrophy: An ultrasound comparison
of conjunctiva and skin. Arch Ophthalmol 85:553, 1971 26. Tieu P, Bach G, Matynia A et al: Four novel mutations underlying mild or intermediate forms of alpha-L-iduronidase
deficiency (MPS IS and MPS IH/S). Hum Mutat 6:55, 1995 27. Wilson P, Suthers G, Callen D et al: Frequent deletions at Xq28 indicate genetic heterogeneity in Hunter syndrome. Hum Genet 86:505, 1991 28. Al-Hazzaa S, Maumenee I, Traboulsi E: Ocular findings in adults with mucopolysaccharidosis II-B (Hunter syndrome
mild). MEJO 1:15, 1993 29. Hobolth N, Pedersen C: Six cases of a mild form of the Hunter syndrome in five generations: Three
affected males with progeny. Clin Genet 13:121, 1978 30. Spranger J, Cantz M, Gehler J et al: Mucopolysaccharidosis II (Hunter disease) with corneal opacities. Eur J Pediatr 129:11, 1978 31. Caruso R, Kaiser-Kupfer M, Muenzer J et al: Electroretinographic findings in the mucopolysaccharidoses. Ophthalmology 93:1612, 1986 32. Beck M: Papilloedema in association with Hunter syndrome. Br J Ophthalmol 67:174, 1983 33. Beck M, Cole G: Disc oedema in association with Hunters syndrome: Ocular histopatho-logical
findings. Br J Ophthalmol 68:590, 1984 34. Braun S, Aronovich E, Anderson R et al: Metabolic correction and cross-correction of mucopolysaccharidosis type
II (Hunter syndrome) by retroviral-mediated gene transfer and expression
of human iduronate-2-sulfatase. Proc Natl Acad Sci USA 90:11830, 1993 35. Van de Kamp J, Niermeiyer M, Von Figura K, Giesberts M: Genetic heterogeneity and clinical variability in the Sanfilippo syndrome (types
A, B, C). Clin Genet 20:152, 1981 36. Kaplan P, Wolfe L: Sanfilippo syndrome type D. J Pediatr 110:267, 1987 37. De Natale P: Sanfilippo B disease: A re-examination of a particular sibship after 12 years. J Inherit Metab Dis 14:23, 1991 38. Sanfilippo J, Podosin R, Langer L et al: Mental retardation associated with acid mucopolysacchariduria (heparitin
sulfate type). J Pediatr 63:837, 1963 39. Scott H, Blanch L, Guo X-H et al: Cloning of the sulphamidase gene and identification of mutations in Sanfilippo
A syndrome. Nature Genet 11:465, 1995 40. Zhao H, Li H, Bach G, Schmidtchen A et al: The molecular basis of Sanfilippo syndrome type B. Proc Natl Acad Sci USA 93:6101, 1996 41. Robertson D, Callen D, Baker E et al: Chromosomal localization of the gene for human glucosamine-6-sulphatase
to 12q14. Hum Genet 79:175, 1988 42. Arbisser A, Donnelly K, Scott C Jr et al:Morquio-like syndrome with beta-galactosidase deficiency and normal hexosamine
sulfatase activity: mucopolysaccharidosis IVB. Am J Med Genet 1:195, 1977 43. DiFerrante N, Ginsburg L, Donnelly P, Diferrante D, Caskey C: Deficiencies of glucosamine-6-sulfate or galactosamine-6-sulfate sulfatases
are responsible for different mucopolysaccharidoses. Science 199:79, 1978 44. Holzgreve W, Grobe H, Von Figura K et al: Morquio syndrome: Clinical findings in 11 patients with MPS IV A and 2 patients
with MPS IV B. Hum Genet 57:360, 1981 45. Sugar J: Corneal manifestations of systemic mucopolysaccharidoses. Ann Ophthalmol 11:531, 1979 46. Beck M, Glossl J, Grubisic A et al: Heterogeneity of Morquio disease. Clin Genet 29:325, 1986 47. Masuno M, Tomatsu S, Nakashima Y et al.Mucopolysaccharidosis IVA: assignment of the human N-acetylgalactosamine-6-sulfate
sulfatase (GALNS) gene to chromosome 16q24. Genomics 16:777, 1996 48. Maroteaux P, Levesque B, Marie J, Lamy M: Une nouvelle dysotose avec elimination urinaire de chrondrotin-sulfate
B. Presses Med 71:1849, 1963 49. O'Brien J, Cautz M, Spranger J: Maroteaux-Lamy disease (mucopolysaccharidosis VI): Subtype A: Deficiency
of N-acetyl-galactosamine-4-sulfatase. Biochem Biophys Res Commun 60:1170, 1974 50. Young R, Kleinman G, Odemann R et al: Compressive myelopathy in Maroteaux-Lamy syndrome: Clinical and pathological
findings. Ann Neurol 8:336, 1980 51. Goldberg M, Scott C, McKusick V: Hydrocephalus and papilledema in the Martoteaux-Lamy syndrome (MPS VI). Am J Ophthalmol 69:969, 1970 52. Schwartz MF, Werblin T, Green W: Occurrence of mucopolysaccharide in corneal grafts in Maroteaux-Lamy syndrome. Cornea 4:58,1985/1986 53. Naumann G: Clearing of cornea after perforating keratoplasty in mucopolysaccharidosis
type VI (Maroteaux-Lamy syndrome). N Engl J Med 312:995, 1985 54. Litjens T, Baker E, Beckman K et al: Chromosomal localization of ARBS, the gene for human N-acetylgalactosamine-4-sulphatase. Hum Genet 82:67, 1989 55. Sly W, Quinton B, McAllister W, Rimoin D: B-glucoronidase deficiency: Report of clinical and radiological and biochemical
features of a new MPS. J Pediatr 82:249, 1973 56. Ward J, Sharpe C, Luthardt F et al: Regional gene mapping of human beta-glucuronidase (GUSB) by dosage analysis: Assignment
to region 7q11.23–7q21 (abstract). Am J Hum Genet 35:56A, 1983 57. Maumenee I: The eye in connective tissues diseases. In Daentl D (ed): Clinical, Structural
and Biochemical Advances in Hereditary Eye Disorders, p 53. New
York: Alan R. Liss, 1980 58. Spranger J, Wiedemann H: The genetic mucolipidoses. Diagnosis and differential diagnosis. Humangenetik 9:113, 1970 59. Kolodny E, Boustany R: Storage disease of reticular endothelial system. In
Nathan D, Oski F (eds): Hematology of Infancy and Childhood, p 1212. Philadelphia: WB
Saunders, 1987 60. Leroy J, Spranger J, Feingold M et al: I-Cell Disease: A Clinical Picture. J Pediatr 79:360, 1971 61. Tondeur M, Vamos-Hurwitz E, Mockel-Pohl S et al: Clinical biochemical and ultrastructured studies in a case of chondrodystrophy
presenting the I-cell phenotype in tissue culture. J Pediatr 79:366, 1971 62. Leroy J, DeMars R: Mutant enzymatic and cytological phenotypes in cultured fibroblasts. Science 157:804, 1967 63. DeMars R, Leroy J: The remarkable cells cultured a human with Hurler's syndrome: An approach
to visual selection for in vitro genetic studies. In Vitro 2:107, 1967 64. Okada S, Owada M, Sakiyama T et al: I-cell disease: Clinical studies of 21 Japanese cases. Clin Genet 28:207, 1985 65. Cipolloni C, Boldrini A, Donti E et al: Neonatal mucolipidosis II (I-cell disease): Clinical, radiological and
biochemical studies in a case. Helv Paediatr Acta 35:85, 1980 66. Whelan D, Chang P, Cockshott P: Mucolipidosis II. The clinical, radiological and biochemical features in
three cases. Clin Genet 24:90, 1983 67. Sprigz R, Doughty R, Spackman T et al: Neonatal presentation of I-cell disease. J Pediatr 93:954, 1978 68. Libert J, Van Hoof F, Farriaux J et al: Ocular findings in I-cell disease (mucolipidosis type II). Am J Ophthalmol 83:617, 1977 69. Mueller O, Wasmuth J, Murray J et al: Chromosomal assignment of N-acetylglucosaminylphosphotransferase, the lysosomal
hydrolase targeting enzyme deficient in mucolipidosis II and
III. Cytogenet Cell Genet 46:664, 1987 70. Ben-Yoseph Y, Mitchell D, Yager R et al: Mucolipidoses II and III variants with normal N-acetylglucosamine 1-phosphotransferase
activity toward alpha-methylmannoside are due to nonallelic
mutations. Am J Hum Genet 50:137, 1992 71. Freisinger P, Padovani JC, Maroteaux P: An atypical form of mucolipidosis III. J Med Genet 29:834, 1992 72. Taylor H, Thomas G, Miller C et al: Mucolipidosis III (Pseudo-Hurler polydystrophy): Cytological and ultrastructural
observation of cultured fibroblast cells. Clin Genet 4:388, 1973 73. Spranger JW, Wiedemann HR. The genetic mucolipidoses. Diagnosis and differential
diagnosis. Humangenetik 9:-113, 1970 74. Kelley T, Thomas G, Taylor H et al: Mucolipidosis III (Pseudo-Hurler polydystrophy): Clinical and laboratory
studies in a series of 12 patients. Johns Hopkins Med J 137:156, 1975 75. Traboulsi E, Maumenee I: Ophthalmologic findings in mucolipidosis III. Am J Ophthalmol 102:529, 1986 76. Bach G: Mucolipidosis type IV. In Goodman R, Motulsky A (eds): Genetic
Diseases Among Ashkenazi Jews, p 187. New York: Raven Press, 1979 77. Ben-Yoseph Y, Momoi T, Hahn L, Nadler H: Catalycally defective ganglioside neuraminidase in mucolipidosis IV. Clin Genet 21:374, 1982 78. Crandall B, Philippart M, Brown W et al: Mucolipidosis IV. Am J Med Genet 12:301, 1982 79. Bach G, Zeigler M, Kohn G: Mucopolysaccharide accumulation in cultured skin fibroblasts derived from
patients with mucolipidosis IV. Am J Hum Genet 29:610, 1977 80. Bach G, Zeigler M, Schaap T et al: Mucolipidosis type IV: Ganglioside sialidase deficiency. Biochem Biophys Res Commun 9:1341, 1979 81. Kenyon K, Maumenee I, Green W et al: Mucolipidosis IV: Histopathology of conjunctiva, cornea and skin. Arch Ophthalmol 97:1106, 1979 82. Merin S, Livni N, Berman E et al: Mucolipidosis IV: Ocular systemic and ultrastructural findings. Invest Ophthalmol 14:437, 1975 83. Abraham F, Brand N, Blumenthal M et al: Retinal function in mucolipidosis IV. Ophthalmologica 191:210, 1985 84. Newell F, Matalon R, Meyer S: A new mucolipidosis with psychomotor retardation, corneal clouding and
retinal degeneration. Am J Ophthalmol 80:440, 1975 85. McKusick V, Francomano C, Antonorakis S: Mendelian Inheritance in Man. Baltimore: Johns
Hopkins University Press, 1994 86. Norden N, Lundblad A, Svensson S et al: A mannose-containing trisaccharide isolated from urines of three patients
with mannosidosis. J Biol Chem 248:6210, 1973 87. Norden N, Lundblad A, Svensson S et al: Characterization of two mannose-containing oligosaccharides isolated from
the urine of patients with mannosidosis. Biochemistry 13:871, 1974 88. Kaneda Y, Hayes H, Uchida T et al: Regional assignment of five genes on human chromosome 19. Chromosoma 95:8, 1987 89. Desnick R, Sharp H, Grabowski G et al: Mannosidosis: Clinical, morphologic, immunologic and biochemical studies. Pediatr Res 19:985, 1976 90. Warner T, Mock A, Nyhan W et al: Alpha-mannosidosis, analysis of urinary oligosaccharides with high performance
liquid chromatography and diagnosis of a case with unusually mild
presentation. Clin Genet 25:248, 1984 91. Chester M, Lundblad A, Ockerman P et al: Mannosidosis. In Durand P, O'Brien
J (eds): Genetic Errors of Glycoprotein Metabolism, p 89. Berlin: Springer-Verlag, 1982 92. Spranger J, Gehler J, Cantz M: The radiographic features of mannosidosis. Radiology 119:401, 1976 93. Yunis J, Lewandowski R, Sanfilippo S et al: Clinical manifestations of mannosidosis—a longitudinal study. Am J Med 61:841, 1976 94. Murphree A, Beaudet A, Palmer E et al: Cataract in mannosidosis. Birth Defects 12:319, 1976 95. Maire I, Zabot M, Mathieu M et al: Mannosidosis tissue culture studies in relation to prenatal diagnosis. J Inherit Metab Dis 1:19, 1978 96. Fukuda M, Tanaka A, Isshiki G: Variation of lysosomal enzyme activity with gestational age in chorionic
villi. J Inherit Metab Dis 13:862, 1990 97. Willems P, Gatti R, Darby J et al: Fucosidosis revisited: A review of 77 patients. Am J Med Genet 38:111, 1991 98. Willems P, Garcia C, De Smedt M et al: Intrafamilial variability in fucosidosis. Clin Genet 34:7, 1988 99. Durand P, Borrone G, Della Cella G: Fucosidosis. J Pediatr 75:665, 1969 100. Michalski J, Wieruszeski J, Alonson C et al: Characterization and 400-MHZ IH-NMR analysis of urinary fucosyl glycoasparagines
in fucosidosis. Eur J Biochem 201:439, 1991 101. Cook P, Noades J, Newton M et al: On the orientation of the Rh:E1-1 linkage group. Ann Hum Genet 41:157, 1977 102. Snodgrass M: Ocular finding in a case of fucosidosis. Br J Ophthalmol 60:508, 1976 103. Smith E, Graham J, Ledman J et al: Fucosidosis. Cutis 19:195, 1977 104. Dvoretzky I, Fisher B: Fucosidosis. Intl J Dermatol 18:213, 1979 105. Spranger J, Cantz M. Mucolipidosis I, the cherry-red spot-myoclonus syndrome
and neuraminidase deficiency. Birth Defects Orig Art Ser 14:105, 1978 106. Kelley T: The mucopolysaccharidosis and mucolipidosis. Clin Orthop 114:116, 1976 107. Cantz M, Gehler J, Spranger J: Mucolipidosis I: Increased sialic acid content and deficiency of an alpha-N-acetylneuraminidase in cultured fibroblasts. Biochem Biophys Res Commun 74:732, 1977 108. Federico A, Cecio A, Battini G et al: Macular cherry red spot and myoclonus syndrome: Juvenile form of sialidosis. J Neurol Sci 48:157, 1980 109. Rapin I, Goldfischer S, Katzman R et al: The cherry-red-spot-myoclonus syndrome. Ann Neurol 3:234, 1978 110. Mueller O, Henry W, Haley L et al: Sialidosis and galactosialidosis: chromosomal assignment of two genes associated
with neuraminidase deficiency disorders. Proc Natl Acad Sci USA 83:1817, 1985 111. Thomas G, Tipton R, Ch'ien L et al: Sialidosis (alpha-N-acetylneuraminidase) deficiency: The enzyme defect in an adult with macular
cherry-red spots and myoclonus without dementia. Clin Genet 13:369, 1978 112. Durand P, Gattlie R, Cavalier S et al: Sialidosis (mucolipidosis I). Helv Acta Paediatr 32:391, 1977 113. Thomas P, Abrams J, Swallow D et al: Sialidosis type 1: Cherry red spots myoclonus syndrome with sialidase deficiency
and altered electrophoretic mobilities of some enzymes known
to be glycoproteins. J Neurol Neurosurg Psychiatr 42:873, 1979 114. Spranger J, Cantz M: Mucolipidosis I, the cherry red spot myoclonus syndrome and neuraminidase
deficiency. Birth Defects Orig Art Ser 14:105, 1978 115. Aula P, Autio S, Ravio K et al: Aspartylglucosaminuria. In Durand P, O'Brien
J (eds): Genetic Errors of Glycoprotein Metabolism, p 123. Berlin: Springer-Verlag, 1982 116. Morris C, Heisterkamp N, Groffen J et al: Chrosomal localization of the human glycoasparaginase gene to 4q32–q33. Hum Genet 88:295, 1992 117. Autio S: Aspartylglycosaminuria: Analysis of thirty-four patients. J Men Def Res Monogr Ser 1:1, 1972 118. Maury C, Palo J: N-Acetylglucosamine-asparagine levels in tissues of patients with aspartyglycosaminuria. Clin Chim Acta 108:293, 1981 119. Desnick R, Wang A: Schindler disease: An inherited neuroaxional dystrophy due to alpha-N-acetylgalactosaminidase deficiency. J Inherit Metab Dis 13:549, 1990 120. deGrout P, Westerveld A, Khan P, Tager J: Localization of a gene for human α-galactosidase B on chromosome 22 (n-acetyl-alpha-d-glucosaminidase). Hum Genet 44:305, 1978 121. Schindler D, Bishop D, Wolfe D et al: Neuroaxonal dystrophy due to lysosomal alpha-N-acetylgalactosaminidase deficiency. N Engl J Med 320:1375, 1989 122. Desnick R, Bishop D: Fabry disease: Alpha-N-acetylgalactosaminidase deficiency. In Stanbury J, Wyngaarden J, Frederickson
D et al (eds): The Metabolic Basis of Inherited Disease, p 751. New
York: McGraw-Hill, 1989 123. Niemann A: Ein unbekanntes Krankheitsbild. Jahrb Kinderheilkd 79:1, 1914 124. Pick L: Uber die lipoidzellige Splenohepatomegalie Typus Niemann-Pick als Stoffwechselerkrankung. Med Klin 23:1483, 1927 125. Carstea E, Polymeropoulos M, Parker C et al: Linkage of Niemann-Pick disease type C to human chromosome 18. Proc Natl Acad Sci USA 90:2002, 1993 126. Crocker A: The cerebral defect in Tay-Sachs disease and Niemann-Pick disease. J Neurochem 7:69, 1961 127. Brady R, Kanfer J, Mock M et al: The metabolism of sphingomylin: II. Evidence of enzymatic deficiency in
Niemann Pick disease. Proc Natl Acad Sci USA 55:366, 1966 128. Poulous A, Shankaran P, Jones C et al: Enzymatic hydrolysis of sphingomyelin lysosomes by normal tissues and tissues
from patients with Niemann-Pick disease. Biochem Biophys Acta 751:428, 1983 129. Vanier M, Rousson R, Garcia I et al: Biochemical studies in Niemann-Pick disease: III. In vitro and in vivo
assays of sphingomyelin degradation in cultured skin fibroblasts and amniotic
fluid cells for diagnosis of the various forms of the disease. Clin Genet 27:20, 1985 130. Brady R: The abnormal biochemistry of inherited disorders of lipid metabolism. Fed Proc 32:1660, 1973 131. Sloan HR, Uhlendorf BW, Kanfer JN et al: Deficiency of sphingomyelin-cleaving enzyme activity in tissue cultures
derived from patients with Niemann-Pick disease. Biochem Biophys Res Comm 34:582, 1969 132. Liscum L, Dahl N: Intracellular cholesterol transport. J Lipid Res 33:1239, 1992 133. Brady R: Sphingomyelin lipidosis: Niemann-Pick disease. In Stanbury J, Wyngaarden
J, Frederickson D et al (eds): The Metabolic Basis of Inherited
Disease, p 831. New York: McGraw Hill, 1983 134. Goodman R: Genetic Disorders Among Jewish People. Baltimore: Johns Hopkins
University Press, 1979 135. Cogan D, Kuwabara T: The sphingolipidosis and the eye. Arch Ophthalmol 79:437, 1968 136. Robb R, Kuwabara T: The ocular pathology of type A Niemann-Pick disease: A light and electron
microscopic study. Invest Ophthalmol 12:366, 1973 137. Walton D, Robb R, Crocker A: Ocular manifestations of group A Niemann-Pick
disease. Am J Ophthalmol 85:-174, 1978 138. Cogan D, Chu F, Barranger J et al: Macula halo syndrome: Variant of Niemann-Pick
disease. Arch Ophthalmol 101:-1698, 1983 139. Grunebaum M: The roentogenographic findings in the acute neuronapathic form of Niemann-Pick
disease. Br J Radiol 49:1018, 1976 140. Harzer K, Ruprecht K, Seuffer-Schulze D et al: Morbus Niemann-Pick type B-enzymatisch gesichert-mot unerwarteter retinaler
Beterligung. Graefes Arch Clin Exp Ophthalmol 206:79, 1973 141. Lipson M, O'Donnell J, JW C et al: Ocular involvement in Niemann-Pick disease type B. J Pediatr 108:582, 1986 142. Hammersen G, Oppermann H, Harms E et al: Oculo-neural involvement in an enzymatically proven case of Niemann-Pick
disease type B. Eur J Pediatr 132:77, 1979 143. Al-Hazzaa S, Ozand P: Niemann Pick disease type B. Middle East J Ophthalmol 3:30, 1995 144. Matthews J, Weiter J, Kolodny E: Macula halos associated with Niemann-Pick
type B disease. Ophthalmology 93:-933, 1986 145. Crocker A, Farber S: Niemann-Pick disease. A review of eighteen patients. Medicine 37:1, 1958 146. Elleder M, Jirasek A: International Symposium Niemann Pick disease. Eur J Pediatr 140:1190, 1983 147. Besley G, Hoogeboom A, Hoogeveen A et al: Somatic cell hybridisation studies showing different gene mutations in
Niemann-Pick variants. Hum Genet 54:409, 1980 148. Wenger D, Barth D, Githens J: Nine case of sphingomyelinlipidosis, a new variant in Spanish-American
children, juvenile variants of Niemann-Pick disease with foamy and sea
blue hitiocytes. Am J Dis Child 131:955, 1977 149. Longstreth W, Daven J, Farrell D et al: Adult dystonic lipidosis: Clinical histologic and biochemical findings
of a neurovisceral storage disease. Neurology 32:1295, 1982 150. Vanier M, Rodrigues-La Frasse C, Rousson R et al: Type C Niemann-Pick disease spectrum of phenotype variation in disruption
of ultracellular LDL derived cholesterol processing. Biochom Biophys Acta 1096:328, 1991 151. Cogan D, Chu F, Bachman D et al: The DAF syndrome. Neuroophthalmology 2:7, 1981 152. Pentechev P, Comly M, Kruth H et al: A defect in cholestrol esterification in Niemann-Pick disease type C patients. Proc Natl Acad Sci USA 82:8247, 1985 153. Argoff C, Kaneski C, Blanchette-Mackie E et al: Type C Nieman Pick disease: Documentation of abnormal LDL processing in
lymphocytes. Biochem Biophys Res Commun 171:38, 1990 154. Neville B, Lake B, Stephens R et al: A neurovisceral storage disease with vertical supranuclear ophthalmoplegia
and its relationship to Niemann-Pick disease: A report of nine patients. Brain 96:97, 1973 155. Dunn H, Sweeney V: Progressive supranuclear palsy in an unusual juvenile variant of Niemann-Pick
disease (abstract). Neurology 21:442, 1971 156. Grover W, Naiman J: Progressive paresis of vertical gaze in lipid storage disease. Neurology 21:896, 1971 157. Butler J, Comly M, Kruth H et al: Niemann-Pick variant disorders: Comparison of errors of cellular cholesterol
hemostasis in group D and group C fibroblasts. Proc Natl Acad Sci USA 84:556, 1987 158. Byers D, Rastogi S, Cook H et al: Defective activity of acyl-CoA: Cholesterol O-acetyl transferase in Niemann-Pick
type C and type D fibroblasts. Biochem J 262:713, 1989 159. Fredrickson D, Sloan H: Sphyngomyelin lipidosis: Niemann-Pick disease. In
Stanbury J, Wyngaarden J, Fredrickson D (eds): The Metabolic Basis
of Inherited Disease, p 1655. New York: McGraw-Hill, 1989 160. Schneider E, Pentchev P, Hibbert S et al: A new form of Niemann-Pick disease characterized by temperature labile
sphingomyelinase. J Med Genet 15:370, 1978 161. Opitz J, Stiles F, Wise D et al: The genetics of angiokeratoma corporis diffusum (Fabry's disease) and
its linkage with Xg (a) locus. Am J Hum Genet 17:325, 1965 162. Kent J: Fabry's disease, alpha-galactosidase deficiency. Science 167:1268, 1970 163. Johnson D, Del Monte M, Collier E et al: Fabry disease: Diagnosis by alpha-galactosidase activity in tears. Clin Chem Acta 63:81, 1975 164. Sorensen S, Hasholt L: Attitudes of persons at risk for Fabry's disease toward predictive
and genetic counselling. J Biosoc Sci 15:89, 1983 165. Brady R, Gal A, Bradley R et al: Enzymatic defect in Fabry's disease: Ceramide trihexosidase deficiency. N Engl J Med 276:1163, 1967 166. Lockman L, Hunninghake D, Krivit W et al: Relief of pain of Fabry's disease by diphenylhydantoin. Neurology 23:871, 1973 167. Matsui S, Murakami E, Takekoshi N: Cardiac manifestations of Fabry's
disease. Nippon Junkankigakushi 41:-1023, 1977 168. Terlinde R, Richard G, Lisch W et al: Ruckbildung der Cornea Verticillata bei Morbus-Fabry durch kontaktlinsen
erste Beobachtungen. Contactologia 4:20, 1982 169. Weicksel J: Angiomatosis, bzw, Angiokeratosis universalis (eine sehr seltene Haut-und-Gaffaskrankheit). Dtsch Med Wochensch 51:898, 1925 170. Gruber H: Cornea verticillata. Ophthalmologica 111:120, 1946 171. Sher N, Letson R, Desnick R: The ocular manifestations in Fabry's disease. Arch Ophthalmol 97:671, 1979 172. Spaeth G, Frost P: Fabry's disease: Its ocular manifestations. Arch Ophthalmol 74:760, 1965 173. Desnick R, Sweeley C: Prenatal detection of Fabry disease. In Dorfman A (ed): Antenatal
Diagnosis, p 185. Chicago: University of Chicago Press, 1971 174. Mayes J, Scheerer J, Sifers R, Donaldson M: Differential assay for lysosomal alpha-galactosidases in human tissues
and its application to Fabry's disease. Clin Chim Acta 112:247, 1981 175. Font R, Fine B: Ocular pathology in Fabry's disease, histochemical electron microscopic
observation. Am J Ophthalmol 73:419, 1972 176. Kleijer W, Hussaarts-Odijk L, Sacks E et al: Prenatal diagnosis of Fabry's disease by direct analysis of chorionic
villi. Prenat Diagn 7:283, 1987 177. Brill N, Mandelbaum F, Libman F: Primary splenomegaly- Gaucher type: Report on one of few cases occurring
in a single generation of one family. Am J Med Sci 129:491, 1905 178. Fried K: Population study of chronic Gaucher's disease. Isr J Med Sci 9:1396, 1973 179. Shafti-Zagardo B, Devine E, Smith M et al: Assignment of the gene for acid beta glucosidase to human chromosome 1. Am J Hum Genet 33:564, 1981 180. Fredrickson S, Sloan H: Glucosyl ceramide lipidosis: Gaucher's disease. In
Stanbury J, Wyngaarden J, Frederickson D (eds): The Metabolic
Basis of Inherited Disease, p 730. New York: McGraw-Hill, 1972 181. Petrohelos M, Tricoulis D, Kotsiras I et al: Ocular manifestation of Gaucher's disease. Am J Ophthalmol 80:1006, 1975 182. Chu F, Rodrigues M, Cogan D et al: The pathology of pingueculae in Gaucher's disease. Ophthalm Pediatr Genet 4:7, 1983 183. Kolodny E, Ullman M, Mankin H et al: Phenotypic manifestations of Gaucher's
disease. Clinical features in 48 biochemically verified type
I patients and comments on type II patients. In Desnick R, Gott S, Grabowski
G (eds): Gaucher Disease: A Century of Delineation and Research, p 33. New
York: Alan R. Liss, 1982 184. Wenger D, Roth S, Kudoh T et al: Biochemical studies in a patient with subacute neuropathic Gaucher disease
without visceral glucosylceramide storage. Pediatr Res 17:344, 1983 185. Erickson A: Gaucher disease, Norrbottnian Type III: Neuropediatric and neurological
aspects of clinical patterns and treatment. Acta Paediatr Scand 326:7, 1986 186. Abrahamov A, Elstein D, Gross-Tsur V et al: Gaucher's disease variant characterised by progressive calcification
of heart valves and unique genotype. Lancet 346:1000, 1995 187. Chabas A, Cormand B, Grinberg D et al: Unusual expression of Gaucher's disease: Cardiovascular calcifications
in three sibs homozygous for the D409H mutation. J Med Genet 32:740, 1995 188. Beutler E, Kuhl W, Trinidad F et al: Beta glucosidase activity in fibroblasts in homozygotes and heterozygotes
for Gaucher's disease. Am J Hum Genet 23:62, 1971 189. Rappeport J, Ginns E: Bone marrow transplantation in severe Gaucher's disease. N Engl J Med 311:84, 1984 190. Von Hirsch T, Peiffer J: Uber histologische Methoden in der differential Diagnose von Leukodystrophien
und Lipoidosen. Arch Psychiatr Nervenker 194:88, 1955 191. Peiffer J: Uber die Metachromatischen leukodystrophien. Arch Psychiatr Nerverkr 199:386, 1959 192. Austin J, Armstrong D, Shearer L: Metachromatic form of diffuse cerebral sclerosis: V. The nature and significance
of low sulfatase activity, a controlled study of brain, liver
and kidney in four patients with metachromatic leukodystrophy (MLD). Arch Neurol 13:593, 1965 193. Austin J, McAfee D, Shearer L: Metachromatic form of diffuse cerebral sclerosis: IV. Low sulfatase activity
in the urine of nine living patients with metachromatic leukodystrophy (MLD). Arch Neurol 12:447, 1965 194. Geurts van Kessel A, Westerveld A, de Groot P et al: localization of the genes coding for human ACO2, ARSA, and NAGA on chromosome 22. Cytogenet Cell Genet 28:169, 1980 195. DeSilva K, Pearce J: Neuropathy of metachromatic leucodystrophy. J Neurol Neurosurg Psychiatr 36:30, 1973 196. Hagberg B: The clinical diagnosis of Krabbe's infantile leucodystrophy. Acta Pediatr Scand 52:213, 1963 197. Cogan D, Kuwabara T, Moser H: Metachromatic leucodystrophy. Ophthalmol 160:2, 1970 198. Libert J, Van Hoof F, Toussaint D et al: Ocular findings in metachromatic leukodystrophy: An electron microscopic
and enzyme study in different clinical and genetic variants. Arch Ophthalmol 97:1495, 1979 199. Goebel H, Shimokawa K, Argyrakis A et al: The ultrastructural of the retina in adult metachromatic leukodystrophy. Am J Ophthalmol 85:841, 1978 200. Goebel H, Busch H: Abnormal lipopigments and lysosomal residual bodies in metachromatic leukodystrophy. Adv Exp Med Biol 266:299, 1989 201. Gordon N: The insidious presentation of the juvenile form of MLD. Postgrad Med J 54:335, 1978 202. McKhann G: Metachromatic leukodystrophy: Clinic and enzymatic parameters. Neuropediatrics 15:4, 1984 203. Carlin L, Roach E, Riela A et al: Juvenile metachromatic leukodystrophy: Evoked potentials and computed tomography. Ann Neurol 13:105, 1983 204. Wulff C, Trojaborg W: Adult metachromatic leukodystrophy: Neurophysiologic findings. Neurology 35:1776, 1985 205. Kappler J, Von Figura K, Gieselmann V: Late onset metachromatic leukodystrophy: Molecular pathology in two siblings. Ann Neurol 31:256, 1992 206. Bosch E, Hart M: Late adult onset metachromatic leukodystrophy: Dementia and polyneuropathy
in 63 year old man. Arch Neurol 35:475, 1978 207. Brismar J: CT and MRI of the brain in inherited neurometabolic disorders. J Child Neurol 7:112, 1992 208. Tagliavini F, Pietrini V, Pilleri G et al: A case report: Adult metachromatic leucodystrophy, clinicopathological
report of two familial cases with slow course. Neuropathol Appl Neurobiol 5:233, 1979 209. Austin J, Armstrong D, Fouch S et al: Metachromatic leukodystrophy (MLD). VIII. MLD in adults: Diagnosis and
pathogenesis. Arch Neurol 18:225, 1968 210. Dayan A: Dichroism of cresyl violet-stained cerebroside sulfate (“Sulfatide”). J Histochem Cytochem 15:421, 1967 211. Takahashi K, Naito M: Lipid storage disease: Part II. Ultrastructural pathology of lipid storage
cells in sphingolipidoses. Acta Pathol Jap 35:385, 1985 212. Markiewicz D, Adamczewska-Goncerzewicz Z, Zelman I et al: A case of MLD with a chronic course (clinical-morphological-biochemical
study). Neuropathol Pol 16:233, 1978 213. Raghavan S, Gajewski A, Kolodny E: Leukocyte sulfatidase for reliable diagnosis of MLD. J Neurochem 36:724, 1981 214. Kudeh T, Wenger D: Diagnosis of MLD after uptake of fatty acid-labeled cerebroside sulfate
into cultured skin fibroblasts. J Clin Invest 70:89, 1982 215. Molzer B, Sundt-Heller R, Kainz-Kraschinsky M et al: Elevated sulfatide excretion in heterozygotes of metachromatic leukodystrophy: Dependence
of arylsulfatase A activity. Am J Med Genet 44:523, 1992 216. Suzuki Y, Suzuki K: Krabbe's globoid cell leukodystrophy: Deficiency of galactocerebrosidase
in serum, leukocyte and fibroblasts. Science 171:73, 1971 217. Zlotogora J, Charkraborty S, Knowleton R et al: Krabbe disease locus mapped to chromosome 14 by genetic linkage. Am J Hum Genet 47:37, 1990 218. Krabbe K: A new familial, infantile form of diffuse brain sclerosis. Brain 39:74, 1916 219. Collier J, Greenfield J: The encephalitis periaxialis of Schilder: A clinical and pathological study
with an account of two cases one of which was diagnosed during life. Brain 47:489, 1924 220. Hagberg B, Kollbberg H, Sourander P et al: Infantile globoid cell leukodystrophy (Krabbe's disease): A clinical
and genetic study of 32 Swedish cases. Neuropaediatrica 1:74, 1970 221. Dunn H, Lake B, Dolman D et al: The neuropathy of Krabbe's infantile cerebral sclerosis (globoid cell
leukodystrophy). Brain 92:329, 1969 222. Suzuki K, Grover W: Krabbe's leukodystrophy (globoid cell leukodystrophy): An ultrastructural
study. Arch Neurol 22:385, 1970 223. Laxdal T, Hallgrimsson K: Krabbe's globoid cell leucodystrophy with hydrocephalus. Arch Dis Child 49:232, 1974 224. Naidu S, Hofmann K, Moser H et al: Galactosylceramidase beta-galactosidase deficiency in association with
cherry-red spot. Neuropediatrics 19:46, 1988 225. Phelps M, Aicardi J, Vanier M: Late onset Krabbe's leukodystrophy, a report of four cases. J Neurol Neurosurg Psychiatr 54:293, 1991 226. Poser C, van Bogaert L: Natural history and evolution of the concept of Schindler's diffuse
sclerosis. Acta Psychiatr Scand 31:285, 1956 227. Loonen M, Van Diggelen O, Janse H et al: Late onset globoid cell leucodystrophy (Krabbe's disease): Clinical
and genetic delineation of two forms and their relation to the early
infantile form. Neuropediatrics 16:137, 1985 228. Basner R, von Figura R, Glossi J et al: Multiple deficiency of mucopolysaccharides sulfatases in mucosulfatidosis. Pediatr Res 13:1316, 1979 229. Fiddler M, Vine D, Shapira E et al: Is multiple sulfatase deficiency due to defective regulation of sulphydrolase
expression. Nature 282:98, 1979 230. Soong B, Cassamassima A, Fink J et al: Multiple sulfatase deficiency. Neurology 38:1273, 1988 231. Burk R, Valle D, Thomas G et al: Early manifestations of multiple sulfatase deficiency. J Pediatr 104:574, 1984 232. Hogan K, Matalon R, Berlow S et al: Multiple sulfatase deficiency: Clinical radiological electrophysiologic
and biochemical features. Neurology 33:245, 1983 233. Bateman J, Phillipart M, Isenberg S: Ocular features of multiple sulfatase deficiency and a new variant of metachromatic
leukodystrophy. J Pediatr Ophthalmol Strabismus 21:133, 1984 234. Harbord M, Guncic J, Chuang S et al: Multiple sulfatase deficiency with early severe retinal degeneration. J Child Neurol 6:229, 1991 235. Al-Aqeel A, Ozand P, Brismar J et al: Saudi variant of multiple sulfatase deficiency. J Child Neurol 7:512, 1992 236. Al-Hazzaa S, Ozand P, Maumenee I: Saudi variant of ocular multiple sulfatase
deficiency (Austin Disease). Presented at Centennial Annual Meeting
of American Academy of Ophthalmology, Chicago, 1996 237. Samuelsson K, Zetterstrom R: Ceramidies in a patient with lipogranulomatosis (Farber's disease) with
chronic course. Scand J Clin Lab Invest 27:393, 1971 238. Sugita M, Dulaney J, Moser H: Ceramidase deficiency in Farber's disease. Science 178:1100, 1972 239. Moser H, Moser A, Chen W et al: Ceramidase deficiency. Farber's disease. In
Stanbury J, Wyngaarden J, Fredrickson D et al (eds): The Metabolic
Basis of Inherited Disease, p 820. New York: McGraw-Hill, 1983 240. Cogan D, Kuwabara T, Moser H et al: Retinopathy in a case of Farber's lipogrannulomatosis. Arch Ophthalmol 75:752, 1960 241. Momoi T, Ben-Yoseph Y, Nadler H: Substrate-specific of acid and alkaline ceramidase in fibroblasts from
patient with Farber's disease and control. Biochem J 205:419, 1982 242. Fensom A, Benson P, Neville B et al: The prenatal diagnosis of Farber's disease. Lancet 2:990, 1979 243. Kerr C, Wells R: Sex-linked ichthyosis. Ann Hum Genet 29:33, 1965 244. Sever R, Frost P, Wienstein G: Eye changes in ichtyosis. JAMA 206:2283, 1968 245. Costagliola C, Fabbrocini G, Illiano G et al: Ocular findings in X-linked ichthyosis: A survey on 38 cases. Ophthalmologica 202:152, 1991 246. Casaroli-Morano R, Ortiz-Stradtmann M, Uxo M et al: Ocular findings associated with congenital x-linked ichthyosis. Ann Ophthalmol 23:167, 1991 247. Okada S, O'Brien J: Generalized gangliosidosis: Beta—galactosidase deficiency. Science 160:1002, 1968 248. Takano T, Yamanouchi Y: Assignment of human beta-galactosidase-A-gene to 3p21.33 by fluorescence
in situ hybridization. Hum Genet 92:403, 1993 249. O'Brien J: Generalized gangliosidosis. J Pediatr 75:167, 1969 250. Gonatas N, Gonatas J: Ultrastructural and biochemical observation on a case of systemic late
infantile lipidosis and its relationship to Tay-Sachs disease and gargoylism. J Neuropathol Exp Neurol 24:318, 1965 251. Lowden JA, Callahan JW, Norman MG et al: Juvenile GM1 gangliosidosis: Occurrence with absence or two beta-galactosidase
components. Arch Neurol 31:200, 1974 252. Derry D, Fawcett J, Andermann F et al: Late infantile systemic lipidosis major monosialoganglisidosis: Delineation
of 2 types. Neurology 18:340, 1968 253. Hooft C, Vlietinck R, Dacremont G et al: GM1 gangliosidosis type II. Eur Neurol 4:1, 1970 254. Suzuki Y, Nakamura N, Fukuoka Shimada F et al: Beta-galactosidase deficiency in jjuvenile and adult patients: Report of
six Japanese cases and review of literature. Hum Genet 36:219, 1977 255. Wenger D, Tarby T, Wharton C: Macular cherry-red spots and myoclonus with dementia: Co-existent neuraminidase
and beta-galactosidase deficiencies. Biochem Biophy Res Commun 82:589, 1978 256. d'Azzo A, Hoogeveen A, Reuser A et al: Molecular defect in combined beta-galactosidase and neuraminidase deficiency
in man. Proc Natl Acad Sci USA 79:4535, 1982 257. Gravel R, Lowden J, Callahan J et al: Infantile sialidosis: A phenocopy of type 1 GM1 gangliosidosis distinguished
by genetic complementation and urinary oligosaccharides. Am J Hum Genet 31:669, 1979 258. Lowden J, Cutz E, Skomorowski M: Infantile type 2 sialidosis with beta-galactosidase
deficiency. In Tetlamanti G, Durand P, Di Donato S (eds): Sialidase
and Sialidosis, p 261. Milan: Edi Ermes, 1981 259. Andria G, Strisciuglio P, Pontgarelli G et al: Infantile neuraminidase
and beta-galactosidase deficiencies (galactosialidosis) with mild clinical
courses. In Durand P, Tetlamanti G, Di Donato S (eds): Sialidases
and Sialidosis, p 379. Milan: Edi Ermes, 1981 260. Strisciuglio P, Sly W, Dodson W et al: Combined deficiency of beta-galactosidase and neuraminidase: Natural history
of the disease in the first 18 years of an American patient with
late infantile onset form. Am J Med Genet 37:573, 1990 261. Suzuki Y, Nanba E, Tsuj A et al: Clinical and genetic hertogeneity in galactosialidosis. Brain Dysfunction 1:285, 1988 262. Suzuki Y, Sakuraba H, Yamanka T et al: Galactosialidosis: A comparative
study of clinical and biochemical data on 22 patients. In Arima M, Suzuki
Y, Yabuuchi H (eds): The Developing Brain and Its Disorders, p 161. Tokyo: University
of Tokyo, 1984 263. Kleijer W, Hoogeveen A, Verheijen F et al: Prenatal diagnosis of sialidosis with combined neurominidase and B-galactosidase
deficiency. Clin Genet 16:60, 1979 264. Takeda K, Nakai H, Hagiwara H et al: Fine assignment of beta hexosaminidase A alpha subunit on 15q23–q24 by
high resolution in situ hybridization. Tohoku J Exp Med 160:203, 1990 265. Fox M, Du Toit D, Warnich L et al: Regional localization of alpha-galactosidase (GLA) to Xpter-q22, hexosaminidase
B (HEXB) to 5q13-qter and arylsulfatase B (ARSB) to 5pter-q13. Cytogenet Cell Genet 38:45, 1984 266. Swallow D, Islam I, Fox M et al: Regional localization of the gene coding for the GM2 activator protein (GM2A) to
chromosome 5q32–33 and confirmation of the assignment
of GM2AP to chromosome 3. Ann Hum Genet 57:187, 1993 267. Tay W: Symmetrical changes in the region of the yellow spot in each eye of an
infant. Trans Ophthalmol Soc UK 1:155, 1881 268. Sachs B: A family form of idiocy, generally fatal, associated with early blindness. J Nerv Ment Dis 21:475, 1896 269. Kivlin J, Snaborn G, Myers G: The cherry red spot in Tay-Sachs and other storage diseases. Ann Neurol 17:356, 1958 270. Collins T: Cited in Kingdom EC: A rare fatal disease of infancy with symmetrical changes
at the macula lutea. Trans Ophthalmol Soc UK 12:126, 1892 271. Cogan D, Kuwabara T: Histochemistry of the retina in Tay-Sachs disease. Arch Ophthalmol 61:414, 1959 272. Sandhoff K, Andreae U, Jatzkewitz H: Deficient hexosamidase activity in an exceptional case of Tay-Sachs disease
with additional storage of kidney globoside in visceral organs. Life Sci 7:283, 1968 273. Sandhoff K, Harzer K, Wassle W et al: Enzyme alterations and lipid storage in three variants of Tay-Sachs disease. J Neurochem 18:2469, 1971 274. Strecker G, Herlant-Peers M, Fournet B et al: Structure of seven oligosaccharides excreted in the urine of a patient
with Sandhoff's disease (GM2 gangliosidosis variant O). Eur J Biochem 81:165, 1977 275. Tremblay M, Szots F: GM2 type 2-gangliosidosis (Sandhoff's disease): Ocular and pathological
manifestations. Can J Ophthalmol 9:338, 1974 276. Brownstein S, Carpenter S, Polomeno R et al: Sandhoff's disease (GM2 gangliosidosis type 2): Histopathology and ultrastructure of the eye. Arch Ophthalmol 98:1089, 1980 277. Hechtman P, Gordon B, Na Ying Kin N: Deficieny of the hexosaminidase A activator protein in a case of GM2 gangliosidosis, variant AB. Pediatr Res 16:217, 1982 278. Burg J, Conzelmann E, Sandhoff K et al: Mapping of the gene coding for the human GM2 activator protein to chromosome 5. Ann Hum Genet 49:41, 1985 279. Kolodny E, Wald I, Moser H et al: GM2 gangliosidosis without deficiency in the artificial substrate cleaving
activity of hexosaminidase A and B. Neurology 23:427, 1973 280. Von Gierke E: Hepato-nephro-megalia glycogenica (Glykogenspeicher-Krankheit der Leber
und Nieren). Beitr-Pathol Anat 82:497, 1929 281. Cori G, Cori C: Glucose-6-phosphatase of the liver in glycogen storage disease. J Biol Chem 199:661, 1952 282. Lei K, Pan C, Shelly L et al: Identification of mutations in the gene for glucose-6-phosphatase, the
enzyme deficient in glycogen storage disease type 1a. J Clin Invest 93:1994, 1994 283. Fine R, Wilson W, Donnell G: Retinal changes in glycogen storage disease type 1. Am J Dis Child 115:328, 1968 284. Hayasaka S, Noda S, Fujii M et al: Inverted eyelashes in patients with type Ia glycogen storage disease. Graefes Arch Clin Exp Ophthalmol 227:209, 1989 285. Golbus M, Simpson T, Koresawa M et al: The prenatal determination of glucose-6-phosphatase activity by fetal liver
biopsy. Prenat Diagn 8:401, 1988 286. Sparkes R, Sparkes M, Funderburk S, Moedjono S: Expression of galactose-1-P uridyltransferase in patients with chromosome
alterations affecting 9p: assignment of the locus to p11–22 (abstract). Cytogenet Cell Genet 25:209, 1979 287. Elsevier S, Kucherlapati R, Nichols E et al: Assignment and regional localization of a gene coding for galactokinase
to human chromosome 17q21–22. Cytogenet Cell Genet 14:287, 1975 288. Lin M, Oizumi J, Ng W et al: Regional mapping of the gene for human UDPGal 4-epimerase on chromosome 1 in
mouse-human hybrids. Cytogenet Cell Genet 24:217, 1979 289. Mason H, Turner M: Chronic galactosemia. Am J Dis Child 50:359, 1935 290. Nadler H, Inouye T, Hsia D: Clinical galactosemia: A study of 55 cases. In
Itsia D (ed): Galactosemia, p 127. Springfield, IL: Charles C. Thomas, 1969 291. Levy N, Krille A, Beutler E: Galactokinase deficiency and cataracts. Am J Ophthalmol 74:41, 1972 292. Beutler E, Matsumoto F, Krille W et al: Galactokinase deficiency as a cause
of cataracts. N Engl J Med 288:-1203, 1973 293. Skalka H, Prchal J: Presenile cataracts formation and decreased activity of galactosemic enzymes. Arch Ophthalmol 98:269, 1980 294. Elman M, Miller M, Matalon R: Galactokinase activity in patients with idiopathic cataracts. Ophthalmology 93:210, 1986 295. Levy N: Etiology and management of infantile cataracts. Ophthalmol Digest 35:41, 1973 296. Kaufman F, Donnell G, Roe T et al: Gonadal function in patients with galactosemia. J Inherit Metab Dis 9:140, 1986 297. Norum K, Gjone E: Familial plasma lecithin cholestrol: Acytransferase deficiency—biochemical
study of a new inborn error of metabolism. Scand J Clin Lab Invest 20:23, 1967 298. Gjone E, Torsvik H, Norum K: Familial plasma cholestrol ester deficiency: A study of erythrocytes. Scand J Clin Lab Invest 21:327, 1968 299. Jacobsen C, Gjone E, Hovig T: Sea blue histiocytes in familial lecithin: cholestrol acyltransferase deficiency. Scand J Haematol 9:106, 1972 300. Gjone E: Familial lecithin: Cholestrol and acyl transferase deficiency: A
clinical survey. Scand J Clin Lab Invest 33:-73, 1974 301. Gjone E: Recent research on lecithin-cholestrol acyltrans-ferase II. Scand J Clin Lab Invest 38:150, 1978 302. Bassen F, Kornzweig A: Malformation of the erythrocytes in a case of atypical retinitis pigmentosa. Blood 5:381, 1950 303. Levy R, Fredrickson D, Laster L: The lipoproteins and lipid transport in abetalipoproteinemia. J Clin Invest 45:531, 1966 304. Shoulders C, Brett D, Bayliss J et al: Abetalipoproteinemia is caused by defects of the gene encoding the 97kDa
subunit of a microsomal triglyceride transfer protein. Hum Molec Genet 2:2109, 1993 305. Wettereau J, Aggerbeck L, Bouma M et al: Absence of microsomal triglyceride transfer protein in individuals with
abetalipoproteinemia. Science 258:999, 1993 306. Singer K, Fisher B, Perlstein M: Acanthocytosis: A generic erythrocyte malformation. Blood 7:577, 1952 307. Lange Y, Steck T: Mechanism of red blood cells acanthocytosis and echinocytosis
in vivo. J Membrane Biol 77:-153, 1984 308. Khachadurian A, Freyha R, Shamma'a M et al: Abetalipoproteinemia and colour blindness. Arch Dis Child 46:871, 1971 309. Dieckert J, White M, Christmann L et al: Angioid streaks associated with abetalipoproteinemia. Ann Ophthalmol 21:173, 1989 310. Mier M, Schwartz S, Boshes B: Acanthocytosis, pigmentary degeneration of the retina and ataxic neuropathies: A
genetically determined syndrome with associated metabolic disorder. J Biol Chem 5:1586, 1960 311. Cohen D, Bosley T, Savino P et al: Primary aberrant regeneration of the oculomotor nerve: Occurrence in a
patient with abetalipoproteinemia. Arch Neurol 42:821, 1985 312. Gouras P, Carr R, Gunkel R: Retinitis pigmentosa in abetalipoproteinemia—effects of vitamin A. Invest Ophthalmol Vis Sci 10:784, 1971 313. Herbert P, Gotto A, Fredrickson D: Familial lipoprotein deficiency (abetaliporpoteinemia, hypolipoproteinemia and Tangier disease). In Stanbury
J, Wyngaarden J, Fredrickson D (eds): The Metabolic Basis of Inherited
Disease, p 544. New York: McGraw-Hill, 1978 314. Bron A: Dyslipoproteinemias and their ocular manifestations. Birth Defects Orig Art Ser 12:257, 1976 315. Van Bogaert L, Scherer H, Epstein E: Une forme cerebrale de la cholesterinose
generalisee. Paris: Mason et Cie, 1937 316. Cali J, Hsieh C, Francke U et al: Mutations in the bile acid biosynthetic enzyme sterol 27-hydroxylase underlie
cerebrotendinous xanthomatosis. J Biol Chem 266:7779, 1991 317. Phillipart M, Van Bogaert L: Cholestanolosic (cerebrotendinous xanthomatosis): A follow-up study on
the original family. Arch Neurol 21:603, 1969 318. McKenna P, Morgan S, Bosanquet R et al: A case of cerebrotendinous xanthomatosis: The sterol contents of a cataractous
lens. Br J Ophthalmol 74:629, 1990 319. Gofman J, Rubin L, McGinley J et al: Hyperlipoproteinemias. Am J Med 17:514, 1954 320. Kwiterovich PJ, Levi R, Fredrickson D: Neonatal diagnosis of familial type-II hyperlipoproteinemia. Lancet 1:118, 1973 321. Goldstein J, Brown M: The LDL receptor locus and the genetics of familial hypercholesterolemia. Annu Rev Genet 13:259, 1979 322. Muller C: Xanthomata, hypercholesterolemia, augria pectori. Acta Med Scan 89:75, 1938 323. Brown M, Kovanen P, Goldstein J et al: Prenatal diagnosis of homozygous familial hypercholesterolemia: Expression
of a genetic receptor disease in utero. Lancet 1:526, 1978 324. McDowell G, Gahl W, Stephenson L et al: Linkage of the gene for cystinosis to markers on the short arm of chromosome 17. Nature Genet 10:246, 1995 325. Gahl W: Cystinosis coming of age. Adv Pediatr 33:95, 1986 326. Cogan D, Kuwabara T, Kinoshita J et al: Ocular manifestations of systemic cystinosis. Arch Ophthalmol 55:36, 1956 327. Kaiser-Kupfer M, Caruso R, Minkler D et al: Long term ocular manifestations in nephropathic cystinosis. Arch Ophthalmol 104:706, 1986 328. Cogan D, Kuwabara T: Ocular pathology of cystinosis with particular reference to the elusiveness
of the corneal crystals. Arch Ophthalmol 63:51, 1960 329. Wong V: Ocular manifestations in cystinosis. Birth Defects Orig Artic Ser 12:181, 1976 330. Katz B, Melles R, Schneider J et al: Corneal thickness in nephropathic cystinosis. Br J Ophthalmol 73:665, 1989 331. Wan W, Minckler D, Rao N: Pupillary-block glaucoma associated with childhood cystinosis. Am J Ophthalmol 101:700, 1986 332. Wong V, Lietman P, Seegmiller J: Alterations of pigment epithelium in cystinosis. Arch Ophthalmol 77:361, 1967 333. Mudd S, Finkelstein J, Irreverre F et al: Homocystinuria: An enzymatic defect. Science 143:1443, 1964 334. Carson N, Neill D: Metabolic abnormalities detected in a survey of mentally backward individuals
in Northern Ireland. Arch Dis Child 37:505, 1962 335. Hu F, Gu Z, Kozich V et al: Molecular basis of cystathioine beta-synthase deficiency in pyridoxine
reponsive and nonresponsive homocystinuria. Hum Mol Gen 2:1857, 1993 336. Aral B, Conde M, London J et al: Two novel mutations (K384E and L539S) in the C-terminal moiety of cystathionine
beta-synthase protein in two French pyridoxine-responsive homocystinuria
patients. Hum Mutat 9:81–82, 1997 337. Mudd S, Skovby F, Levy H et al: The natural history of homocystinuria due to cystathionine beta-snythase
deficiency. Am J Hum Genet 37:1, 1985 338. Nelson L, Maumenee I: Ectopia lentis. Surv Ophthalmol 27:143, 1982 339. Ramsey M, Yanoff M, Fine B: The ocular histopathology of homocystinuria. A light and electron microscopic
study. Am J Ophthalmol 74:377, 1972 340. Ramsey M, Dickson D: Lens fringe in homocystinuria. Br J Ophthalmol 59:338, 1975 341. Colditz P, Yu J, Billson F et al: Tyrosinaemia II. Med J Aust 141:244, 1984 342. Barton D, Yang-Feng T, Francke U: The human tyrosine aminotransferase gene mapped to the long arm of chromosome 16 (region 16q22–q24) by somatic hybrid analysis and in situ
hybridization. Hum Genet 72:221, 1986 343. Richner H: Horhautaffektion bei Keratoma palmare et plantare heriditarium. Klin
Monatsbl Augenheinkld 100:-580, 1938 344. Hanhart E: Neue Sonderfornen von Keratosis palmo-plantaris, u.a. eine regelmaessigdominante
mit system atisieren Lipomen, ferner 2 einfach-rezessive mit
Schwachsinn und z.T. mit Hornhautveraenderungen des Auges. Dermatologica 94:286, 1947 345. Heidemann D, Dunn S, Bawle E et al: Early diagnosis of tyrosinemia type II. Am J Ophthalmol 107:559, 1989 346. Bohnert A, Anton-Lamprecht I: Richner-Hanhart's syndrome: Ultrastructural abnormalities of epidermal
keratinization indicating a causal relationship to high intracellular
tyrosine levels. J Invest Dermatol 79:68, 1982 347. Goldsmith L: Tyrosinemia and related disorders. In Stanbury J, Wyngaarden
J, Fredrickson D et al (eds): The Metabolic Basis of Inherited Disease, p 547. New
York: McGraw-Hill, 1989 348. Gramet C, Lods F: Syndrome de Richner-Hanhart sans atteinte cutanee. Bull Soc Ophthalmol Fr 84:129, 1984 349. Westall R, Dancis J, Miller S: Maple syrup urine disease. Am J Dis Child 94:571, 1957 350. Menkes J, Hurst P, Craig J: A new syndrome: Progressive familial infantile cerebral dysfunction associated
with an unusual urinary substance. Pediatrics 14:462, 1954 351. Mantovani J, Naidich T, Prensky A et al: MSUD: Presentation with pseudotumor cerebri and CT abnormalities. J Pediatr 96:279, 1980 352. Zee D, Freeman J, Holtzman N: Ophthalmoplegia in maple syrup urine disease. J Pediatr 84:113, 1974 353. Millington D, Kodo N, Norwood D et al: Tandem mass spectrometry: A new method for acylcarnitine profiling with
potential for neonatal screening for inborn errors of metabolism. J Inherit Metab Dis 13:321, 1990 354. Boedeker C: Ueber das Alcapton ein neuer Beitrog zur Froge: Welche staffe des Haus
konnen Kupferreduction bewirken. Rat Med 7:130, 1859 355. Pollak M, Chou Y-H, Cerda J et al: Homozygosity mapping of the gene for alkaptonuria to chromosome 3q2. Nature Genet 5:201, 1993 356. O'Brien W, LoDubn B, Bunim J: Biochemical pathological and clinical aspects of alcaptonuria, ochronosis
and ochronosis arthropathy. Am J Med 34:813, 1963 357. Srsen S, Vondracek J, Srsnova K et al: Analysis of the life span of alcaptonuric patients. J CSS Lek Cesk 124:1288, 1985 358. Gaines JJ: The pathology of alkaptonuric ochronosis. Hum Pathol 20:40, 1989 359. Smith J: Ochronosis of the sclera and cornea complicating alkaptonuria: Review of
the literature and report of four cases. JAMA 120:1282, 1942 360. Wolff J, Barshop B, Nyhan W et al: Effects of ascorbic acid in alkaptonuria: Alterations in benzoquinone acetic
acid and an ontogenic effect in infancy. Pediatr Res 26:140, 1989 361. Bennett M, Gray R, Isherwood D et al: The diagnosis and biochemical investigation of a patient with a short-chain
fatty acid oxidation defect. J Inherit Metab Dis 8:135, 1985 362. Amendt B, Greene C, Sweetman L et al: Short-chain-acyl-co-enzyme A dehydrogenase deficiency: Clinical and biochemical
studies in two patients. J Clin Invest 79:1303, 1987 363. Turnbull D, Shepherd I, Bartlett K et al: Short-chain acyl-CoA dehydrogenase deficiency: Fatty acid oxidation—Clinical, biochemical
and molecular aspects. Progress Clin Biol Res 321:313, 1990 364. Barton D, Yang-Feng T, Finnocchiaro G et al: Short-chain-acyl-CoA dehydrogenase (SCADS) maps to chromosome 12 (q22-qter) and
electron transfer flavoprotein (ETFA) to 15 (q23–q25). Cytogenet Cell Genet 46:577, 1987 365. Rhead W: Inborn errors of fatty acid oxidation in man. Clin Biochem 24:319, 1991 366. Stanley C: New genetic defects in mitochondrial fatty acid oxidation and carnitine
deficiency. Adv Pediatr 34:59, 1987 367. Coates P, Hale D, Finocchiaro G et al: Genetic deficiency of short-chain
acyl-co-enzyme: A dehydrogenase in cultured fibroblasts from a patient
with muscle carnitine deficiency and severe skeletal muscle weakness. J
Clin Invest 81:-175, 1988 368. Turnbull D, Bartlett K, Stevens D et al: Short-chain acyl-CoA dehydrogenase deficiency associated with a lipid storage
myopathy and secondary carnitine deficiency. N Engl J Med 311:1232, 1984 369. Amendt BA, Greene C, Sweetman et al.Short-chain acyl-coenzyme A dehydrogenase deficiency: Clinical and biochemical
studies in two patients. J Clin Invest 79:1303–1309, 1987 370. Hale D, Bennett M: Fatty acid oxidation disorders, a new class of metabolic diseases. J Pediatr 121:1, 1992 371. Burlina A, Zacchello F, Dionisi-Vici C et al: New clinical phenotype of branched-chain acyl-Co-A oxidation defect. Lancet 338:1522, 1991 372. Burlina A, Dionisi-Vici C, Bennett M et al: A new syndrome with ethylmalonic-aciduria and normal fatty and oxidation
I fibroblasts. J Pediatr 124:79, 1994 373. Ozand P, Rashed M, Millington D et al: Ethylmalonic aciduria: an organic acidemia with CNS involvement and vasculopathy. Brain 16:12, 1994 374. Cogan D, Schulman J, Porter R et al: Epileptiform ocular movements with methylmalonic aciduria and homocystimuria. Am J Ophthalmol 90:251, 1980 375. Robb R, Dowton S, Fulton A et al: Retinal degeneration in Vitamin B12 disorder associated with methymalonic
aciduria and sulfur amino acid abnormalities. Am J Ophthalmol 97:691, 1984 376. Carmel R, Bedros A, Mace J et al: Congenital methylmalonic aciduria-homocystinuria with megaloblastic anemia: Observations
on response to hydroxcobalamin and the effect of homocyteine
and methronine on the deoxyuridine suppression test. Blood 55:570, 1980 377. Watkins D, Rosenblatt D: Functional methionine synthase deficiency (CblE and CblG): Clinical and
biochemical heterogenetity. Am J Med Genet 34:427, 1989 378. Traboulsi E, Silva J, Geraghty M et al: Ocular histopathologic characteristics of cobalamin C type vitamin B12 defect
with methylmalonic aciduria and homocystinuria. Am J Ophthalmol 113:269, 1992 379. Rosenberg L: Disorders of propionate, methylmalonate and cobalamin metabolism. In
Stanbury J, Wyngaarden J, Fredrickson D et al (eds): The Metabolic
Basis of Inherited Disease, p 411. New York: McGraw-Hill, 1983 380. Zeman W, Dyken P: Neuronal ceroid lipofuscinosis (Batten's disease): Relationship to
amaurotic family idiocy? Pediatrics 44:570, 1969 381. Rider J, Rider D: Batten disease: Past, present and future. Am J Med Genet 5:21, 1988 382. Hofman I: The Batten-Spielmeyer-Vogt Disease. Doorn, The Netherlands: Bartimeus
Foundation, 1990 383. Dyken P: Reconsideration of the classification of the neuronal ceroid lipofuscinosis. Am J Med Genet 5:69, 1988 384. Kohlschutter A, Gardiner R, Goebel H: Human forms of neuronal ceroid lipofuscinosis (Batten disease): Consensus
on diagnostic criteria, Hamburg 1992. J Inherit Metab Dis 16:241, 1993 385. Garborg I, Torvik A, Hals J et al: Congenital neuronal ceroid lipofuscinosis A case report. Acta Pathol Microbiol Immunol 95:119, 1987 386. Boustany R-M, Alroy J, Kolodny E: Clinical classification of neuronal ceroid-lipofuscinosis subtypes. Am J Med Genet 5:47, 1988 387. Goebel H: Neuronal ceroid lipofuscinoses: The current status. Brain Dev 14:203, 1992 388. Berkovic S, Carpenter S, Andermann F et al: Kufs' disease: A critical reappraisal. Brain 111:27, 1988 389. Constantinidis J, Wisniewski K, Wisniewski T: The adult and a new late adult forms of neuronal ceroid lipofuscinosis. Acta Neuropathol 83:461, 1992 390. Santavuori P: Neuronal ceroid lipofuscinoses in childhood. Brain Dev 10:80, 1988 391. Santavuori P, Vanhanen S, Sainio K et al: New aspects in the diagnosis of neuronal ceroid lipofuscinosis. Brain Dysfunction 4:211, 1991 392. Eiberg M, Gardiner R, Mohr J: Batten disease (Spielmeyer-Sjogren disease) and haptoglobin (HP): Indication
of linkage and assignment to chromosome 16. Clin Genet 36:217, 1989 393. Callen D, Baker E, Lane S et al: Regional mapping of the Batten disease locus (CLN3) to human chromosome 16p12. Am J Hum Genet 49:1372, 1991 394. Jarvela I, Schleutker J, Haataja L et al: Infantile form of neuronal ceroid lipofuscinosis (CLN1) maps to the short
arm of chromosome 1. Genomics 9:170, 1991 395. Vesa J, Hallsten E, Verkruyse L et al: Mutations in the palmitoyl protein thioesterase gene causing infantile
neuronal ceroid lipofuscinosis. Nature 376:584, 1995 396. Williams R, Vesa J, Jarvela I et al: Genetic heterogeneity in neuronal ceroid lipofuscinosis (NCL): Evidence
that the late-infantile subtype (Jansky-Bielschowsky disease; CLN2) is
not an allelic form of the juvenile or infantile subtypes. Am J Hum Genet 53:931, 1993 397. Savukoski M, Kestila M, Williams R et al: Defined chromosomal assignment of CLN5 demonstrates that at least four
genetic loci are involved in the pathogenesis of human ceroid lipofuscinoses. Am J Hum Genet 55:695, 1994 398. Boustany R, Kolodny E: Neurological progress. The neuronal ceroid-lipofuscinoses: A review. Rev Neurol (Paris) 145:105, 1989 399. Boustany R-M: Neurology of the neuronal ceroid lipofuscinoses: Late infantile and juvenile
types. Am J Med Genet 42:533, 1992 400. Palmer D, Fearnley I, Walker J et al: Mitochondrial ATP synthase subunit c storage in the ceroid lipofuscinoses (Batten
disease). Am J Med Genet 42:561, 1992 401. Kominami E, Ezaki J, Muno D et al: Specific storage of subunit c of mitochondrial ATP synthase in lysosomes
of neuronal ceroid lipofuscinosis (Batten's disease). J Biochem 111:278, 1992 402. Tyynela J, Palmer D, Baumann M et al: Storage of saposins A and D in infantile neuronal ceroid lipofuscinosis. FEBS Lett 330:8, 1993 403. Santavuori P, Haltia M, Rapola J et al: Infantile type of so-called neuronal ceroid lipofuscinosis: I. A clinical
study of 15 patients. J Neurol Sci 18:257, 1973 404. Goebel H, Zeman W, Damaske E: An ultrastructural study of the retina in the Jansky-Bielschowsky type
of neuronal ceroid lipofucsinosis. Am J Ophthalmol 83:70, 1977 405. Schochet S Jr, Font R, Morris H: Jansky-Bielschowsky form of neuronal ceroid lipofuscinosis: Ocular pathology
of-the Batten-Vogt syndrome. Arch Ophthalmol 98:1083,-1980 406. Copenhaver R, Goodman G: The electroretinogram in infantile, late infantile and juvenile amaurotic
family idiocy. Arch Ophthalmol 63:203, 1960 407. Vogt H: Uber familiare omurotische Idiote und verwandte Krankenheitsbilder. Monatschrr Psychiatr Neurol 18:161, 1905 408. Spielmeyer W: Uber familiore amurotische Idiotie. Neurol Cbl 24:620, 1905 409. Spalton D, Taylor D, Sanders M: Juvenile Batten's disease: An ophthalmological assessment of 26 patients. Br J Ophthalmol 64:726, 1980 410. Traboulsi E, Green W, Luckenbach M et al: Neuronal ceroid lipofuscinosis: Ocular histopathologic and electron microscopic
studies in the late infantile, juvenile, and adult forms. Graefes Arch Clin Exp Ophthalmol 225:391, 1987 411. Goebel H, Fix J, Zeman W: The fine structure of the retina in neuronal
ceroid lipofuscinosis. Am J Ophthalmol 77:-25, 1974 412. Pardo C, Rabin B, Palmer D et al: Accumulation of the adenosine triphosphate synthase subunit c in the mnd
mutant mouse: A model for neuronal ceroid lipofuscinosis. Am J Pathol 144:829, 1994 413. Raininko R, Santavuori P, Heiskala H et al: CT findings in neuronal ceroid lipofuscinosis. Neuropediatrics 21:95, 1990 414. Libert J: Diagnosis of lysosomal storage diseases by the ultrastructural study of
conjunctival biopsies. Pathol Ann 15:37, 1980 415. Brod R, Packer A, Van Dyk H: Diagnosis of neuronal ceroid lipofuscinosis by ultrastructural examination
of peripheral blood lymphocytes. Arch Ophthalmol; 105:1388, 1987 416. Conradi N, Uvebrant P, Hokegard K et al: First-trimester diagnosis of juvenile neuronal ceroid lipofuscinosis by
demonstration of fingerprint inclusions in chorionic villi. Prenat Diagn 9:283, 1989 417. Rapola J, Salonen R, Ammala P et al: Prenatal diagnosis-of the infantile type of neuronal ceroid lipofuscinosis
by electron microscopic investigations of human chorionic villi. Prenat Diagn 10:553, 1990 418. Jarvela I, Rapola J, Peltonen L et al: DNA-based prenatal diagnosis of the infantile form of neuronal ceroid lipofuscinosis (INCL; CLN1). Prenat Diagn 11:323, 1991 419. Uvebrant P, Bjork E, Conradi N et al: Successful DNA-based prenatal exclusion of juvenile neuronal ceroid lipofuscinosis. Prenat Diagn 13:651, 1993 420. Hellsten E, Vesa J, Jarvela I et al: Refined assignment of the infantile neuronal ceroid lipofuscinosis (INCL) locus
at 1p32 and the current status of prenatal and carrier diagnosis. J Inherit Metab Dis 16:335, 1993 421. Gibbs K, Walshe J: Biliary excretion of copper in Wilson's disease. Lancet 2:538, 1980 422. Petrukhin K, Lutsenko S, Chernov I et al: Characterization of the Wilson
disease gene encoding a P-type copper transporting ATPase: Genomic organization, alternative
splicing and structure function predictions. Hum
Mol Genet 3:-1647, 1994 423. Chowrimootoe G, Ahmed H, Seymour C: New insights-into the pathogenesis of copper toxicosis in Wilson's
dis—ease: Evidence for copper incorporation and defective canalicular
transport of caeruloplasmin. Biochem J 315:851, 1996 424. Sass-Kortsak A: Wilson's disease: A treatable cause of liver disease in children. Pediatr Clin North Am 22:963, 1975 425. Strickland G, Leu M: Wilson's disease: Clinical and laboratory manifestations in 40 patients. Medicine 54:113, 1975 426. Scheinberg I, Sternlieb I: Wilson's Disease. Philadelphia: WB Saunders, 1984 427. Lowe C, Terrey M, MacLachan E: Organic aciduria, decreased renal ammonia
production, hydrophthalmos and mental retardation: A clinical entity. Am
J Dis Child 83:-164, 1952 428. Zhang X, Jefferson A, Auethavekiat V et al: The protein deficient in Lowe syndrome is a phosphatidylinositol-4,5-biphosphate 5-phosphatase. Proc Natl Acad Sci USA 92:4853, 1995 429. Menkes J, Alter M, Steigleder G et al: A sex-linked recessive disorder with retardation of growth, peculiar hair-and
focal and cerebellar degeneration. Pediatrics 29:764,-1962 430. Grover W, Johnson W, Henkin R: Clinical and biochemical aspects of trichopoliodystrophy. Ann Neurol 5:65, 1979 431. Horn N. Menkes' X-linked disease: Prenatal diagnosis and carrier detection. J Inherit Metab Dis 6:59, 1983 432. Billings D, Degnan M: Kinky hair syndrome: A new case and review. Am J Dis Child 121:447, 1971 433. Singh S, Bresnan M: Menkes kinky hair syndrome (trichopoliodystrophy): Low copper level in
the blood, hair and urine. Am J Dis Child 125:572, 1973 434. Danks D, Campbell P, Stevens B et al: Menke's kinky hair syndrome: An inherited defect in copper absorption
with widespread effects. Pediatrics 50:188, 1972 435. Van Bogaert L, Bertrand I: Sur une idiotie familiale avec degenerescence spongieuse de nevraxe. Acta Neurol Belg 49:572, 1949 436. Kaul R, Gao G, Balamurugan K et al: Cloning of the human aspartoacylase, DNA and a common missence mutation
in Canavan disease. Nat Genet 5:118, 1993 437. Kaul R, Balamurugan K, Gao G et al: Canavan disease: genomic organization and localization of human ASPA to 17p13-ter
and conservation of the ASPA gene during evolution. Genomics 21:364, 1994 438. Matalon R, Michals K, Sebesta D et al: Aspartoacylase deficiency and N-acetylaspartic aciduria in patients with
Canavans disease. Am J Med Genet 29:463, 1988 439. Divry P, Mathieu M: Aspartoacylase deficiency and N-acetylaspartic aciduria in patients with
Canavan disease. Am J Med Genet 32:550, 1989 440. Ungar M, Goodman R: Spongy degeneration of the brain in Israel. A retrospective study. Clin Genet 23:23, 1983 441. Kelley R, Stamas J: Quantification of N-acetyl-L-aspartic acid in urine by isotope dilution
gas chromatography-mass spectrometry. J Inherit Metab Dis 15:97, 1992 442. Borash V, Flhor D, Morag B et al: A radiometric assay for aspartoacylase activity in human fibroblasts: Application
for the diagnosis of Canavan's disease. Clin Chim Acta 201:175, 1991 |