| Combined disorders of eyes and kidneys may result from a metabolic or a
developmental defect or from vascular or autoimmune disease, infection, tumoral
process, or use of toxic products. The term oculorenal syndromes refers to a large and still expanding group of inherited and noninherited
malformations and multisystemic diseases with peculiar ocular and
renal features. Table 1 shows a classification of this group of disorders according to the present
etiologic knowledge. The classic and a few recently recognized oculorenal
syndromes are discussed here in detail. TABLE 1. An Etiologic Classification of the Oculorenal Syndromes
Chromosomal Abnormality Syndromes
Trisomy 13 syndrome
Trisomy 18 syndrome
Partial trisomy 10q syndrome
Cat-eye syndrome
Cri-du-chat syndrome
WAGR syndrome (aniridia—Wilms' tumor association)
Deletion of the long arm of chromosome 13 (13q— syndrome)
Deletion of the short arm of chromosome 18 (18p— syndrome)
Turner syndrome
Syndromes With Mendelian Mode of Inheritance
Autosomal Dominant Syndromes
Acrorenal syndrome
Apert's syndrome
Alagille's syndrome
Beckwith-Wiedemann syndrome
Charcot-Marie-Tooth disease
Coloboma-brachydactyly association type Sorsby
Familial amyloidosis type 3
Nail-patella syndrome
Neurofibromatosis
Papillorenal syndrome
Tuberous sclerosis
Von Hippel-Lindau syndrome
Autosomal Recessive Syndromes
Alström syndrome
Bardet-Biedl syndrome
Carbohydrate-deficient glycoprotein (CDG) syndromes
Cockayne's syndrome
Cystinosis
Choroidal coloboma—mental retardation association
Fraser-cryptophthalmos syndrome
Fryns syndrome
Galactosemia
Hunter's syndrome
Hypercalciuria—macular coloboma association
Jeune's syndrome
Mainzer-Saldino syndrome
Meckel-Gruber syndrome
Pierson's syndrome
Potter's syndrome
Primary hyperoxaluria
Senior-Loken syndrome
Sialidosis and galactosialidosis
Sickle cell anemia
Smith-Lemli-Opitz syndrome
Wilson's disease
Zellweger syndrome
X-linked Syndromes
Alport's syndrome
Fabry's disease
Lenz microphthalmos syndrome
Lowe oculocerebrorenal syndrome
Other Disorders (Multifactorial, Teratogenic, or Unknown Etiology)
Congenital rubella
Cornelia de Lange syndrome
Diabetes mellitus
Goldenhar syndrome, CHARGE and VATER associations
Membranoproliferative glomerulonephritis type II
Nephronophthisis-mitochondropathy association syndrome
Wildervanck syndrome
CHROMOSOMAL ABNORMALITY SYNDROMES WAGR Syndrome Deletion of band p13 of chromosome 11 produces the WAGR syndrome consisting
of Wilms' tumor, sporadic aniridia, genitourinary malformations, and mental retardation.9,10 Wilms' tumor is an embryonic neuroblastoma and is the most common malignant
renal tumor in both children and adolescents.11 About 6% of all primary renal tumors regardless of age are Wilms' tumors. Although
most Wilms' tumors occur as an isolated sporadic event, there
are several syndromic associations of Wilms' tumor, including WAGR
syndrome.10 A Wilms' tumor suppressor gene has been localized to band p13 of chromosome 11 near
the gene responsible for sporadic aniridia. Sporadic aniridia
is present in 1% of children with Wilms' tumor and results from
a 11p13 deletion that includes both genes. The Wilms' tumor suppressor
gene appears to play an important role in the normal development and
maturation of the kidneys and gonads. Any newborn with nonfamiliar aniridia
should be examined by a geneticist and followed by a medical team
familiar with detection and management of Wilms' tumor.11,12 SYNDROMES WITH MENDELIAN MODE OF INHERITANCE Autosomal Dominant Syndromes ACRO-RENAL-OCULAR SYNDROME. The acro-renal-ocular syndrome is an autosomal
dominant dysmorphogenetic syndrome with high penetrance and variable
expression. The main components of the syndrome are hand, urinary tract, and
ocular anomalies. In addition, there may be perceptive deafness, cardiac
anomalies, and anal stenosis.13,14 The radial ray abnormalities of the hand vary in expression from mild
thenar hypoplasia or inability to flex the interphalangeal joint of the
thumb to hypoplastic thumbs or prominent upper limb abnormalities. The
urinary tract anomalies include unilateral renal agenesis, renal ectopia, malrotation, and
bilateral renal hypoplasia. Duane's anomaly
is the most common ocular abnormality in patients affected by the acro-renal-ocular
syndrome. Microcornea and uveal and optic nerve colobomas
have been reported on occasion.14,15 NAIL-PATELLA SYNDROME. Nail-patella syndrome (hereditary osteo-onychodysplasia, HOOD) is
an autosomal dominant disorder of nails, skeleton, and
kidney. The nail-patella locus has been assigned to the distal end
of the long arm of chromosome 9 (9q34). The collagen gene COL 5A1, which
encodes the pro alpha 1(V) chain, maps in the same region, providing
evidence that the nail-patella syndrome could be an inherited connective
tissue disorder attributable to mutations of the COL 5A1 gene. The nails are hypoplastic or absent, particularly affecting the thumb and
index finger. Characteristic skeletal abnormalities include small or
absent patellae, iliac horns, and hypoplasia of the capitellum and lateral
epicondyle of the distal humerus together with deformity of the
radial head. Recurrent subluxations of knees and elbows, flexion deformities
of the elbows, and Madelung's deformity with volar position
of the wrists often occur in patients with nail-patella syndrome but
rarely are incapacitating. Nephropathy is usually mild and manifests as proteinuria or intermittent
nephrotic syndrome, but end-stage renal failure may occur and nephropathy
may be the most serious complication in some families. Typical renal
lesions are focal glomerular basement membrane thickening with subendothelial
fibrillar electron-dense deposits and moth-eaten appearance
of the glomerular basement membrane. A cloverleaf dark pigmentation of the central area of the iris with scalloped
iris collarette (Lester's sign of the iris16) is a peculiar finding in some patients with the nail-patella syndrome. Keratoconus, microcornea, sclerocornea, microphakia, and cataracts have
also been described.17 PAPILLORENAL SYNDROME. The papillorenal syndrome has been defined by Bron18 as a dominantly inherited disorder with a bilateral dysplasia of the optic
discs associated with a severe form of glomerulonephritis that may
lead to renal failure. The disc anomaly ranges from morning glory anomaly
or the Handmann anomaly to coloboma or optic pit (Fig. 2). Optic nerve function may be impaired, and in addition there is a risk
of visual loss as a result of serous macular detachment. The pathogenesis
of the renal changes and the genetic mechanism linking the renal
and ocular abnormalities are unknown. 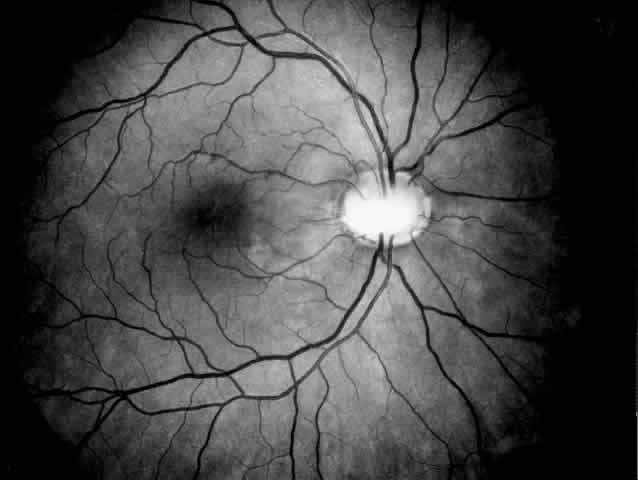 Fig. 2. Papillorenal syndrome with the Handmann optic disc anomaly and macular
changes as a result of chronic macular detachment in a renal transplant
patient. An optic pit and severe hypertensive changes were observed
in his brother, who also received a renal transplant (see Fig. 1). Poor renal function had been recorded in six other family members in
three generations. Fig. 2. Papillorenal syndrome with the Handmann optic disc anomaly and macular
changes as a result of chronic macular detachment in a renal transplant
patient. An optic pit and severe hypertensive changes were observed
in his brother, who also received a renal transplant (see Fig. 1). Poor renal function had been recorded in six other family members in
three generations.
|
VON HIPPEL-LINDAU DISEASE. Von Hippel-Lindau disease is a dominantly inherited
familial cancer syndrome affecting eyes, brain, and kidneys. The
characteristic manifestations are capillary angiomas of the retina
and optic nerve head; cerebellar, medullary, and spinal hemangioblastomas; renal
cell carcinoma; pheochromocytoma; and renal, pancreatic, and
epididymal cysts. The penetrance of von Hippel-Lindau disease is age
and tumor dependent. The cumulative risk of a patient with von Hippel-Lindau
disease developing retinal angioma, cerebellar hemangioblastoma, and
renal cell carcinoma at age 30 years are 44%, 38%, and 5%, respectively, rising
to 84%, 70%, and 69%, respectively, at age 60 years.19 Von Hippel-Lindau disease maps to the region of chromosome 3 associated
with renal cell carcinoma, and there is convincing evidence that the
gene for von Hippel-Lindau disease functions as a tumor suppressor gene
of the retinoblastoma type.19 Presymptomatic diagnosis of von Hippel-Lindau disease with flanking DNA
markers has become available, but the number of families in which presymptomatic
diagnosis is possible is still limited, and presymptomatic
diagnosis remains impossible for relatives of isolated cases. All affected
patients and at-risk relatives require lifelong ocular screening
and investigation for extraocular complications.19 The disease is often lethal. Renal cell carcinoma is the most frequent
cause of death, followed by cerebellar hemangioblastoma.20 Renal cell carcinoma often presents as hematuria, obstructive nephropathy, or
an abdominal mass.21 Delayed diagnosis and surgical removal of the tumor may result in death
from metastasis or uremia. Cerebellar hemangioblastoma may present as
signs of increased intracranial pressure or cerebellar dysfunction. Symptomatic
lesions should be excised. Pheochromocytoma tends to cluster
in certain predisposed families. The more common cysts and angiomatous
tumors of several visceral organs rarely cause symptoms. Screening
of extraocular complications requires annual physical examination, renal
ultrasound examination, and urinary estimation of vanillylmandelic
acids or catecholamines. In addition, magnetic resonance imaging of the
brain and computed tomography of the kidneys should be performed in
all symptomatic patients because they appear to be more sensitive diagnostic
tests. In the Cambridge screening protocol, abdominal computed
tomographic scans and brain magnetic resonance imaging or computed tomographic
scans are also planned at regular intervals in asymptomatic
individuals.19,20 Retinal angiomatosis is the most frequent complication and often the initial
manifestation of von Hippel-Lindau disease. The screening protocol
for von Hippel-Lindau disease in affected patients and at-risk relatives
should include at least annual direct and indirect ophthalmoscopy
and, preferably, fluorescein angiography or angioscopy to permit early
detection and treatment of retinal angiomas. Tumors to 3 mm respond
well to laser photocoagulation.22 Larger peripheral angiomas and those with exudative detachment or arising
close to the ora serrata require cryocoagulation or other surgical
treatment modalities.19 Vitreoretinal surgery may be indicated in cases with extensive epiretinal
fibrosis and traction retinal detachment. In addition, radiation therapy
may be used for optic disc hemangiomas and for tumors not responding
to cryotherapy.19,22 Autosomal Recessive Syndromes BARDET-BIEDL SYNDROME AND RELATED DISORDERS. The cardinal features of Bardet-Biedl
syndrome are retinal dystrophy, polydactyly, obesity, mental
retardation, and hypogenitalism. Renal and urinary tract abnormalities
are common. Incomplete manifestation of the cardinal signs is frequent, but
retinal dystrophy is a essential feature for the Bardet-Biedl
syndrome.23 The electroretinogram is a sensitive detector of retinopathy in patients
with Bardet-Biedl syndrome and may allow identification of the photoreceptor
cell degeneration (cone-rod dystrophy) in young children with
normal fundus appearance.24,25 Fundus changes appear at the end of the first decade, or in the second
or third decade of life, and usually present as atypical central and
peripheral pigmentary abnormalities, attenuated retinal vasculature, and
pale discs. Loss of visual acuity is usually mild in the first decade
of life and marked in the second and third decades. In addition to classic Bardet-Biedl syndrome, several variants and related
disorders with or without ocular abnormalities have been described.23 Those with ocular abnormalities include Laurence-Moon syndrome, Biemond
II syndrome, and Alström syndrome. Patients with Laurence-Moon
syndrome share most features of Bardet-Biedl patients but are spastic
and do not have polydactyly. Patients with Biemond II syndrome have an
iris coloboma and lack pigmentary retinopathy. Alström syndrome
can be distinguished from Bardet-Biedl syndrome by severe visual loss
in the first decade of life, nerve deafness, and diabetes mellitus. Slowly
progressive nephropathy and renal failure are very common as well
in Alström syndrome as in Bardet-Biedl syndrome, and renal failure
is probably the most frequent cause of death.26 CARBOHYDRATE-DEFICIENT GLYCOPROTEIN SYNDROMES. The carbohydrate-deficient
glycoprotein syndromes are a group of genetic multisystemic diseases
with characteristic deficiency of several carbohydrates in a number
of glycoproteins. The severity of symptoms is variable, and several subtypes
have been identified.27,28 These syndromes are mainly nervous system disorders, but ocular and renal
involvement are part of the syndromes. The disease may present in infancy or early childhood with combinations
of failure to thrive, developmental delay and strokelike episodes, esotropia, lipoatrophic
changes, liver dysfunction, or pericardial effusions.27,28 In older children and in adults the combination of mental retardation, olivopontocerebellar
atrophy, peripheral neuropathy, and photoreceptor
cell degeneration29–31 is strongly suggestive of carbohydrate-deficient glycoprotein syndrome. Renal
cysts are common.31,32 CYSTINOSIS. Cystinosis is an autosomal recessive lysosomal storage disorder. In
affected patients a primary defective transport of cystine out
of lysosomes causes intracellular cystine accumulation and deposition
of cystine crystals in many body tissues, including the kidneys and
the eyes. The infantile form of nephropathic cystinosis is the most common
and severe variant. In late-onset or adolescent cystinosis the symptoms
are similar but usually milder. Benign or adult cystinosis is a
rare nonnephropathic variant. Corneal crystal deposition invariably occurs
in all types of cystinosis, which allows easy clinical diagnosis
of the disorder based on the biomicroscopic observation of refractile
crystals. The diagnosis may be confirmed by measuring the white blood
cell cystine content. Cystinosis has an autosomal recessive pattern of
inheritance, and the various forms of cystinosis appear to result from
different defects in the same gene.33 Heterozygotes can be detected by measuring the cystine content of polymorphonuclear
leukocytes, and prenatal diagnosis of nephropathic cystinosis
can be made using cultured amniocytes or chorionic villi. The systemic features of nephropathic cystinosis usually present in the
first year of life with failure to thrive and dehydration due to proximal
renal tubular dysfunction. Rickets and corneal crystals often appear
by 1 to 2 years of age. The typical phenotype of patients with nephropathic
cystinosis includes short stature and light complexion. As a
result of progressive renal failure, end-stage renal disease generally
occurs by the end of the first decade of life, necessitating dialysis
or renal transplantation. In young children, growth can be improved
and renal deterioration delayed by cysteamine, a cystine-depleting agent.34 Since the advent of renal transplantation many cystinotic patients live
to adulthood and become susceptible to new complications caused by long-standing
cystine accumulation in nonrenal organs. Cerebral atrophy
is a common finding on computed tomographic scans of older cystinotic
patients, but severe involvement of the central nervous system is rare. A
large spectrum of neurologic features has been described, including
nonabsorptive hydrocephalus and a peculiar form of encephalopathy, leading
to a “pseudo-bulbar state.”33,35 Hypohidrosis, hypothyroidism, late sexual maturation, insulin-dependent
diabetes, liver enlargement with portal hypertension, and severe epistaxis
have been observed in several patients who survived into their
second and third decades.34,35 All cystinotic patients have ocular involvement, and older patients with
nephropathic cystinosis are at risk of severe ocular complications. In
nephropathic cystinosis, crystal deposits usually appear in the cornea
within the first year of life. Initially, crystals can be identified
in the anterior peripheral part of the cornea by slit lamp biomicroscopy. With
time, progressive accumulation of crystals occurs throughout
the corneal stroma, inducing photophobia even in young children and
provoking a hazy, ground-glass appearance of the cornea in older patients.36 Crystal deposits are also formed in conjunctiva, iris, and retinal pigment
epithelium.37 Focal degeneration of the retinal pigment epithelium with patchy depigmentation
of the fundus may appear early in life and is generally present
by age 7.37 The fundus lesions are bilateral and symmetric and involve mainly the
periphery, although some patients also develop atrophic macular changes (Fig. 3). Abnormal retinal function with reduced or extinguished responses on
electroretinograms and decreased visual acuity are frequent complications
in older cystinotic patients.37,38 Several other complications have been described in patients with nephropathic
cystinosis, including superficial punctate keratopathy, recurrent
erosions, corneal vascularization, band keratopathy, tight miosis, posterior
synechiae, and pupillary-block glaucoma.35,37–39 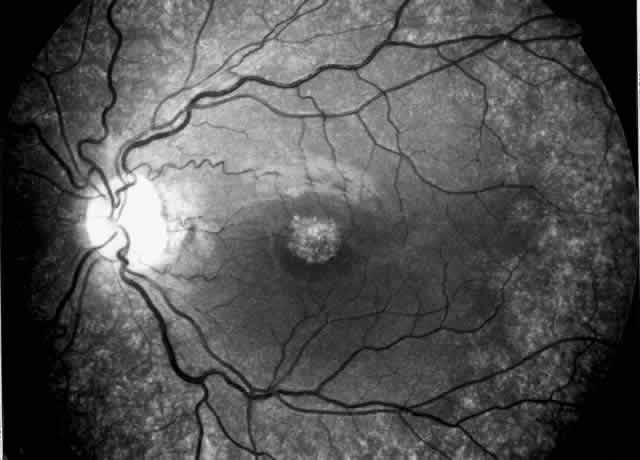 Fig. 3. Cystinotic fundus changes in a 19-year-old patient, demonstrating a pale
optic disc and numerous peripheral and macular small white spots at
the level of the retinal pigment epithelium. Fluorescein angiography confirmed
marked degenerative changes of the retinal pigment epithelium
with macular, peripapillary, and peripheral window defects. Fig. 3. Cystinotic fundus changes in a 19-year-old patient, demonstrating a pale
optic disc and numerous peripheral and macular small white spots at
the level of the retinal pigment epithelium. Fluorescein angiography confirmed
marked degenerative changes of the retinal pigment epithelium
with macular, peripapillary, and peripheral window defects.
|
Oral administration of cysteamine does not appear to improve the corneal
deposits, and the effectiveness in improving other nonrenal complications
is uncertain. However, frequent instillation of topical cysteamine (0.1% to 0.5%) has
been shown to clear the cornea in young patients, to
reduce the amounts of intracorneal cystine crystals in older patients, to
diminish photophobia and blepharospasm, and to improve visual
function.40,41 Corneal complications may require symptomatic therapy including artificial
tears or bandage contact lenses, and, rarely, penetrating keratopathy
may be indicated.36 Nephronophthisis (nephronophthisis-medullary cystic disease complex) includes
a group of hereditary disorders characterized by urinary concentrating
defects and progressive renal failure. The renal disease can be
the only abnormality or be a part of multiorgan involvement with mainly
skeletal and ocular abnormalities. The fundamental defect in nephronophthisis
appears to be production of abnormal tubular basement membranes.42 Patients with nephronophthisis have multiple cysts at the corticomedullary
junction and in the medulla that can be visualized on ultrasonography
and computed tomography of the kidneys. The clinical course is variable, but
tubulointerstitial nephropathy frequently leads to progressive
renal insufficiency, requiring dialysis or renal transplantation. A number of extrarenal abnormalities have been described in patients with
nephronophthisis, singly or in various combinations.43 The term renal-retinal dysplasiarefers to the syndromes with associated retinal abnormalities. The ocular
manifestations frequently resemble Leber's amaurosis or present
as retinitis pigmentosa or congenital stationary nightblindness. Senior-Loken
syndrome is characterized by nephronophthisis and retinal degeneration-type
Leber's amaurosis with flat or severely reduced electroretinograms
from early age. The fundus appearance varies from near
normal to manifest retinal degeneration with changes typical of retinitis
pigmentosa or atypical features or marbleized fundus.44 Abnormal electroretinograms have been recorded in patients with nephronophthisis
without clinical evidence of ocular involvement45 and in some carriers.43 Mainzer-Saldino syndrome is the association of nephronophthisis, tapetoretinal
degeneration resembling Leber's amaurosis, cone-shaped epiphyses, and
cerebellar ataxia. Boichis syndrome is the association of
nephronophthisis with liver fibrosis and may occur in combination with
deafness, cerebellar ataxia, and tapetoretinal degeneration. Nephronophthisis
and retinal degeneration have been reported in association with
asphyxiating thoracic dystrophy (Jeune's syndrome) and with mitochondrial
cytopathy and features of Kearns-Sayre syndrome. Primary hyperoxaluria type I is an autosomal recessively inherited metabolic
disorder caused by a deficiency of the peroxisomal enzyme alanine
glyoxylate aminotransferase (AGT) in liver. The defective transamination
of glyoxylate causes overproduction and urinary excretion of oxalate
and glycolate and leads to recurrent calcium oxalate urolithiasis
and nephrocalcinosis, end-stage renal failure, and systemic oxalosis with
precipitation of calcium oxalate crystals throughout the body. A similar
but frequently milder pattern of disease may occur in the rare
types II and III of primary hyperoxaluria and in secondary hyperoxaluria
resulting from increased intake or absorption of dietary oxalate or
precursors of oxalate. Three clinically distinguishable variants of primary hyperoxaluria type
I have been recognized.46 Infantile oxalosis, the most severe form, generally presents before the
age of 4 months with manifestations of progressive renal insufficiency, including
failure to thrive, vomiting, pallor, anemia, metabolic acidosis, and
convulsions. The diagnosis of infantile oxalosis can be established
by the finding of increased urinary and plasma values of oxalate
and glycolate and by renal ultrasound examination demonstrating
increased echogenicity due to nephrocalcinosis. Death occurs within the
first year of life. The juvenile form of primary hyperoxaluria is the
most common variant, presenting between 2 years and 18 years with repeated
urolithiasis and a variable degree of renal failure. The clinical
course is similar to the adult form. The adult variant is characterized
by repeated attacks of renal colic with hematuria and spontaneous
passing of stones and progressive renal damage as a result of obstruction
and urinary tract infections. As renal insufficiency occurs, the
effect of oxalate retention is superimposed on overproduction, leading
to systemic oxalosis with deposition of calcium oxalate crystals in many
tissues, including the eyes. The most severe extrarenal complications
are oxalotic osteodystrophy, peripheral vascular insufficiency, and
heart block. Nearly one third of older patients with type I hyperoxaluria
respond to pyridoxine treatment. Renal transplantation is life saving
in oxalotic patients with end-stage renal disease, but it leaves
the patient exposed to the risk of recurrent stone formation and nephrocalcinosis. Liver
transplantation replaces the enzyme-deficient organ
and may offer definitive treatment if combined with renal transplantation
or performed before irreversible renal damage has occurred.47 Ocular manifestations are variable and include changes of retinal pigment
epithelium, retinal vascular obstruction, and optic nerve atrophy. Calcium
oxalate crystals are deposited predominantly in the retinal pigment
epithelium of the posterior pole and may induce pronounced reactive
lesions but generally do not cause severe visual loss.48 The abnormalities reported in patients with infantile oxalosis include
scattered crystalline flecks, subretinal small black ringlets, large
geographic macular lesions, and optic atrophy, which is rare but invariably
associated with severe visual impairment.49 Bilateral symmetric retinopathy has been reported in 30% of patients with
primary hyperoxaluria, and the presence of oxalate retinopathy was
positively correlated with infantile onset and a severe systemic course
of the disease.49 In patients with the juvenile or adult variants the most common abnormalities
are crystalline spots or yellowish flecks in the posterior pole, and
rare manifestations include macular black lesions, retinal periarterial
crystal deposition, retinal vascular occlusions, and neovascularization.50,51 Zellweger (cerebrohepatorenal) syndrome is a childhood multisystemic disorder
caused by peroxisomal malfunction. The primary defect is impaired
biogenesis of peroxisomes, with resultant peroxisomal enzyme defects
and several biochemical abnormalities, including deficiency of ether
glycolipids and accumulation of very long chain fatty acids, pipecolic
acid, phytanic acid, and bile precursors.52,53 Organs that have an abundance of peroxisomes are principally affected, and
death generally occurs within the first year as a result of gross
defects of early brain development and major complications. Zellweger
syndrome is an autosomal recessive disease. Heterozygotes have lenticular
opacities with curvilinear cortical condensations, which can be observed
after maximal pupillary dilatation, and these characteristics
may be used for detection of the carrier status.54 Prenatal diagnosis of Zellweger syndrome can be made using cultured amniocytes
or chorionic villi. Infants with Zellweger syndrome are severely hypotonic at birth, have a
characteristic face with a tall forehead, hypoplastic supraorbital ridges, and
epicanthal folds and have a combination of neurologic, hepatic, renal, cardiac, skeletal, and ocular abnormalities.52 The renal abnormalities include multiple cortical cysts, proteinuria, and
aminoaciduria. The ocular abnormalities of Zellweger syndrome are prominent and have dysgenetic
and degenerative components.52 Retinal dystrophy and extinguished electroretinograms are consistent features
of Zellweger syndrome. Lens opacities at the corticonuclear interface
appear to be characteristic abnormalities.54 Opticus hypoplasia, microphthalmos, malformation of the anterior chamber, glaucoma, and
corneal clouding can occur in Zellweger syndrome. X-linked Syndromes Classic Alport's syndrome is characterized by progressive hematuric
nephritis, specific ultrastructural changes in the glomerular basement
membrane, progressive perceptive high-tone hearing loss, and ocular
signs. The abnormal structure and function of the glomerular basement
membrane have been attributed to inadequate production of the alpha 5 chain
of collagen IV. The gene COL 4A5, which encodes the alpha 5 chain
of type IV collagen, has been localized to the long arm of the X chromosome
in the Xq21-q22 region, and a variety of mutations in the gene
have been identified in families with Alport's syndrome. The cardinal clinical manifestation of Alport's nephritis is chronic
hematuria; proteinuria is a frequent finding. Renal biopsy shows diagnostic
ultrastuctural changes with thinning and splitting of the glomerular
basement membrane. Affected males are more likely to develop end-stage
renal disease and deafness than are females. Ocular signs are
significantly linked to poor renal function.55,56 Variants of Alport's syndrome include Alport's syndrome without
hearing loss or ocular lesions56,57 and the variants with associated leiomyomatosis or macrothrombocytopenia. Alport's syndrome may affect the cornea, lens, and retina, the most
characteristic ocular signs being posterior polymorphous dystrophy, anterior
lenticonus, and superficial perimacular flecks (Fig. 4). There is growing evidence that the ocular abnormalities, like the glomerular
lesions, result from a common defect in basement membrane formation.56,58,59 Changes are uncommon and subtle in young patients with Alport's syndrome
and seem to increase in frequency and severity with age.60 The corneal changes associated with Alport's syndrome include endothelial
vesicles compatible with posterior polymorphous dystrophy, subepithelial
opacities,58 corneal arcus, and recurrent corneal epithelial erosions. 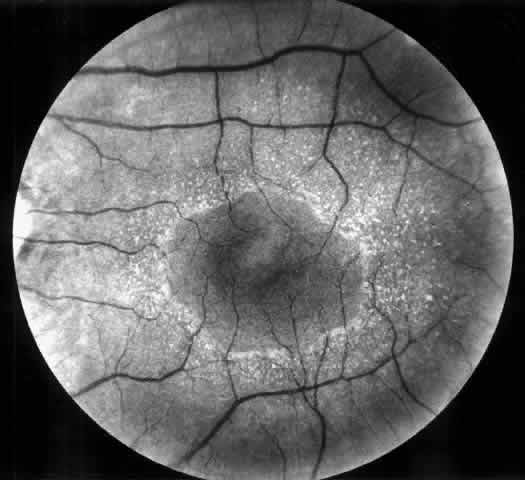 Fig. 4. Superficial perimacular flecks in a 15-year-old boy with normal vision. Alport's
nephritis was diagnosed at the age of 3 years and progressed
to end-stage renal disease at the age of 14 years. Perceptive high-tone
hearing loss was detected at the age of 11 years. The proband's
mother has had persistent microscopic hematuria since the age of 20 years
but had no other manifestations of the disease. Fig. 4. Superficial perimacular flecks in a 15-year-old boy with normal vision. Alport's
nephritis was diagnosed at the age of 3 years and progressed
to end-stage renal disease at the age of 14 years. Perceptive high-tone
hearing loss was detected at the age of 11 years. The proband's
mother has had persistent microscopic hematuria since the age of 20 years
but had no other manifestations of the disease.
|
Anterior lenticonus is a common finding in patients with Alport's
syndrome. The “oil droplet” retinoscopy reflex is an early
sign of anterior lenticonus. Advanced stages are characterized by increasing
myopia, visual loss, and abnormal slit lamp examination showing
protrusion of the lens and thinning of the capsule over the conus, with
or without anterior subcapsular cataract or capsular rupture. Less
specific or rare lenticular abnormalities reported in patients with Alport's
syndrome include several other types of cataracts, spherophakia, and
posterior lenticonus. Superficial perimacular flecks do not interfere with vision but are a reliable
indicator of Alport's syndrome and are often associated with
renal deterioration.55,56,60 Midperipheral retinal flecks and pigment epithelial lesions are also specific
for Alport's syndrome. Macular pigmentation and weakened
foveal reflex have been observed occasionally, and there have been individual
reports of macular hole, retinal detachment, and optic disc drusen
in patients with Alport's syndrome. Electrophysiologic abnormalities
are unusual in classic Alport's syndrome. Retinitis pigmentosa
and cone dystrophy, however, have been described in a few families
with hereditary nephritis. No significant eye changes have been reported in patients with Alport's
syndrome variant affected by familial nephritis without deafness.56,57 Cataracts have been observed in the leiomyomatosis- and macrothrombocytopenia-associated
variants. Fabry's disease is an X-linked glycosphingolipid storage disease characterized
by specific skin and eye lesions and a high risk of developing
renal disease and cardiovascular complications. The basic abnormality
is α-galactosidase A deficiency as a result of mutation in the
encoding X-linked gene. The enzymatic deficiency leads to intracellular
glycosphingolipid accumulation in many tissues and primarily affects
the vascular endothelium. In the hemizygous males the manifestations
of the disease are severe, while the heterozygous females often are
healthy carriers or have only mild involvement. Confirmation of Fabry's
disease can be obtained by enzyme analysis and study of ultrastructural
changes in bioptic specimens (cellular vacuolation and characteristic
inclusion bodies). Prenatal diagnosis of α-galactosidase
deficiency is available. Signs of the disease often appear in late childhood and become more profuse
during the third and fourth decade. Episodes of burning pain in the
limbs are usually the initial symptoms and are often associated with
fever and increased sedimentation rate. At the same time clusters of
angiectases appear on the lower trunk and mucous membranes. Renal disease
and renovascular hypertension often develop. Death usually occurs
in mid life as a result of renal failure, stroke, or complications of
hypertrophic cardiomyopathy, atrioventricular block, or coronary artery
disease. Whirl-like opacification of the cornea (cornea verticillata) is a subtle
but diagnostic sign in Fabry's disease.61,62 The corneal epithelial deposits are present in nearly all hemizygotes
and heterozygotes and can be detected from the age of 6 years. Other characteristic
abnormalities are anterior and posterior capsular lens opacities, aneurysmal
dilatations of the conjunctival vessels, and tortuosity
of the retinal vessels.61 Ischemic optic neuropathy and retinal artery occlusion have been reported
as ocular complications of Fabry's disease. The Lowe oculocerebrorenal syndrome is characterized by congenital cataracts, mental
retardation, muscular hypotonia, and renal tubular dysfunction. The
disorder is X-linked, and the locus has been mapped to Xq25–q26. Although
the biochemical defect in Lowe oculocerebrorenal
syndrome is not known, there is evidence suggesting that a defect of
mitochondrial metabolism could be involved in the pathogenesis. Affected males have a characteristic hypotonic facial appearance and generalized
hypotonia leading to joint dislocations and scoliosis.63 Areflexia, mental retardation, and seizures are common findings in Lowe
syndrome. Patchy or diffuse white matter abnormalities have been characterized
using magnetic resonance imaging. Renal tubular dysfunction
presents within the first year of life and leads to rickets and renal
failure. Cataracts with a small discoid lens and peculiar capsular and epithelial
changes are diagnostic for Lowe syndrome and are present at birth in
nearly all affected males.63 Changes include a lack of demarcation between the nucleus and cortex, posterior
lenticonus adherent to condensed anterior vitreous, chamber
angle abnormalities associated with congenital glaucoma, miotic pupils
with adhesions to the anterior lens surface, and corneal keloids.63 Heterozygote females develop progressive lens changes, a sign that can
be used for carrier detection. Carriers can be diagnosed in the second
decade of life, on the basis of presence of several punctate cortical
opacities. These cortical dots increase in number with the age of the
carriers, and in older carriers subcapsular plaques may be observed
as well.64 OTHER DISORDERS (MULTIFACTORIAL, TERATOGENIC, OR UNKNOWN ORIGIN) Diabetes Mellitus The microangiopathy of diabetes mellitus mainly affects retina and kidney. With
time, nearly all diabetics have clinically evident diabetic retinopathy, while
manifestations of diabetic nephropathy occur in a subset
of patients. Diabetes mellitus causes renal hyperperfusion, glomerular
hyperfiltration, microalbuminuria, and structural changes with glomerular
membrane thickening and mesangial expansion. The early phase
of diabetic nephropathy is characterized by persistent microalbuminuria. Overt
diabetic nephropathy with frank proteinuria occurs in nearly 40% of
insulin-dependent diabetics approximately 17 years after the onset
of diabetes. Poor glycemic control and hypertension appear to be
risk factors for diabetic nephropathy. Once proteinuria appears, renal
function inexorably declines, with 50% of patients reaching end-stage
renal disease within 7 years of the onset of proteinuria.65 In addition to increased risk of progressing to overt diabetic nephropathy, insulin-dependent
diabetics with microalbuminuria suffer a high
cardiovascular mortality rate. In patients with non-insulin-dependent
diabetes mellitus, persistent microalbuminuria also predicts progressive
nephropathy and cardiovascular mortality. Strict diabetic control and
treatment of hypertension have been shown to protect renal function. Once
diabetic nephropathy is established, blood pressure control and
low-protein diets may retard the progression of renal failure. Angiotensin-converting
enzyme inhibitors decrease albuminuria in patients with
diabetic nephropathy and, in addition, have a protective effect on
the blood-retina barrier.66 Renal transplantation is the preferred treatment for end-stage renal failure
in diabetic patients and provides a better quality of life than
dialysis.67,68 Simultaneous pancreas transplantation can further enhance the quality
of life but may potentially cause additional complications. Diabetic retinopathy is characterized by increased vascular permeability
leading to retinal edema and capillary closure leading to retinal ischemia
and new vessel growth. With time, diabetic retinopathy occurs in
nearly all diabetics; many will develop proliferative retinopathy. In
patients with diabetic nephropathy, retinopathy is always present and
proliferative retinopathy is common. However, 35% of patients with proliferative
retinopathy have no signs of diabetic nephropathy, and these
patients will probably never develop nephropathy.69,70 Retinopathy tends to deteriorate as renal failure develops, particularly
in patients with poorly controlled blood pressure and in patients in
whom no retinal treatment has been given before development of renal
failure. Hypertension changes the funduscopic aspect and the course of
background retinopathy by producing diffuse macular edema, cotton-wool
spots, and numerous flame-shaped hemorrhages, particularly in the peripapillary
area and along the main retinal veins (Fig. 5). In addition, hypertension accelerates the evolution of background to
proliferative retinopathy. Treatment of hypertension and of end-stage
renal failure will improve the retinopathy, particularly macular edema, and
stabilize vision.68 It is currently accepted that preservation of vision correlates well with
blood pressure control and that patients with end-stage renal disease
suffering from diabetic retinopathy now enjoy an equivalent visual
prognosis whether treated by dialysis or given a kidney transplant.71 Since the progression of diabetic retinopathy is independent of diabetic
nephropathy and not reversed by treatment of nephropathy, further follow-up
and treatment of diabetic retinopathy is imperative. Patients
with persistent macular edema may benefit from macular photocoagulation. In
diabetics with active proliferative retinopathy, panretinal photocoagulation
and vitrectomy may improve the visual prognosis by inducing
involutional retinopathy and by removing vitreous hemorrhages and
vitreoretinal traction. Moreover, diabetic patients with end-stage renal
disease are predisposed to cataract, which may require surgical intervention. The
progress made in improving the visual prognosis in diabetic
end-stage renal disease reflects the synergistic efforts made by
internists and ophthalmologists and emphasizes the importance of team
approach in preventing blindness.71 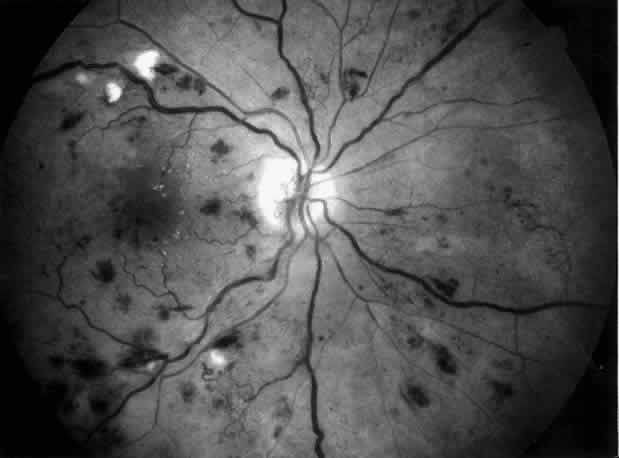 Fig. 5. Combined diabetic and hypertensive retinopathy in a 52-year-old patient
with long-standing diabetes and end-stage renal disease. Note optic disc
and retinal new vessels, diffuse retinal edema, numerous hemorrhages, and
several cotton-wool spots along the main veins. Fig. 5. Combined diabetic and hypertensive retinopathy in a 52-year-old patient
with long-standing diabetes and end-stage renal disease. Note optic disc
and retinal new vessels, diffuse retinal edema, numerous hemorrhages, and
several cotton-wool spots along the main veins.
|
Membranoproliferative Glomerulonephritis Type II A systemic disease of unknown origin, membranoproliferative glomerulonephritis (MPGN) type
II, affects mainly the glomerular basement membrane
and the complex of choriocapillaris, Bruch's membrane, and retinal
pigment epithelium. Electron-dense deposits are characteristically
observed within the lamina densa of the glomerular basement membrane
and have been described in Bruch's membrane and choriocapillaris
by Duvall-Young and co-workers, who also made the first clinical reports
of fundus changes with drusen-like deposits and mottled pigmentation.72,73 MPGN represents a group of disorders characterized by proliferation of
mesangial cells, increased mesangial matrix, and irregular thickening
of the glomerular capillary wall. Several immunofluorescence and electron
microscopic patterns have been identified. In MPGN type II (dense
deposit disease), the lamina densa of the glomerular basement membrane
is largely replaced by electron-dense material, which does not include
immunoglobulin or complement. Similar dense deposits are also found
in the basement membranes of Bowman's capsule, of the tubules, and
of the spleen and in Bruch's membrane and choriocapillaris. The
renal disease frequently shows a progressive course, with onset usually
occurring in childhood. Type II MPGN almost invariably recurs morphologically
in renal allografts. Other clinical features of MPGN type II
include chronic hypocomplementemia with increased susceptibility for
infections, partial lipodystrophy, and a higher incidence of diabetes
mellitus. Drusen-like lesions and retinal pigment epithelium damage have also been
recognized as a feature of MPGN type II.72–79 In a fluorescein angiographic study of 26 patients who had biopsy-proven
MPGN type II, specific fundus lesions were identified in 24 patients (92%).79 Two adolescents with a history of renal disease of 13 months and 2 months
had normal fundi. Small-sized lesions similar to small hard drusen
were observed in all 24 patients with a history of renal disease lasting
for 16 months or more (Fig. 6). In all 15 subjects with a history of renal disease of at least 12 years, larger
drusen-like lesions were also noticed. In all 11 patients
with renal disease persisting for 18 years or more, drusen occupied most
of the fundus and areas of geographic atrophy were seen as well. Foci
of new vessels and disciform scarring were observed in eight eyes of
five patients with a renal history of 15 years or more (Fig. 7). Most eyes that did not show subretinal neovascularization had normal
or nearly normal vision and visual fields. Three patients, however, exhibited
ocular symptoms, which were related to pronounced macular atrophic
changes, hypertensive retinopathy, and cataracts. The type of fundus
lesions was statistically correlated (p<0.0001) with the duration of the renal disease, but not with age, sex, or
renal insufficiency. Fundus changes between first and last visit as
well as cross-sectional studies suggest a slow progression of retinal
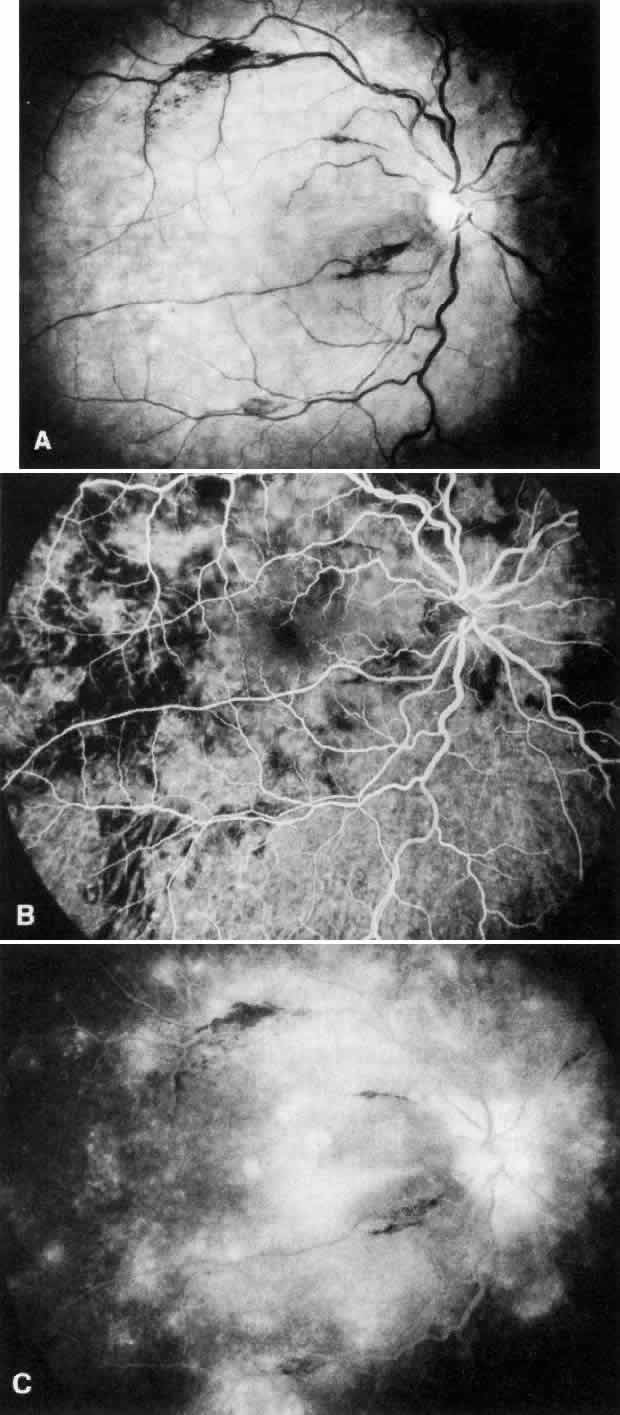
disease, which is probably independent of treatment and age of the patient.77–79 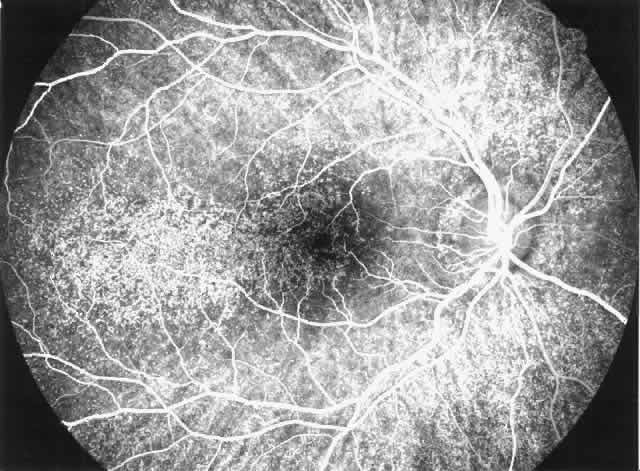 Fig. 6. Specific fundus lesions of membranoproliferative glomerulonephritis type
II in a 12-year-old child with renal disease since the age of 3 years. The
fluorescein angiogram shows numerous small lesions similar to hard
drusen. (Leys A, Vanrenterghem Y, Van Damme B et al: Fundus changes in membranoproliferative
glomerulonephritis type II: A fluorescein angiographic study
of 23 patients. Graefes Arch Clin Exp Ophthalmol 229:406, 1991) Fig. 6. Specific fundus lesions of membranoproliferative glomerulonephritis type
II in a 12-year-old child with renal disease since the age of 3 years. The
fluorescein angiogram shows numerous small lesions similar to hard
drusen. (Leys A, Vanrenterghem Y, Van Damme B et al: Fundus changes in membranoproliferative
glomerulonephritis type II: A fluorescein angiographic study
of 23 patients. Graefes Arch Clin Exp Ophthalmol 229:406, 1991)
|
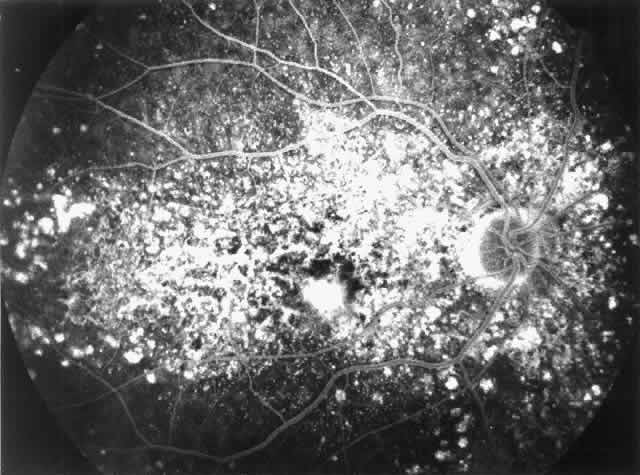 Fig. 7. Fluorescein angiographic changes in a 32-year-old patient with renal signs
of membranoproliferative glomerulonephritis type II since the age
of 9 years. Numerous small and larger drusen-like lesions, atrophic changes, and
a small infrafoveolar subretinal neovascular membrane that
was successfully treated with argon laser coagulation can be seen. (Leys A, Michielsen B, Leys M et al: Subretinal neovascular membranes associated
with chronic membranoproliferative glomerulonephritis type II. Graefes
Arch Clin Exp Ophthalmol 228:499, 1990) Fig. 7. Fluorescein angiographic changes in a 32-year-old patient with renal signs
of membranoproliferative glomerulonephritis type II since the age
of 9 years. Numerous small and larger drusen-like lesions, atrophic changes, and
a small infrafoveolar subretinal neovascular membrane that
was successfully treated with argon laser coagulation can be seen. (Leys A, Michielsen B, Leys M et al: Subretinal neovascular membranes associated
with chronic membranoproliferative glomerulonephritis type II. Graefes
Arch Clin Exp Ophthalmol 228:499, 1990)
|
|