1. Abramson DH, Jereb B, Ellsworth RM: External beam radiation for retinoblastoma. Bull NY Acad Med 57: 787, 1981 2. Gallie BL, Muncaster M, Hamel P et al: Retinoblastoma: a model for the role of recessive mutations in cancer initiation
and progression. Can J Oncol 2:50, 1992 3. Gallie BL, Dunn JM, Chan HL et al: The genetics of retinoblastoma: relevance to the patient. Pediatr Clin North Am 38:299, 1991 4. Goddard AD, Phillips RA, Greger V et al: Use of the RB1 cDNA as a diagnostic probe in retinoblastoma families. Clin Genet 37:117, 1990 5. Albert D: Historic review of retinoblastoma. Ophthalmology 94:654, 1987 6. Hilgartner HL: Report of a case of double glioma treated by X-ray. Tex Med J 18:322, 1903 7. Moore RF: Value and technique of use of radon in certain intraocular conditions. Trans Ophthalmol Soc UK 53: 215, 1933 8. Reese AB, Merriam GL, Martin AG: Treatment of bilateral retinoblastoma by irradiation and surgery: report
on 15 year results. Am J Ophthalmol 32:175, 1949 9. Stallard HB: Radiotherapy of malignant intraocular neoplasms. Br J Ophthalmol 32:618, 1948 10. Kupfer C: Retinoblastoma treated with intravenous nitrogen mustard. Am J Ophthalmol 36:1721, 1953 11. Weve HJM: The diathermy treatment of intraocular tumors. Trans Ophthalmol Soc Austr 13:47, 1953 12. Melchers MJ: Diathermy treatment of intraocular tumors. Drukkeris Fa Schotamus
and Jens Utredht, 1953 13. Ellsworth RM: Treatment of retinoblastoma. Am J Ophthalmol 66:49, 1968 14. Meyer-Schwickerwath G: The preservation of vision by treatment of intraocular tumors with light
coagulation. Arch Ophthalmol 66:458, 1961 15. Abramson DH: Retinoblastoma 1990: diagnosis, treatment and implications. Pediatr Ann 19:387, 1990 16. Hart P: The role of the retinoblastoma gene in tumor pathogenesis. Clin Oncol 4:125, 1992 17. Brooks L, Meyer D, Shields JA et al: Removal of radiation-induced cataracts in patients treated for retinoblastoma. Arch Ophthalmol 108:1701, 1990 18. Zimmerman LE: Retinoblastoma and retinocytoma. In Spencer WH (ed): Ophthalmic
Pathology: An Atlas and Textbook, Vol 2, pp 1292–1351. Philadelphia, WB
Saunders, 1985 19. Gately DP, Howell SB: Cellular accumulation of the anticancer agent cisplatin: a review. Br J Cancer 67:1171, 1993 20. Knudson AG: Retinoblastoma: a prototypic hereditary neoplasm. Semin Oncol 5:57, 1971 21. Pendergrass TW: Incidence of retinoblastoma in the United States. Arch Ophthalmol 98:1204, 1980 22. Maat-Kievit JA, Depkes D, Hartwig NG et al: A large retinoblastoma detected in a fetus at 21 weeks of gestation. Prenat Diagn 13:377, 1993 23. Pierro L, Brancato R, Capoferri C: Prenatal detection and early diagnosis of hereditary retinoblastoma in
a family. Ophthalmologica 207:106, 1993 24. Abramson DH, Servodidio CA: Retinoblastoma in the first year of life. Ophthalmic Paediatr Genet 13:191, 1992 25. Matsunaga E, Minoda K, Sasaki MS: Parental age and seasonal variation in the births of children with sporadic
retinoblastoma: a mutation-epidemiologic study. Hum Genet 84:155, 1990 26. Tamboli A, Podgor MJ, Horm JW: The incidence of retinoblastoma in the United States: 1974 through 1985. Arch Ophthalmol 108:128, 1990 27. Sorahan T, Stewart AM: Retinoblastoma and fetal irradiation. BMJ 307:870, 1993 28. Lerche W: Merkwurdige Entartung des linken Augapfels bei allen (3) mannlichen Kindern
einer Familie. Vermischte Abwandlungen aus dem Jahre 1:188, 1821 29. Munier F, Spence MA, Pescia G et al: Paternal selection favoring mutant alleles of the retinoblastoma susceptibility
gene. Hum Genet 89:508, 1992 30. Jay M, Cowell J, Hungerford J: Register of retinoblastoma: preliminary results. Eye 2:102, 1988 31. Wardrop J: Observation on fungus haematodes or soft cancer. Edinburgh, George
Ramsey and Co, 1809 32. Comings DE: A general theory of carcinogenesis. Proc Natl Acad Sci USA 70:3324, 1973 33. Gallie BL, Phillips RA: Retinoblastoma: a model of oncogenesis. Ophthalmology 91:666, 1984 34. Friend SH, Bernards R, Rogelj S et al: A human DNA segment with properties of the gene that predisposes to retinoblastoma
and osteosarcoma. Nature 323:643, 1986 35. McGee TL, Yandell DW, Dryja TP: Structure and partial genomic sequence of the human retinoblastoma susceptibility
gene product. Gene 80:119, 1989 36. Dunn JM, Phillips RA, Becker AJ, Callie BL: Identification of germline and somatic mutations affecting the retinoblastoma
gene. Science 241:1797, 1988 37. Yokota J, Akiyama T, Fung YKT et al: Altered expression of the retinoblastoma gene in small-cell carcinoma of
the lung. Oncogene 3:471, 1989 38. Oscier D, Chapman R, Fitchett M et al: Deletion of retinoblastoma gene in B-cell chronic lymphocytic leukemia. Blood 76:241, 1990 39. T'Aang A, Varley JM, Chakraborty S et al: Structural rearrangement of the retinoblastoma gene in human breast carcinoma. Science 242:263, 1988 40. Dunn JM, Phillips RA, Zhu X et al: Mutations in the RB1 gene and their effects on transcription. Mol Cell Biol 9:4596, 1989 41. Verhoeff FH, Jackson E: Minutes of the proceedings of the 62nd annual meeting. Trans Am Ophthalmol Soc 24:38, 1926 42. Warneford S, Townsend M, Rowe PD et al: Overexpression of the retinoblastoma gene in a familial adrenocortical
carcinoma. Cell Growth Differ 2:439, 1991 43. Naumova A, Hansen M, Strong L et al: Concordance between parental origin of chromosome 13q loss and chromosome 6p
duplication in sporadic retinoblastoma. Am J Hum Genet 54:274, 1994 44. Ohnishi Y, Shigeto M, Ishibashi T, Hirata J: Familial pericentric inversion of chromosome 11 in a child with sporadic
unilateral retinoblastoma. Ophthalmic Paediatr Genet 11:281, 1990 45. Musarella MA, Gallie BL: A simplified scheme for genetic counseling in retinoblastoma. J Pediatr Ophthalmol Strabismus 24:124, 1987 46. Zacksenhaus E, Bremner R, Jiang Z et al: Unraveling the function of the retinoblastoma gene. Adv Cancer Res 61:115, 1993 47. Antelman D, Machemer T, Huyghe BG et al: Inhibition of tumor cell proliferation in vitro and in vivo by exogenous
p110RB, the retinoblastoma tumor suppressor protein. Oncogene 10:697, 1995 48. Cavanaugh AH, Hempel WM, Taylor LJ et al: Activity of RNA polymerase I transcription factor UBF blocked by Rb gene
product. Nature 374(6518):177, 1995 49. Haye C, Dejardins L, Schlienger P et al: Treatment of bilateral retinoblastoma stage V at the Curie Foundation. Ophthalmic Paediatr Genet 8:73, 1987 50. Kodlilinye HC: Retinoblastoma in Nigeria: problems of treatment. Am J Ophthalmol 63:469, 1967 51. Schultz KR, Ranada S, Neglia JP, Ravindranath Y: An increased relative frequency of retinoblastoma at a rural regional referral
hospital in Miraj, Maharachtra, India. Oncology 72:222, 1993 52. Fontanesi J, Pratt C, Meyer D et al: Asynchronous bilateral retinoblastoma: The St. Jude Children's Hospital
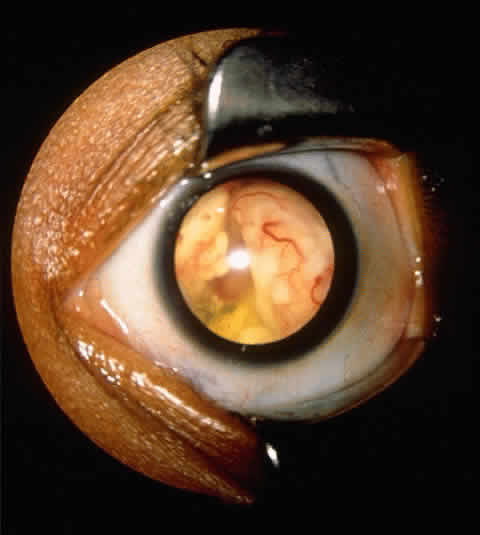
experience. Ophthalmic Genet 16:109, 1995 53. Mathew L, Miale TD, Rao S et al: Retrospective analysis of 58 children with retinoblastoma. Ophthalmol Paediatr Genet 4:67, 1984 54. Shields JA, Shields CL, Suvarnamani C et al: Retinoblastoma manifesting as orbital cellulitis. Am J Ophthalmol 112:442, 1991 55. Shields CL, Shields JA, Shields MB: Prevalence and mechanism of secondary intraocular pressure elevation in
eyes with intraocular tumors. Ophthalmology 94:839, 1987 56. Shields JA, Shields CL, Eagle RC, Blair CJ: Spontaneous pseudohypopyon secondary to diffuse infiltrating retinoblastoma. Arch Ophthalmol 106:1302, 1988 57. Byrnes GA, Shields CL, Shields JA et al: Retinoblastoma presenting with spontaneous hyphema and dislocated lens. J Pediatr Ophthalmol Strabismus 30:334, 1993 58. Howard GM: Spontaneous hyphema in infancy and childhood. Arch Ophthalmol 69:583, 1968 59. Jakobiec FA, Tso MOM, Zimmerman LE, Danis P: Retinoblastoma and intracranial malignancy. Cancer 39:2048, 1977 60. Pesin SR, Shields JA: Seven cases of trilateral retinoblastoma. Am J Ophthalmol 107:121, 1989 61. Guyton AC, Hall JE: Reproduction and hormonal function of the male (and
the pineal gland). In Guyton AC, Hall JE (eds): Textbook of Medical Physiology, 9th
ed, pp 1015–1016. Philadelphia, WB Saunders, 1996 62. Sivak JGL: The vertebrate median eye. Vision Res 14: 137, 1974 63. Blach LE, McCormick B, Abramson DH, Ellsworth RM: Trilateral retinoblastoma—incidence and outcome: a decade of experience. Int J Radiol Oncol 29:729, 1994 64. Bader JL, Meadows AT, Zimmerman LE et al: Bilateral retinoblastoma with ectopic intracranial retinoblastoma: trilateral
retinoblastoma. Cancer Genet Cytogenet 5: 203, 1982 65. Katayama Y, Tsubokawa T, Yamamoto T, Nemoto N: Ectopic retinoblastoma within the 3rd ventricle: case report. Neurosurgery 28:158, 1991 66. Palazzi M, Abramson D, Ellsworth RH: Endophytic vs. exophytic unilateral retinoblastoma: is there any real difference? J Pediatr Ophthalmol Strabismus 27:255, 1990 67. Nicholson DH, Norton EWD: Diffuse infiltrating retinoblastoma. Trans Am
Ophthalmol Soc LXXCVIII:265, 1980 68. Gallie BL, Ellsworth RM, Abramson DH, Phillips RA: Retinoma: spontaneous regression of retinoblastoma or benign manifestation
of the mutation? Br J Cancer 45: 515, 1982 69. Eagle RC, Shields JA, Donoso L, Milner RS: Malignant transformation of spontaneously regressed retinoblastoma, retinoma/retinocytoma
variant. Ophthalmology 96: 1389, 1989 70. Assaf AA, Phillips CI: Spontaneous regression of unilateral retinoblastoma in a father of three
sons with bilateral retinoblastoma. Ophthalmic Paediatr Genet 6:179, 1985 71. Allderdice PW, Davis JG, Miller OJ et al: The 13 q-deletion syndrome. Am J Hum Genet 21:499, 1969 72. Grabowski EF, Abramson DH: Intraocular and extraocular retinoblastoma. Hematol Oncol Clin North Am 1:1721, 1987 73. Mafee MF, Goldberg MF, Greenwald MF et al: Retinoblastoma and simulating lesions: role of CT and MRI imaging. Radiol Clin North Am 25:667, 1987 74. Shields JA, Shields CL, Eagle R et al: Calcified intraocular abscess simulating retinoblastoma. Am J Ophthalmol 114:227, 1992 75. Char DH, Hedges TR III, Norman D: Retinoblastoma: CT diagnosis. Ophthalmology 91:1347, 1984 76. Shields JA, Leonard BC, Michelson JB, Sarin LK: B-scan ultrasonography in the diagnosis of atypical retinoblastomas. Can J Ophthalmol 11:42, 1976 77. Senft SH, Hidayat AA, Cavender JC: Atypical presentation of Coats' disease. Retina 14:36, 1994 78. Boal DK: Case 4: Noncalcified retinoblastoma. AJR 162: 1474, 1994 79. Abramson DH, Piro PA, Ellsworth RM et al: Lactate dehydrogenase levels and isozyme patterns: Measurements in the
aqueous humor and serum of retinoblastoma patients. Arch Ophthalmol 97:870, 1979 80. Akhtar M, Ashraf AM, Sabbah R et al: Aspiration cytology of retinoblastoma: light and electron microscopic correlations. Diagn Cytopathol 4:306, 1988 81. O'Hara BJ, Ehya H, Shields JA et al: Fine needle aspiration biopsy in pediatric ophthalmic tumors and pseudotumors. Acta Cytol 37:125, 1993 82. Karcioglu ZA, Gordon RA, Karcioglu GL: Tumor seeding in ocular fine needle aspiration biopsy. Ophthalmology 92:1763, 1985 83. Abramson DH, Greenfield DS, Ellsworth RM et al: Neuron-specific enolase and retinoblastoma. Retina 9:148, 1989 84. Bomanji J, Hungerford JL, Kingston JE et al: 123I metaiodobenzylguanidine (MIBG) scintigraphy of retinoblastoma: preliminary
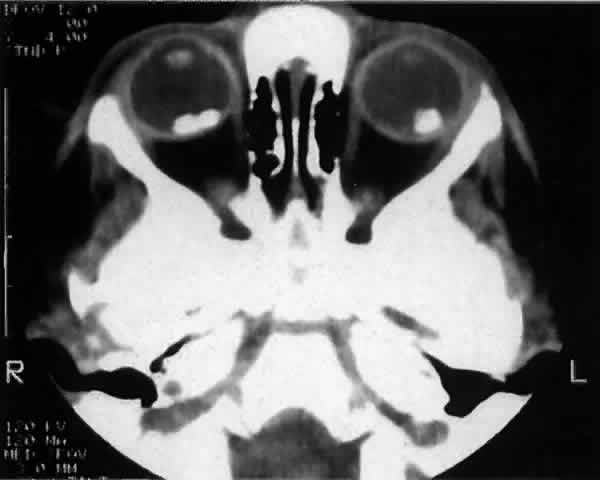
experience. Br J Ophthalmol 73:146, 1989 85. Mafee MF, Goldberg MF, Cohen SB et al: Magnetic resonance imaging versus computed tomography of leukocoric eyes
and use of in vitro proton magnetic resonance spectroscopy of retinoblastoma. Ophthalmology 96:965, 1989 86. Hopping W: The new Essen prognosis classification for conservative sightsaving
treatment of retinoblastoma in intraocular tumors. In Lommatzsch
PK, Blodi FC (eds): Intraocular Tumors: International Symposium Under
the Auspices of the European Ophthalmological Society, pp 497–505. Berlin, Springer-Verlag, 1983 87. Howarth C, Meyer D, Hustu HO et al: Stage-related combined modality treatment of retinoblastoma: results of
a prospective study. Cancer 45:851, 1980 88. McCartney ACE, Olver JM, Kingston JE, Hungerford JL: Forty years of retinoblastoma: into the fifth age. Eye 2(suppl):13, 1988 89. Katsetos CD, Herman MM, Frankfurther A et al: Neuron-associated class III B-tubulin isotype microtubule associated protein 2, and synaptophysin in
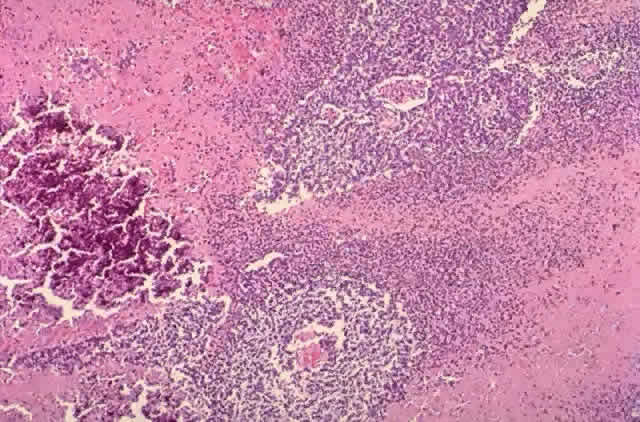
human retinoblastoma in situ. Lab Invest 64:45, 1991 90. Nork TM, Schwartz TL, Doshi HM, Millecchia LL: Retinoblastoma: cell of origin. Arch Ophthalmol 113:791, 1995 91. Flexner S: A peculiar glioma (neuroepithelioma) of the retina. Bull Johns Hopkins 2:115, 1891 92. Wintersteiner H: Die Neuroepithelioma Retinae: Eine Anatomiche und Klinische
Studie. Leipzig, Dentisae, 1897 93. Howard MA, Dryja TP, Walton DS, Albert DM: Identification and significance of multinucleate tumor cells in retinoblastoma. Arch Ophthalmol 107:1025, 1989 94. Fontanesi J, Rao BN, Fleming ID et al: Pediatric brachytherapy: The St. Jude Children's Research Hospital
experience. Cancer 74:733, 1994 95. Tajima Y, Munakata S, Ishida Y et al: Photoreceptor differentiation of retinoblastoma: an electron microscopic
study of 29 cases. Pathol Int 44:837, 1994 96. Sanders BM, Draper GJ, Kingston JE: Retinoblastoma in Great Britain 1969-80: incidence, treatment and survival. Br J Ophthalmol 72:576, 1988 97. Zelter M, Schwartz L, Gonzalez G et al: Retinoblastoma: results of treatment and evaluation of vision. Proc Am Soc Clin Oncol 5:203, 1986 98. Stevenson KE, Hungerford J, Garner A: Local extraocular extension of retinoblastoma following intraocular surgery. Br J Ophthalmol 73:739, 1989 99. Magramm I, Abramson DH, Ellsworth RM: Optic nerve involvement in retinoblastoma. Ophthalmology 96:217, 1989 100. Shields CL, Shields JA, Baez K et al: Optic nerve invasion of retinoblastoma: metastatic potential and clinical
risk factors. Cancer 73:692, 1994 101. Baez KA, Ulbig MW, Cater J et al: Iris neovascularization, increased intraocular pressure and vitreous hemorrhage
as risk factors for invasion of the optic nerve and choroid in
children with retinoblastoma. Ophthalmologie 91:796, 1994 102. Chintagumpala MM, Boniuk M, Steinkuller P, Mahoney DH: Retinoblastoma: treatment and follow-up of 35 patients from a single institution. Proceedings of the Society of Pediatric Hematology and Oncology 2:30, 1993 103. Doz F, Khelfaoui F, Mosseri V et al: The role of chemotherapy in orbital involvement of retinoblastoma: the
experience of a single institution with 33 patients. Cancer 74:722, 1994 104. Hungerford J, Kingston JE, Plowman PN: Orbital recurrence of retinoblastoma. Ophthalmic Paediatr Genet 8: 63, 1987 105. Abramson DH, Ellsworth RM, Zimmerman LE: Nonocular cancer in retinoblastoma survivors. Trans Am Acad Ophthalmol Otolaryngol 81:454, 1976 106. Derkinderen DJ, Koten JW, Wolterbeek R et al: Non-ocular cancer in hereditary retinoblastoma survivors and relatives. Ophthalmic Paediatr Genet 8:23, 1987 107. Roarty JD, McLean IW, Zimmerman LE: Incidence of second neoplasms in patients with bilateral retinoblastoma. Ophthalmology 95:1583, 1988 108. Draper GJ, Sanders BM, Kingston JE: Second primary neoplasms in patients with retinoblastoma. Br J Cancer 53:661, 1986 109. Hausmann N, Stefani FH: Second nonocular tumors in cured unilateral retinoblastoma patients. J Cancer Res Clin Oncol 117:4, 1991 110. Helton KJ, Fletcher BD, Kun LE et al: Bone tumors other than osteosarcoma after retinoblastoma. Cancer 71:2847, 1993 111. Ellsworth RM: Treatment of retinoblastoma. In Haik GM (ed): Transactions
of the New Orleans Academy of Ophthalmology, pp 55–59. New Orleans, NOAO, 1986 112. Strong LC, Herson J, Haas C et al: Cancer mortality in relatives of retinoblastoma patients. J Natl Cancer Inst 73:303, 1984 113. Nelson SC, Friedman HS, Oakes WJ et al: Successful therapy for trilateral retinoblastoma. Am J Ophthalmol 114:23, 1992 114. Weiss AH, Karr DJ, Kalina RE et al: Visual outcomes of macular retinoblastoma after external beam radiation
therapy. Ophthalmology 101:1244, 1994 115. Servidio CA, Abramson DH, Boxrud C, Ellsworth RM: Nursing implications of visual fields in successfully treated retinoblastoma
patients. Insight 18:10, 1993 116. Holbeck S, Ehlers N: Long-term visual results in eyes cured for retinoblastoma by radiation. Acta Ophthalmologica 67:560, 1989 117. Abramson DH, Ellsworth RM, Rozakis GW: Cryosurgery for retinoblastoma. Arch Ophthalmol 100:1253, 1982 118. Abramson DH: The focal treatment of retinoblastoma with emphasis on xenon arc photocoagulation. Acta Ophthalmologica 194:1, 1989 119. Howard GM: Invasion of the choroid and sclera by retinoblastoma following photocoagulation. Trans Am Acad Ophthalmol 70:984, 1966 120. Shields JA, Shields CL, Parsons H, Giblin ME: The role of photocoagulation in the management of retinoblastoma. Arch Ophthalmol 108:205, 1990 121. Ohnishi Y, Yamana Y, Minei M: Photoradiation therapy using argon laser and a hematoporphyrin derivative
for retinoblastoma: a preliminary report. Jpn J Ophthalmol 30:409, 1986 122. Venter DJ, Bevan KL, Ludwig RL et al: Retinoblastoma gene deletions in human glioblastoma. Oncogene 6:445, 1991 123. Dougherty TJ, Kaufman JE, Goldfarb KA et al: Photoradiation therapy for the treatment of malignant tumors. Cancer Res 38:401, 1978 124. Murphree L, Steele D, Malogolowkin M, Sato J: Thermochemotherapy for retinoblastoma: protocol, instrumentation and outcome. Proceedings of the
International Society for Genetic Eye Disease, Niagara On The Lake, Ontario, Canada, June 23–24, 1994 125. Shields JA, Shields CL: Current management of retinoblastoma. Mayo Clin Proc 69:50, 1994 126. Shields JA, Shields CL, Sivalingam V: Decreasing frequency of enucleation in patients with retinoblastoma. Am J Ophthalmol 108:185, 1989 127. Donoso LA, Shields CL, Lee EY: Immunohistochemistry of retinoblastoma: a review. Ophthalmol Paediatr Genet 10:3, 1989 128. Abramson DH, Ellsworth RM, Tretter P et al: Simultaneous bilateral radiation for advanced retinoblastoma. Arch Ophthalmol 99:1763, 1981 129. Abramson DH, Ellsworth RM, Tretter P et al: Treatment of bilateral groups I through III retinoblastoma with bilateral
radiation. Arch Ophthalmol 99:1761, 1981 130. Abramson DH, Marks RF, Ellsworth RM et al: The management of unilateral retinoblastoma without primary enucleation. Arch Ophthalmol 100:1249, 1982 131. Harris AMP, Nortje CJ, Lucchesi MV: Some effects of radiation therapy during early childhood on facial growth
and tooth development. J Dent Assoc S Africa 41:681, 1986 132. Lam BL, Judisch GF, Sobol WM, Blodi CF: Visual prognosis in macular retinoblastoma. Am J Ophthalmol 110:229, 1990 133. Monge OR, Flage T, Hatlevoll R, Vermund H: Sightsaving therapy in retinoblastoma: experience with external megavoltage
radiotherapy. Acta Ophthalmologica 64:414, 1986 134. Messmer EP, Sauerwein W, Heinrich T et al: New and recurrent tumor foci following local as well as external beam radiation
in eyes of patients with hereditary retinoblastoma. Graefes Arch Clin Exp Ophthalmol 228:426, 1990 135. Abramson DH, McCormick B, Fass D et al: Retinoblastoma: the long-term appearance of radiated intraocular tumors. Cancer 67:2753, 1991 136. Messmer EP, Fritze H, Mohr C et al: Long-term effects in patients with bilateral retinoblastoma: ocular and
midfacial findings. Graefes Arch Clin Exp Ophthalmol 229: 309, 1991 137. Mohr C, Fritze H, Messmer E, Heinrich T: Zur Frage der Wachstumshemmung in Mittelgesicht nach fruhkindlicher Retinoblastomtherapie. Dtsch Z Mund Kiefer Gesichts Chir 14:391, 1990 138. Salmonsen PC, Ellsworth RM, Kitchin FD: The occurrence of new retinoblastomas after treatment. Ophthalmology 86:837, 1979 139. Schipper J, Tan Kewp van Peperzeel JA: Treatment of retinoblastoma by precision megavoltage radiation therapy. Radiother Oncol 3:97, 1985 140. Desjardin L, Levy S, Lavit A et al: An experience of the use of radioactive plaques after failure of external
beam radiation in the treatment of retinoblastoma. Ophthalmic Paediatr Genet 14:39, 1993 141. Shields JA, Giblin ME, Shields CL et al: Episcleral plaque radiotherapy for retinoblastoma. Ophthalmology 96:530, 1989 142. Bentel GC, Halperin EC, Buckley EC: 125I embedded in an orbital prosthesis for retreatment of recurrent retinoblastoma. Med Dosimetry 18:1, 1993 143. Fass D, McCormick B, Abramson D, Ellsworth R: Cobalt60 plaques in recurrent retinoblastoma. Int J Radiat Oncol Biol Phys 21:625, 1991 144. Shields CL, Hernandez C, Shields JA et al: Letter to the editor: Plaque radiotherapy of retinoblastoma. Ophthalmology 100:1277, 1993 145. Chan HSL, Canton MD, Gallie BL: Chemosensitivity and multidrug resistance to antineoplastic drugs in retinoblastoma
cell lines. Anticancer Res 9:469, 1989 146. Inomata M, Kaneko A: Chemosensitivity profiles of primary and cultured human retinoblastoma
cells in a human tumor clonogenic assay. Jpn J Cancer Res 78:858, 1987 147. Chan HSL, Thorner PS, Haddad G, Gallie BL: Multidrug-resistant phenotype in retinoblastoma correlates with P-glycoprotein
expression. Ophthalmology 98:1425, 1991 148. Advani SH, Rao SR, Iyer RS et al: Pilot study of sequential combination chemotherapy in advanced and recurrent
retinoblastoma. Med Pediatr Oncol 22:125, 1994 149. Kingston JE, Hungerford JL, Plowman PN: Chemotherapy in metastatic retinoblastoma. Ophthalmic Paediatr Genet 8:69, 1987 150. Donaldson SS, Smith LM: Retinoblastoma: biology, presentation, and current management. Oncology 3:45, 1989 151. Petersen RA, Friend SH, Albert DM: Prolonged survival of a child with metastatic retinoblastoma. J Pediatr Ophthalmol Strabismus 24:247, 1987 152. Doz F, Neuenschwander S, Plantaz D et al: Etoposide and carboplatin in extraocular retinoblastoma: a study by the
Societe Francaise d'Oncologie Pediatrique. J Clin Oncol 13:902, 1995 153. Zelter M, Damel A, Gonzalez G, Schwartz L: A prospective study on the treatment of retinoblastoma in 72 patients. Cancer 68:1685, 1991 154. Uchida I, Kinouchi K, Tashiro C: A new photoradiation therapy and anesthesia. Anesth Analg 70:222, 1990 155. Mohney BG, Robertson DM: Ancillary testing for metastasis in patients with newly diagnosed retinoblastoma. Am J Ophthalmol 118:707, 1994 156. Yandell DW, Campbell TA, Siri H et al: Oncogenic point mutations in the human retinoblastoma gene: their application
to genetic counseling. N Engl J Med 321:1689, 1989 157. DePotter P, Shields CL, Shields JA, Singh AD: Use of hydroxyapatite ocular implants in the pediatric population. Arch Ophthalmol 112:208, 1994 158. Shields JA, Shields CL, DePotter P: Enucleation technique for children with retinoblastoma. J Pediatr Ophthalmol Strabismus 29:213, 1992 159. Chang MW, Barr E, Seltzer J et al: Cytostatic gene therapy for vascular proliferative disorders with a constitutively
active form of the retinoblastoma gene product. Science 267:518, 1995 |