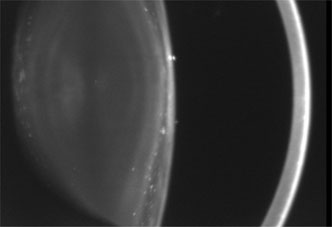
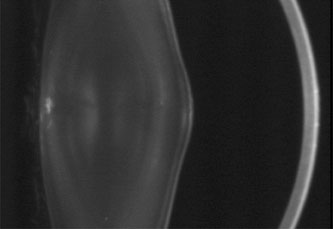
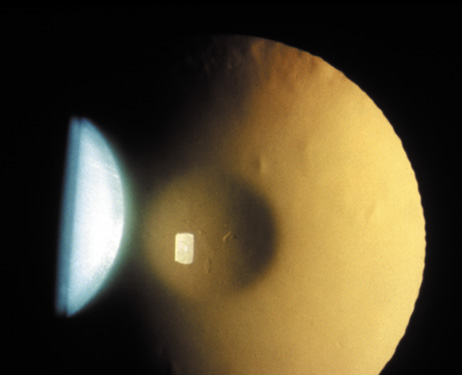
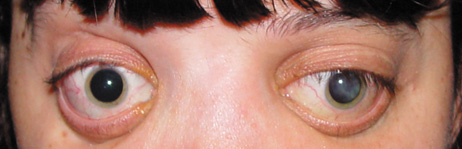
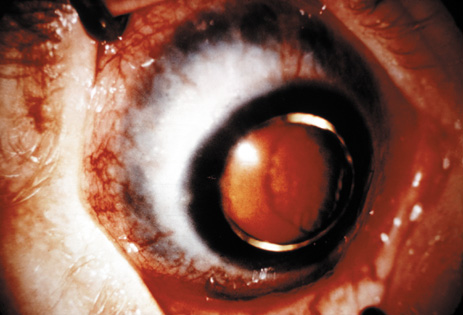
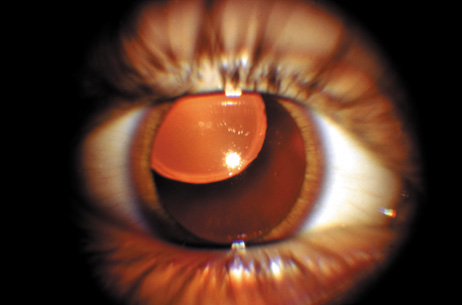
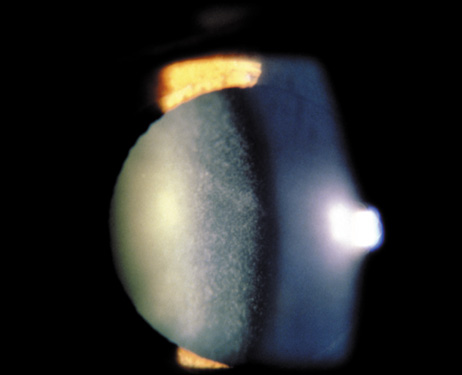
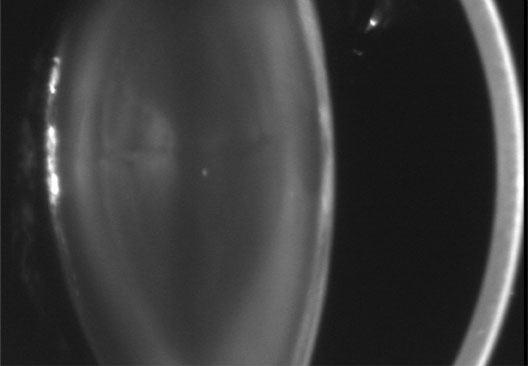
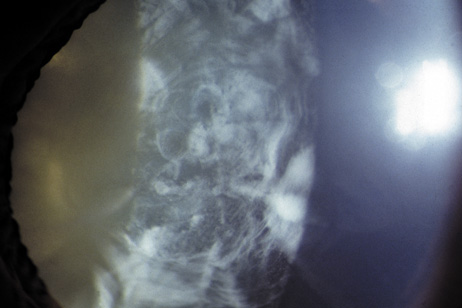
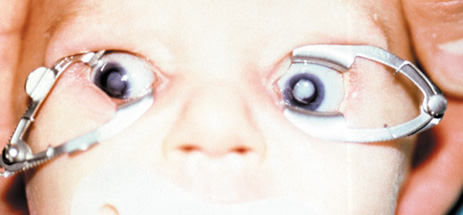
1. Lowe CU, Terry M, MacLachlan EA: Organic aciduria, decreased renal ammonia production, hydrophthalmos, and
mental retardation: a clinical entity. Am J Dis Child 83:164–184, 1952 2. Reilly DS, Lewis RA, Nussbaum RL: Genetic and physical mapping of Xq24–q26 markers flanking the Lowe
oculocerebrorenal syndrome. Genomics 8:62–70, 1990 3. Gropman A, Levin S, Yao L, et al: Unusual renal features of Lowe syndrome in a mildly affected boy. Am J Med Genet 95:461–466, 2000 4. Endres W, Schaub J, Stefani F, et al: Cataract in a fetus at risk for oculo-cerebro-renal syndrome (Lowe). Klin Wochenschr 55:141–144, 1977 5. Tripathi RC, Cibis GW, Tripathi BJ: Pathogenesis of cataracts in patients with Lowe's syndrome. Ophthalmology 93:1046–1051, 1986 6. Kruger S, Wilson MJ, Hutchinson A, et al: Cataracts and glaucoma in patients with oculocerebrorenal syndrome. Arch Ophthalmol 121:1234–1237, 2003 7. Lin T, Lewis RA, Nussbaum RL: Molecular confirmation of carriers for Lowe syndrome. Ophthalmology 106:119–122, 1999 8. Baser ME, Kuramoto L, Joe H, et al: Genotype-phenotype correlations for cataracts in neurofibromatosis 2. J Med Genet 40:758–760, 2003 9. Ragge NK, Baser ME, Klein J, et al: Ocular abnormalities in neurofibromatosis 2. Am J Ophthalmol 120:634–641, 1995 10. Zhu D, Alcorn DM, Antonarakis S, et al: Assignment of the Nance-Horan syndrome to the distal short arm of the X
chromosome. Hum Genet 86:54–58, 1990 11. Brooks SP, Ebenezer ND, Poopalasundaram S, et al: Identification of the gene for Nance-Horan syndrome (NHS). J Med Genet 41:768–771, 2004 12. Lewis RA, Nussbaum RL, Stambolian D: Mapping X-linked ophthalmic diseases. IV. Provisional assignment of the
locus for X-linked congenital cataracts and microcornea (the Nance-Horan
syndrome) to Xp22.2–p22.3. Ophthalmology 97:110–120, discussion 20–21, 1990 13. Amaya L, Taylor D, Russell-Eggitt I, et al: The morphology and natural history of childhood cataracts. Surv Ophthalmol 48:125–144, 2003 14. Francis PJ, Berry V, Hardcastle AJ, et al: A locus for isolated cataract on human Xp. J Med Genet 39:105–109, 2002 15. Dharmaraj S, Leroy BP, Sohocki MM, et al: The phenotype of Leber congenital amaurosis in patients with AIPL1 mutations. Arch Ophthalmol 122:1029–1037, 2004 16. Koenekoop RK: An overview of Leber congenital amaurosis: a model to understand human
retinal development. Surv Ophthalmol 49:379–398, 2004 17. Acland GM, Aguirre GD, Ray J, et al: Gene therapy restores vision in a canine model of childhood blindness. Nat Genet 28:92–95, 2001 18. Cox DW, Moore SD: Copper transporting P-type ATPases and human disease. J Bioenerg Biomembr 34:333–338, 2002 19. Becker M, Rohrschneider K: Ocular manifestations of Wilson disease. Ophthalmologe 94:865–870, 1997 20. Obara H, Ikoma N, Sasaki K, Tachi K: Usefulness of Scheimpflug photography to follow up Wilson's disease. Ophthalmic Res 27(Suppl 1):100–103, 1995 21. Girelli D, Olivieri O, De Franceschi L, et al: A linkage between hereditary hyperferritinaemia not related to iron overload
and autosomal dominant congenital cataract. Br J Haematol 90:931–934, 1995 22. Bonneau D, Winter-Fuseau I, Loiseau M, et al: Bilateral cataract and high serum ferritin: a new dominant genetic disorder? J Med Genet 32:778–779, 1995 23. Beaumont C, Leneuve P, Devaux I, et al: Mutation in the iron responsive element of the L ferritin mRNA in a family
with dominant hyperferritinaemia and cataract. Nat Genet 11:444–446, 1995 24. Lieuallen K, Christensen M, Brandriff B, et al: Assignment of the human lens fiber cell MP19 gene (LIM2) to chromosome 19q13.4 and
adjacent to ETFB. Somat Cell Mol Genet 20:67–69, 1994 25. Craig J, Clark J, McLeod J, et al: Hereditary hyperferritinemia-cataract syndrome: Prevalence, lens morphology, spectrum
of mutations, and clinical presentations. Arch Ophthalmol 121:1753–1761, 2003 26. Girelli D, Corrocher R, Bisceglia L, et al: Molecular basis for the recently described hereditary hyperferritinemia-cataract
syndrome: a mutation in the iron-responsive element of ferritin
L-subunit gene (the “Verona mutation”). Blood 86:4050–4053, 1995 27. Mumford A, Vulliamy T, Lindsay J, Watson A: Hereditary hyperferritinemia-cataract syndrome: Two novel mutations in
the L-ferritin iron-responsive element. Blood 91:367–368, 1998 28. Brooks D, Manova-Todorova K, Farmer J, et al: Ferritin crystal cataracts in hereditary hyperferritinemia cataract syndrome. Invest Ophthalmol Vis Sci 43:1121–1126, 2002 29. Pitt DB, O'Day J: Phenylketonuria does not cause cataracts. Eur J Pediatr 150:661–664, 1991 30. Zwaan J: Eye findings in patients with phenylketonuria. Arch Ophthalmol 101:1236–1237, 1983 31. Parks MM, Schwilk NF: Bilateral lamellar cataracts in the case of phenylketonuria. Am J Ophthalmol 79:479, 1975 32. Liquori C, Ricker K, Moseley M, et al: Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science 293:864–867, 2001 33. Ranum L, Day J: Dominantly inherited, non-coding microsatellite expansion disorders. Curr Opin Genet Dev 12:266–271, 2002 34. Shun-Shin G, Vrensen G, Brown N, et al: Morphologic characteristics and chemical composition of Christmas tree
cataract. Invest Ophthalmol Vis Sci 34:3489–3496, 1993 35. Pepin B, Mikol J, Goldstein B, et al: Familial mitochondrial myopathy with cataract. J Neurol Sci 45:191–203, 1980 36. Kornfeld S, Sly WS: I-cell disease and pseudo-Hurler polydystrophy: disorders of lysosomal
enzyme phosphorylation and localisation. In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds). Metabolic and molecular bases of inherited disease. New York: McGraw-Hill, 2001 37. Seyrantepe V, Poupetova H, Froissart R, et al: Molecular pathology of NEU1 gene in sialidosis. Hum Mutat 22:343–352, 2003 38. Bunge S, Clements P, Byers S, et al: Genotype-phenotype correlations in mucopolysaccharidosis type I using enzyme
kinetics, immunoquantification and in vitro turnover studies. Biochim Biophys Acta 1407:249–256, 1998 39. Peters C, Shapiro E, Anderson J, et al: Hurler syndrome: II. Outcome of HLA-genotypically identical sibling and
HLA-haploidentical related donor bone marrow transplantation in fifty-four
children. The Storage Disease Collaborative Study Group. Blood 91:2601–2608, 1998 40. Gullingsrud E, Krivit W, Summers C: Ocular abnormalities in the mucopolysaccharidoses after bone marrow transplantation. Longer
follow-up. Ophthalmology 105:1099–1105, 1998 41. Brooks D: Alpha-L-iduronidase and enzyme replacement therapy for mucopolysaccharidosis
I. Expert Opin Biol Ther 2:967–976, 2002 42. Gosele S, Dithmar S, Holz F, Volcker H: Late diagnosis of Morquio syndrome. Clinical histopathological findings
in a rare mucopolysaccharidosis. Klin Monatsbl Augenheilkd 217:114–117, 2000 43. Cahane M, Treister G, Abraham F, Melamed S: Glaucoma in siblings with Morquio syndrome. Br J Ophthalmol 74:382–383, 1990 44. Olsen H, Baggesen K, Sjolie A: Cataracts in Morquio syndrome (mucopolysaccharidosis IV A). Ophthalmic Paediatr Genet 14:87–89, 1993 45. Kasmann-Kellner B, Weindler J, Pfau B, Ruprecht K: Ocular changes in mucopolysaccharidosis IV A (Morquio A syndrome) and
long-term results of perforating keratoplasty. Ophthalmologica 213:200–205, 1999 46. Galanos J, Nicholls K, Grigg L, et al: Clinical features of Fabry's disease in Australian patients. Intern Med J 32:575–584, 2002 47. Orssaud C, Dufier J, Germain D: Ocular manifestations in Fabry disease: a survey of 32 hemizygous male
patients. Ophthalmic Genet 23:129–139, 2003 48. Thomas PK, Abrams JD, Swallow D, Stewart G: Sialidosis type 1: Cherry red spot-myoclonus syndrome with sialidase deficiency
and altered electrophoretic mobilities of some enzymes known
to be glycoproteins. I. Clinical findings. J Neurol Neurosurg Psychiatry 42:873–880, 1979 49. Beccari T, Stinchi S, Orlacchio A: Lysosomal alpha-D-mannosidase. Biosci Rep 19:157–162, 1999 50. Letson RD, Desnick RJ: Punctate lenticular opacities in type II mannosidosis. Am J Ophthalmol 85:218–224, 1978 51. Maumenee IH: Classification of hereditary cataracts by linkage analysis. Ophthalmology 86:1554–1558, 1979 52. Dagher H, Buzza M, Colville D, et al: A comparison of the clinical, histopathologic, and ultrastructural phenotypes
in carriers of X-linked and autosomal recessive Alport's syndrome. Am J Kidney Dis 38:1217–1228, 2001 53. Zhou W, Hirsch M, Junk AK, et al: Evaluation of lenticonus in Alport's syndrome: Quantitative Scheimpflug
analysis. Ophthalmologica 217:189–193, 2003 54. Junk AK, Stefani F, Ludwig K: Bilateral anterior lenticonus: Scheimpflug documentation and ultrastructural
confirmation of Alport syndrome in the lens capsule. Arch Ophthalmol 118:895–897, 2000 55. Streeten B, Robinson M, Wallace R, Jones D: Lens capsule abnormalities in Alport's syndrome. Arch Ophthalmol 105:1693–1697, 1987 56. Pajari H, Setaelae K, Heiskari N, et al: Ocular findings in 34 patients with Alport syndorme: correlation of the
findings to mutations in COL4A5 gene. Acta Ophthalmol Scand 77:214–217, 1999 57. Olitsky SE, Waz WR, Wilson ME: Rupture of the anterior lens capsule in Alport syndrome. J AAPOS 3:381–382, 1999 58. Colville D, Savige J: Alport syndrome. A review of the ocular manifestations. Ophthalmic Genet 18:161–173, 1997 59. Deutsch S, Rideau A, Bochaton-Piallat M, et al: Asp1424Asn MYH9 mutation results in an unstable protein responsible for
the phenotypes in May-Hegglin anomaly/Fechtner syndrome. Blood 102:529–534, 2003 60. Seri M, Pecci A, Di Bari F, et al: MYH9-related disease: May-Hegglin anomaly, Sebastian syndrome, Fechtner
syndrome, and Epstein syndrome are not distinct entities but represent
a variable expression of a single illness. Medicine (Baltimore) 82:203–215, 2003 61. Bleik JH, Traboulsi EI, Maumenee IH: Familial posterior lenticonus and microcornea. Arch Ophthalmol 110:1208, 1992 62. Cenedella R: Cholesterol and cataracts. Surv Ophthalmol 40:320–337, 1996 63. Opitz J, Gilbert-Barness E, Ackerman J, Lowichik A: Cholesterol and development: The RSH (“Smith-Lemli-Opitz”) syndrome
and related conditions. Pediatr Pathol Mol Med 21:153–181, 2002 64. Loeffler J, Utermann G, Witsch-Baumgartner M: Molecular prenatal diagnosis of Smith-Lemli-Opitz syndrome is reliable
and efficient. Prenat Diagn 22:827–830, 2002 65. Atchaneeyasakul L-O, Linck L, Connor W, et al: Eye findings in 8 children and a spontaneously aborted fetus with RSH/Smith-Lemli-Opitz
syndrome. Am J Med Genet 80:501–505, 1998 66. Moghadasian M, Salen G, Frohlich J, Scudamore C: Cerebrotendinous xanthomatosis. Arch Neurol 59:527–529, 2002 67. van Heijst A, Verrips A, Wevers R, et al: Treatment and follow-up of children with cerebrotendinous xanthomatosis. Eur J Pediatr 157:313–316, 1998 68. Cruysberg J, Wevers R, van Engelen B, et al: Ocular and systemic manifestations of cerebrotendinous xanthomatosis. Am J Ophthalmol 120:597–604, 1995 69. Dotti M, Rufa A, Federico A: Cerebrotendinous xanthomatosis: Heterogeneity of clinical phenotype with
evidence of previously undescribed ophthalmological findings. J Inherit Metab Dis 24:696–706, 2001 70. Milunsky JM, Maher TA, Metzenberg AB: Molecular, biochemical, and phenotypic analysis of a hemizygous male with
a severe atypical phenotype for X-linked dominant Conradi-Hunermann-Happle
syndrome and a mutation in EBP. Am J Med Genet 166A:249–254, 2003 71. Has C, Bruckner-Tuderman L, Muller D, et al: The Conradi-Hunermann-Happle syndrome (CDPX2) and emopamil binding
protein: novel mutations, and somatic and gonadal mosaicism. Hum Mol Genet 9:1951–1955, 2000 72. Cenedella R, Sexton P: Probing cataractogenesis associated with mevalonic aciduria. Curr Eye Res 17:153–158, 1998 73. Dollfus H, Porto F, Caussade P, et al: Ocular manifestations in the inherited DNA repair disorders. Surv Ophthalmol 48:107–122, 2003 74. Cockayne EA: Dwarfism with retinal atrophy and deafness. Arch Dis Child 11:1–8, 1936 75. Traboulsi EI, De Becker I, Maumenee IH: Ocular findings in Cockayne syndrome. Am J Ophthalmol 114:579–583, 1992 76. Lehmann AR, Francis AJ, Giannelli F: Prenatal diagnosis of Cockayne's syndrome. Lancet 1:486–488, 1985 77. Itin PH, Pittelkow MR: Trichothiodystrophy: Review of sulfur-deficient brittle hair syndromes
and association with the ectodermal dysplasias. J Am Acad Dermatol 22:705–717, 1990 78. Vermeulen W, Bergmann E, Auriol J, et al: Sublimiting concentration of TFIIH transcription/DNA repair factor
causes TTD-A trichothiodystrophy disorder. Nat Genet 26:257–258, 2000 79. Botta E, Nardo T, Broughton BC, et al: Analysis of mutations in the XPD gene in Italian patients with trichothiodystrophy: site
of mutation correlates with repair deficiency, but gene
dosage appears to determine clinical severity. Am J Hum Genet 63:1036–1048, 1998 80. Taylor EM, Broughton BC, Botta E, et al: Xeroderma pigmentosum and trichothiodystrophy are associated with different
mutations in the XPD (ERCC2) repair/transcription gene. Proc Natl Acad Sci U S A 94:8658–8663, 1997 81. Broughton BC, Berneburg M, Fawcett H, et al: Two individuals with features of both xeroderma pigmentosum and trichothiodystrophy
highlight the complexity of the clinical outcomes of mutations
in the XPD gene. Hum Mol Genet 10:2539–2547, 2001 82. Berg E, Chuang TY, Cripps D: Rothmund-Thomson syndrome. A case report, phototesting, and literature
review. J Am Acad Dermatol 17:332–338, 1989 83. Wang LL, Levy ML, Lewis RA, et al: Clinical manifestations in a cohort of 41 Rothmund-Thomson syndrome patients. Am J Med Genet 102:11–17, 2001 84. Vennos EM, Collins M, James WD: Rothmund-Thomson syndrome: Review of the world literature. J Am Acad Dermatol 27:750–762, 1992 85. Bachrati CZ, Hickson ID: RecQ helicases: Suppressors of tumorigenesis and premature aging. Biochem J 374:577–606, 2003 86. Goto M: Werners syndrome: from clinics to genetics. Clin Exp Rheumatol 18:760–766, 2000 87. Jonas JB, Rupprecht KW, Schmitz-Valckenberg P, et al: Ophthalmic surgical complications in Werner's syndrome: Report on 18 eyes
of nine patients. Ophthalmic Surg 18:760–764, 1987 88. Gray MD, Shen JC, Kamath-Loeb AS, et al: The Werner syndrome protein is a DNA helicase. Nat Genet 17:100–103, 1997 89. Marcus DM, Shore JW, Albert DM: Anophthalmia in the focal dermal hypoplasia syndrome. Arch Ophthalmol 108:96–100, 1990 90. Philippe J, Cherifi M, Fournier D, et al: Anhydrotic ectodermal dysplasia. Apropos of a case with severe ocular complications. J Fr Ophtalmol 11:287–292, 1988 91. Smahi A, Courtois G, Rabia SH, et al: The NF-kappaB signalling pathway in human diseases: From incontinentia
pigmenti to ectodermal dysplasias and immune-deficiency syndromes. Hum Mol Genet 11:2371–2375, 2002 92. Puel A, Picard C, Ku CL, et al: Inherited disorders of NF-kappaB-mediated immunity in man. Curr Opin Immunol 16:34–41, 2004 93. Chen CC, Huang JL, Yang KD, Chen HJ: Atopic cataracts in a child with atopic dermatitis: A case report and review
of the literature. Asian Pac J Allergy Immunol 18:69–71, 2000 94. Mullner-Eidenbock A, Moser E, Klebermass N, et al: Ocular features of the congenital cataracts facial dysmorphism neuropathy
syndrome. Ophthalmology 111:1415–1423, 2004 95. Lagier-Tourenne C, Tranebaerg L, Chaigne D, et al: Homozygosity mapping of Marinesco-Sjogren syndrome to 5q31. Eur J Hum Genet 11:770–778, 2003 96. Williams T, Buchhalter JR, Sussman MD: Cerebellar dysplasia and unilateral cataract in Marinesco-Sjogren syndrome. Pediatr Neurol 14:158–161, 1996 97. Shimizu T, Matsuishi T, Yamashita Y, et al: Marinesco-Sjogren syndrome: Can the diagnosis be made prior to cataract
formation? Muscle Nerve 20:909–910, 1997 98. Hallermann W: Vogelgesicht und Cataracta Congenita. Klin Monatsbl Augenheilkd 113:315–318, 1948 99. Streiff EB: Mandibulofacial dysmorphia with ocular abnormalities. Ophthalmologica 120:79–83, 1950 100. Rohrbach JM, Djelebova T, Schwering MJ, Schlote T: [Hallermann-Streiff syndrome: should spontaneous resorption of the
lens opacity be awaited?]. Klin Monatsbl Augenheilkd 216:172–176, 2000 101. Pizzuti A, Flex E, Mingarelli R, et al: A homozygous GJA1 gene mutation causes a Hallermann-Streiff/ODDD spectrum
phenotype. Hum Mutat 23:286, 2004 102. Muller U, Steinberger D, Kunze S: Molecular genetics of craniosynostotic syndromes. Graefes Arch Clin Exp Ophthalmol 235:545–550, 1997 103. Rochels R, Schmitt EJ: The development of eye symptoms in dysostosis craniofacialis crouzon—A
contribution to pathogenesis. Klin Padiatr 193:17–19, 1981 104. Passos-Bueno MR, Armelin LM, Alonso LG, et al: Craniosynostosis associated with ocular and distal limb defects is very
likely caused by mutations in a gene different from FGFR, TWIST, and
MSX2. Am J Med Genet 113:200–206, 2002 105. Caputo AR, Wagner RS, Reynolds DR, et al: Down syndrome. Clinical review of ocular features. Clin Pediatr (Phil) 28:355–358, 1989 106. Plotz FB, van Essen AJ, Bosschaart AN, Bos AP: Cerebro-costo-mandibular syndrome. Am J Med Genet 62:286–292, 1996 107. Rogers NK, Strachan IM: Pierre Robin anomalad, maculopathy, and autolytic cataract. J Pediatr Ophthalmol Strabismus 32:391–392, 1995 108. Webb AC, Markus AF: The diagnosis and consequences of Stickler syndrome. Br J Oral Maxillofac Surg 40:49–51, 2002 109. Schlote T, Volker M, Knorr M, Thiel HJ: Lens coloboma and lens dislocation in Stickler (Marshall) syndrome. Klin Monatsbl Augenheilkd 210:227–228, 1997 110. Vu CD, Brown JJ, Korkko J, et al: Posterior chorioretinal atrophy and vitreous phenotype in a family with
Stickler syndrome from a mutation in the COL2A1 gene. Ophthalmology 110:70–77, 2003 111. Cantani A, Gagliesi D: Rubinstein-Taybi syndrome. Review of 732 cases and analysis of the typical
traits. Eur Rev Med Pharmacol Sci 2:81–87, 1998 112. van Genderen MM, Kinds GF, Riemslag FC, Hennekam RC: Ocular features in Rubinstein-Taybi syndrome: investigation of 24 patients
and review of the literature. Br J Ophthalmol 84:1177–1184, 2000 113. Derbent M, Agras PI, Gedik S, et al: Congenital cataract, microphthalmia, hypoplasia of corpus callosum and
hypogenitalism: report and review of Micro syndrome. Am J Med Genet 128A:232–234, 2004 114. Jonas JB MU, Budde WM: Ocular findings in cerebro-oculo-facial-skeletal syndrome (Pena-Shokeir-II
syndrome). Eur J Ophthalmol 13:209–211, 2003 115. Hennekam RC, van de Meeberg AG, van Doorne JM, et al: Martsolf syndrome in a brother and sister: Clinical features and pattern
of inheritance. Eur J Pediatr 147:539–543, 1988 116. Shapiro I, Borochowitz Z, Degani S, et al: Neu-Laxova syndrome: Prenatal ultrasonographic diagnosis, clinical and
pathological studies, and new manifestations. Am J Med Genet 43:602–605, 1992 117. Urban MD, Schosser R, Spohn W, et al: New clinical aspects of hereditary mucoepithelial dysplasia. Am J Med Genet 39:338–341, 1991 118. da Cunha RP, Moreira JB: Ocular findings in Down's syndrome. Am J Ophthalmol 122:236–244, 1996 119. Sybert VP, McCauley E: Turner's syndrome. N Engl J Med 351:1227–1238, 2004 120. Koole FD, Velzeboer CM, van der Harten JJ: Ocular abnormalities in Patau syndrome (chromosome 13 trisomy syndrome). Ophthalmic Paediatr Genet 11:15–21, 1990 121. Smith AC, Dykens E, Greenberg F: Behavioral phenotype of Smith-Magenis syndrome (del 17p11.2). Am J Med Genet 81:179–185, 1998 122. Chen RM LJ, Greenberg F, Lewis RA: Ophthalmic manifestations of Smith-Magenis syndrome. Ophthalmology 103:1084, 1996 123. Rubin SE, Nelson LB, Pletcher BA: Anterior polar cataract in two sisters with an unbalanced 3;18 chromosomal
translocation. Am J Ophthalmol 117:512–515, 1994 124. Johnson BL, Cheng KP: Congenital aphakia: A clinicopathologic report of three cases. J Pediatr Ophthalmol Strabismus 34:35–39, 1997 125. Rosias PR, Sijstermans JM, Theunissen PM, et al: Phenotypic variability of the cat eye syndrome. Case report and review
of the literature. Genet Couns 12:273–282, 2001 126. Meins M, Burfeind P, Motsch S, et al: Partial trisomy of chromosome 22 resulting from an interstitial duplication
of 22q11.2 in a child with typical cat eye syndrome. J Med Genet 40:E62, 2003 127. Funke B, Pandita RK, Morrow BE: Isolation and characterization of a novel gene containing WD40 repeats
from the region deleted in velo-cardio-facial/DiGeorge syndrome on
chromosome 22q11. Genomics 73:264–271, 2001 128. Nelson LB, Spaeth GL, Nowinski TS, et al: Aniridia. A review. Surv Ophthalmol 28:621–642, 1984 129. Wolf MT, Lorenz B, Winterpacht A, et al: Ten novel mutations found in Aniridia. Hum Mutat 12:304–313, 1998 130. Gupta SK, De Becker I, Tremblay F, et al: Genotype/phenotype correlations in aniridia. Am J Ophthalmol 126:203–210, 1998 131. Ricardi VM, Sujansky E, Smith AC, Franke U: Chromosomal imbalance in the aniridia-Wilms's tumor association: 11p
interstitial deletion. Pediatrics 61:604–610, 1978 132. Wu-Chen WY, Christiansen SP, Berry SA, et al: Ophthalmic manifestations of Wolf-Hirschhorn syndrome. J AAPOS 8:345–348, 2004 133. Mayer UM, Bialasiewicz AA: Ocular findings in a 4 p-deletion syndrome (Wolf-Hirschhorn). Ophthalmic Paediatr Genet 10:69–72, 1989 134. Cornish K, Bramble D: Cri du chat syndrome: Genotype–phenotype correlations and recommendations
for clinical management. Dev Med Child Neurol 44:494–497, 2002 135. Farrell JW, Morgan KS, Black S: Lensectomy in an infant with cri du chat syndrome and cataracts. J Pediatr Ophthalmol Strabismus 25:131–134, 1988 136. Kitsiou-Tzeli S, Dellagrammaticas HD, Papas CB, et al: Unusual ocular findings in an infant with cri-du-chat syndrome. J Med Genet 20:304–307, 1983 137. Grotsky H, Hsu LY, Hirschhorn K: A case of cri-du-chat associated with cataracts and transmitted from a
mother with a 4–5 translocation. J Med Genet 8:369–371, 1971 138. Auffarth GU, Tetz MR, Krastel H, et al: Complicated cataracts in various forms of retinitis pigmentosa. Type and
incidence. Ophthalmologe 94:642–646, 1997 139. Refsum S: Heredoataxia hemeralopica polyneuritiformis. Nordisk Med 28:2682–2685, 1945 140. Claridge KG, Gibberd FB, Sidey MC: Refsum disease: The presentation and ophthalmic aspects of Refsum disease
in a series of 23 patients. Eye 6:371–375, 1992 141. Marcaud V, Defontaines B, Jung P, Degos CF: Refsum's disease: Evolution 35 years after diagnosis. Rev Neurol (Paris) 158:225–229, 2002 142. Espinos C, Perez-Garrigues H, Beneyto M, et al: Sydromic hereditary deafness. Usher's syndrome. Oto-neurologic and
genetic factors. An Otorrinolaringol Ibero Am 26:83–95, 1999 143. Beales PL, Elcioglu N, Woolf AS, et al: New criteria for improved diagnosis of Bardet-Biedl syndrome: Results of
a population survey. J Med Genet 36:437–446, 1999 144. Hallgren B: Retinitis pigmentosa in combination with congenital deafness and vestibulocerebellar
with psychiatric abnormality in some cases: A clinical and
genetic study. Acta Genet Stat Med, 1958;ZS8:97–104 145. Urquidi GA, Topaz AM: Hallgren's syndrome in one family: Retinitis pigmentosa, congenital
deafness and ataxia. Acta Neurol Latinoam 25:75–79, 1979 146. Folz SJ, Trobe JD: The peroxisome and the eye. Surv Ophthalmol 35:353–368, 1991 147. Hittner HM, Kretzer FL, Mehta RS: Zellweger syndrome. Lenticular opacities indicating carrier status and
lens abnormalities characteristic of homozygotes. Arch Ophthalmol 99:1977–1982, 1981 148. Cohen SM, Green WR, de la Cruz ZC, et al: Ocular histopathologic studies of neonatal and childhood adrenoleukodystrophy. Am J Ophthalmol 95:82–96, 1983 149. Yap S: Classical homocystinuria: vascular risk and its prevention. J Inherit Metab Dis 26:259–265, 2003 150. Sulochana KN, Amirthalakshmi S, Vasanthi SB, et al: Homocystinuria with congenital/developmental cataract. Indian J Pediatr 67:725–728, 2000 151. Ades LC, Holman KJ, Brett MS, et al: Ectopia lentis phenotypes and the FBN1 gene. Am J Med Genet 126A:284–289, 2004 152. Traboulsi EI, Whittum-Hudson JA, Mir SH, Maumenee IH: Microfibril abnormalities of the lens capsule in patients with Marfan syndrome
and ectopia lentis. Ophthalmic Genet 21:9–15, 2000 153. Faivre L, Dollfus H, Lyonnet S, et al: Clinical homogeneity and genetic heterogeneity in Weill-Marchesani syndrome. Am J Med Genet 123A:204–207, 2003 154. Mao JR, Bristow J: The Ehlers-Danlos syndrome: On beyond collagens. J Clin Invest 107:1063–1069, 2001 155. Cikrit DF, Glover JR, Dalsing MC, Silver D: The Ehlers-Danlos specter revisited. Vasc Endovascular Surg 36:213–217, 2002 156. Al-Hussain H, Zeisberger SM, Huber PR, et al: Brittle cornea syndrome and its delineation from the kyphoscoliotic type
of Ehlers-Danlos syndrome (EDS VI): Report on 23 patients
and review of the literature. Am J Med Genet 124A:24–34, 2004 157. Borger PH, van Leeuwen R, Hulsman CA, et al: Is there a direct association between age-related eye diseases and mortality? The
Rotterdam Study. Ophthalmology 110:1292–1296, 2003 158. Valero MP, Fletcher AE, De Stavola BL, et al: Vitamin C is associated with reduced risk of cataract in a Mediterranean
population. J Nutr 132:1299–1306, 2002 159. Chung SS, Ho EC, Lam KS, Chung SK: Contribution of polyol pathway to diabetes-induced oxidative stress. J Am Soc Nephrol 14(8 suppl 3):s233–s236, 2003 160. Biswas S, Harris F, Singh J, Phoenix D: Role of calpains in diabetes mellitus-induced cataractogenesis: a mini
review. Mol Cell Biochem 261:151–159, 2004 161. Worgul BV, Medvedovsky CM, Merriam GR: Cortical cataract development—The expression of primary damage to
the lens epithelium. Lens Eye Toxicity Res 6:559–571, 1989 162. Merriam GR Jr, Worgul BV: Experimental radiation cataract—Its clinical relevance. Bull N Y Acad Med 59:372–392, 1983 163. Orts Vila P, Devesa Torregrosa P, Belmonte Martinez J: Juvenile ciabetic cataract. A rare finding which lead us to the diagnosis
of this illness. Arch Soc Esp Oftalmol 78:389–391, 2003 164. Klein BE, Klein R, Moss SE: Prevalence of cataracts in a population-based study of persons with diabetes
mellitus. Ophthalmology 92:1191–1196, 1985 165. Nitiyanant W, Tandhanand S, Mahtab H, et al: The Diabcare-Asia 1998 study—Outcomes on control and complications
in type 1 and type 2 diabetic patients. Curr Med Res Opin 18:317–327, 2002 166. Hennis A, Wu SY, Li X, et al: Lens opacities and mortality: The Barbados Eye Studies. Ophthalmology 108:498–504, 2001 167. Merin S, Crawford JS: Hypoglycemia and infantile cataract. Arch Ophthalmol 86:495–498, 1971 168. Grunt JA, Howard RO: Eye findings in children with ketotic hypoglycemia. Can J Ophthalmol 7:151, 1972 169. Wets B, Milot JA, Polomeno RC, Letarte J: Cataracts and ketotic hypoglycemia. Ophthalmology 89:999–1002, 1982 170. Gable EM, Brandonisio TM: Ocular manifestations of Donohue's syndrome. Optom Vis Sci 80:339–343, 2003 171. Al-Till M, Jarrah NS, Ajlouni KM: Ophthalmologic findings in fifteen patients with Wolfram syndrome. Eur J Ophthalmol 12:84–88, 2002 172. Tyfield L, Reichardt J, Fridovich-Keil J, et al: Classical galactosemia and mutations at the galactose-1phosphate uridyl
transferase (GALT) gene. Hum Mutat 13:417–430, 1999 173. Bosch A, Bakker H, van Gennip A, et al: Clinical features of galactokinase deficiency: A review of the literature. J Inherit Metab Dis 25:629–634, 2002 174. Reich S, Hennermann J, Vetter B, et al: An unexpectedly high frequency of hypergalactosemia in an immigrant Bosnian
population revealed by newborn screening. Pediatr Res 51:598–601, 2002 175. Misumi H, Wada H, Kawakami M, et al: Detection of UDP-galactose-4-epimerase deficiency in a galactosemia screening
program. Clin Chim Acta 116:101–105, 1981 176. Viestenz A, Gusek-Schneider GC, Junemann AG, et al: Early childhood cataract in hereditary UDP-galactose-4-epimerase deficiency—A
case report. Klin Monatsbl Augenheilkd 218:121–124, 2001 177. Fujii H: Glucose-6-phosphate dehydrogenase. Nippon Rinsho 53:1221–1225, 1995 178. Assaf AA, Tabbara KF, el-Hazmi MA: Cataracts in glucose-6-phosphate dehydrogenase deficiency. Ophthalmic Paediatr Genet 14:81–86, 1993 179. Babalola OE, Danboyi P, Abiose AA: Hereditary congenital cataracts associated with sickle cell anaemia in
a Nigerian family. Trop Doct 30:12–14, 2000 180. Faig J, Kalinyak J, Marcus R, Feldman D: Chronic atypical seizure disorder and cataracts due to delayed diagnosis
of pseudohypoparathyroidism. West J Med 157:64–65, 1992 181. Schiekofer S, Heilmann P, Nawroth PP, Schilling T: The “needle man”: more than 40,000 injections in 40 y. Dtsch Med Wochenschr 127:2447–2448, 2002 182. Rajendram R, Deane JA, Barnes M, et al: Rapid onset childhood cataracts leading to the diagnosis of autoimmune
polyendocrinopathy-candidiasis-ectodermal dystrophy. Am J Ophthalmol 136:951–952, 2003 183. Mornet E: Hypophosphatasia: The mutations in the tissue-nonspecific alkaline phosphatase
gene. Hum Mutat 15:309–315, 2000 184. Roxburgh S: Atypical retinitis pigmentosa with hypophosphatasia. Trans Ophthalmol Soc UK 103:513–516, 1983 185. Minassian DC, Mehra V, Verrey JD: Dehydrational crisis: A major risk factor in blinding cataract. Br J Ophthalmol 73:100–105, 1989 186. Zodpey S, Ughade S, Khanolkar V, Shrikhande S: Dehydrational crisis from severe diarrhoea and risk of age-related cataract. J Indian Med Assoc 97:13–15, 24, 1999 187. Mahto RS: Ocular features of hypothyroidism. Br J Ophthalmol 56:546–550, 1972 188. Frohman LP: Systemic disease and neuro-ophthalmology: annual update 2000 (part
II). J Neuroophthalmol 21:74–82, 2001 189. Kiyosawa M, Baba T: Ophthalmological findings in patients with Takayasu disease. Int J Cardiol 66(Suppl 1):141–147, discussion 9, 1998 190. Klein BE, Klein R, Lee KE, Meuer SM: Socioeconomic and lifestyle factors in the 10-year incidence of age related
cataracts. Am J Ophthalmol 136:506–512, 2003 191. Hiratsuka Y, Li G: Alcohol and eye diseases: A review of epidemiologic studies. J Stud Alcohol 62:397–402, 2001 192. Miller D: A case of anorexia nervosa in a young woman with development of cataracts. Trans Ophthalmol Soc UK 78:217, 1958 193. Abraham SF, Banks CN, Beaumont PJ: Eye signs in patients with anorexia nervosa. Aust J Ophthalmol 8:55–57, 1980 194. Weintraub JM, Willett WC, Rosner B, et al: A prospective study of the relationship between body mass index and cataract
extraction among US women and men. Int J Obes Relat Metab Disord 26:1588–1595, 2002 195. McGhee CN, Dean S, Danesh-Meyer H: Locally administered ocular corticosteroids: Benefits and risks. Drug Saf 25:33–55, 2002 196. Gillies MC, Simpson JM, Billson FA, et al: Safety of an intravitreal injection of triamcinolone: Results from a randomized
clinical trial. Arch Ophthalmol 122:336–340, 2004 197. Zonana-Nacach A, Barr SG, Magder LS, Petri M: Damage in systemic lupus erythematosus and its association with corticosteroids. Arthritis Rheum 43:1801–1808, 2000 198. Smeeth L, Boulis M, Hubbard R, Fletcher AE: A population based case-control study of cataract and inhaled corticosteroids. Br J Ophthalmol 87:1247–1251, 2003 199. Leung AT, Cheng AC, Chan WM, Lam DS: Chlorpromazine-induced refractile corneal deposits and cataract. Arch Ophthalmol 117:1662–1663, 1999 200. Kaufman PL EK, Neider MW: Echothiophate iodide cataracts in monkeys. Occurrence despite loss of accommodation
induced by retrodisplacement of ciliary muscle. Arch Ophthalmol 101:125–128, 1983 201. Kaida T, Ogawa T, Amemiya T: Cataract induced by short-term administration of large doses of busulfan: A
case report. Ophthalmologica 213:397–399, 1999 202. Odrich S, Worgul BV, Merriam GR, et al: Mutagen induced micronucleation in the lens epithelium. Invest Ophthalmol Vis Sci 276, 1986 203. Herman DC, Dyer JA: Anterior subcapsular cataracts as a possible adverse ocular reaction to
isotretinoin. Am J Ophthalmol 103:236–237, 1987 204. Heuberger A, Buchi ER: Irreversible cataract as a possible side effect of isoretinoin. Klin Monatsbl Augenheilkd 204:465–467, 1994 205. Fraunfelder FT, LaBraico JM, Meyer SM: Adverse ocular reactions possibly associated with isotretinoin. Am J Ophthalmol 100:534–537, 1985 206. Garbe E, Suissa S, LeLorier J: Exposure to allopurinol and the risk of cataract extraction in elderly
patients. Arch Ophthalmol 116:1652–1656, 1998 207. Goldberg DE, Wang H, Azen SP, Freeman WR: Long term visual outcome of patients with cytomegalovirus retinitis treated
with highly active antiretroviral therapy. Br J Ophthalmol 87:853–855, 2003 208. Merriam GR Jr, Focht EF: A clinical study of radiation cataracts and the relationship to dose. AJR Am J Roentgenol 77:759–785, 1957 209. Klein BEK, Klein R, Linton KLP, Franke T: Diagnostic X-ray exposure and lens opacities: The Beaver Dam eye study. Am J Public Health 83:588–590, 1993 210. Otake M, Finch S, Chosi K, et al: Radiation related ophthalmological changes and aging among Hiroshima and
Nagasaki A-bomb survivors: A reanalysis. Radiat Res 131:315–324, 1992 211. Worgul BV, Kundiyev YI, Sergiyenko NM, et al: Low-dose radiation causes cataracts—A follow-up study of Chernobyl
clean-up workers and its implications regarding permissible eye exposures. Radiat Res (submitted) 212. Ferreiro I, Melendez J, Regalado J, et al: Factors influencing the sequelae of high tension electrical injuries. Burns 24:649–653, 1998 213. Portellos M, Orlin SE, Kozart DM: Electric cataracts. Arch Ophthalmol 114:1022–1023, 1996 214. Gilbert C, Foster A: Childhood blindness in the context of VISION 2020—The right to sight. Bull World Health Organ 79:227–232, 2001 215. Stegmann BJ, Carey JC: TORCH infections. Toxoplasmosis, other (syphilis, varicella-zoster, parvovirus
B19), rubella, cytomegalovirus (CMV), and
herpes infections. Curr Womens Health Rep 2:253–258, 2002 216. Vutova K, Peicheva Z, Popova A, et al: Congenital toxoplasmosis: eye manifestations in infants and children. Ann Trop Paediatr 22:213–218, 2002 217. Malathi J, Therese KL, Madhavan HN: The association of rubella virus in congenital cataract—A hospital-based
study in India. J Clin Virol 23:25–29, 2001 218. Cushnir VN, Slepova OS, Cruglova TB, et al: The role of HBV-infection in development of cataracts in children and adults. Oftalmologia 41:318–322, 1997 219. Siegel M: Congenital malformations following chickenpox, measles, mumps, and hepatitis. Results
of a cohort study. JAMA 226:1521–1524, 1973 220. Lambert SR, Taylor D, Kriss A, et al: Ocular manifestations of the congenital varicella syndrome. Arch Ophthalmol 107:52–56, 1989 221. Raghu H, Subhan S, Jose RJ, et al: Herpes simplex virus-1—Associated congenital cataract. Am J Ophthalmol 138:313–314, 2004 222. Lorenz B, Schroeder J, Reischl U: First evidence of an endogenous Spiroplasma sp. infection in humans manifesting as unilateral cataract associated
with anterior uveitis in a premature baby. Graefes Arch Clin Exp Ophthalmol 240:348–353, 2002 223. O'Neill JF: The ocular manifestations of congenital infection: A study of the early
effect and long-term outcome of maternally transmitted rubella and toxoplasmosis. Trans Am Ophthalmol Soc 96:813–879, 1998 224. Sauerbrei A, Wutzler P: The congenital varicella syndrome. J Perinatol 20(8 Pt 1):548–554, 2000 225. Singh-Parikshak R, Bothun ED, Superstein R, et al: Sequestration and late activation of lenticular candida abscess in premature
infants. Arch Ophthalmol 122:1393–1395, 2004 226. Margo CE, Hamed LM: Ocular syphilis. Surv Ophthalmol 37:203–220, 1992 |