The most commonly encountered ophthalmic disorders belong to the craniofacial dysplasias. The craniosynostoses include Apert's syndrome, Crouzon's syndrome, Pfeiffer's syndrome, and other isolated suture abnormalities such as plagiocephaly. The clefting syndromes include cleft lip and palate, oblique facial clefts, and mandibulofacial deformities (Goldenhar's syndrome, Treacher Collins syndrome, and facial microsomia).
Hypertelorism may present as an isolated anomaly but frequently is a component of syndromes affecting the midface such as frontonasal dysplasia or the craniosynostoses.
CRANIOSYNOSTOSES
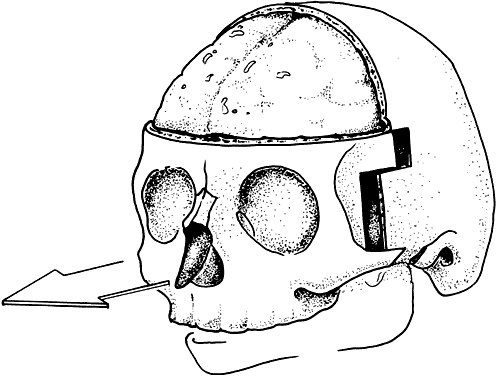
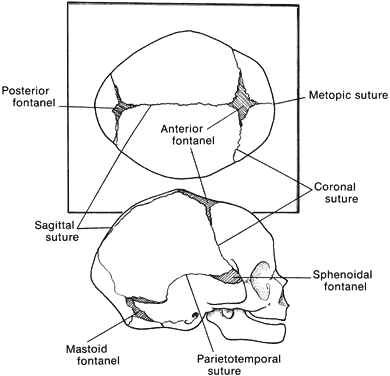
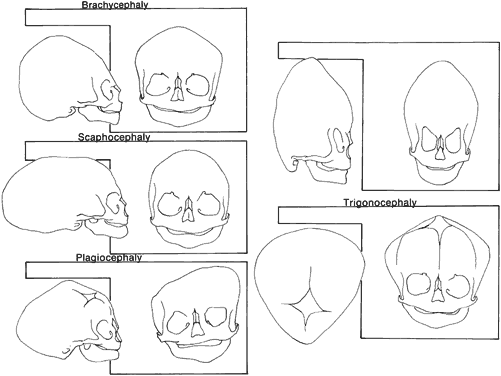
Craniosynostosis (craniostenosis) is the premature closure of one or more cranial sutures.14–21 The dysmorphologic makeup observed in craniostenosis is a direct result of premature suture closure and, chronologically, is the initial manifestation. The diagnosis usually is obvious. The malformation varies, depending on the site and the number of sutures involved in the synostosis and on subsequent alterations of growth. In fetuses of 14 weeks' gestation, that is, about 7.5 cm crown-to-rump length, most of the sutures are formed.22,23 The metopic suture (between the two frontal bones) closes before birth.23 The posterior fontanel normally closes by 2 months of age, the anterolateral suture between 3 and 6 months, and the anterior and posterolateral sutures during the first year of life.23 The sagittal suture closes by 2 years, and the coronal suture closes by 3 years (Fig. 1). The cause of premature suture closure is not known. Either an abnormal fusion of bone centers occurs, or there is malfunction of the underlying dura mater, which normally ensures suture patency.22–26 Subsequent cranial and facial growth are influenced by any prematurely closed suture. Virchow demonstrated in 1851 that, after premature suture closure, growth proceeds parallel to the suture and is inhibited perpendicular to it (Virchow's law).27 Closure of the sagittal suture results in an elongated skull (scaphocephaly). Single coronal, sagittal, metopic, or lambdoidal suture closure leads to a variety of abnormal shapes such as brachycephaly, scaphocephaly, trigonocephaly, or plagiocephaly (Fig. 2). Classification of craniosynostosis has been divided into three main groups: (1) craniofacial dysostosis (Crouzon's syndrome), (2) acrocephalosyndactyly (Apert's syndrome), and (3) simple or single craniosynostosis (plagiocephaly). Classification systems can be confusing because of the overlapping clinical and genetic manifestations.3–5 Although a functional classification based on pathomorphogenic features can be used, diagnosis by syndrome has been the traditional method of classification.7 The most commonly encountered craniofacial synostoses are Crouzon's and Apert's syndromes.
|
|
Crouzon's Syndrome
Crouzon's syndrome originally was described in 1912 in a mother and her daughter under the name of a hereditary craniofacial dysostosis.20 It is characterized by craniostenosis of the acrobrachycephalic or oxycephalic type with associated hypoplasia of the maxilla and exophthalmos. A progressive craniosynostosis often is observed at birth and usually is completed by 2 to 3 years of age. Although there is variable presentation with multiple suture involvement and no characteristic calvarial shape, 96% to 98% of patients show radiographic evidence of coronal and sagittal sutural fusion. This pattern of fusion leads to a more predictable skull shape, which features a shortened calvarium, steep forehead, and flattened occiput6,15–17,19,22,28,29 (Fig. 3). Neurologic complications of early suture closure occur commonly and include headaches (29% to 50%), increased intracranial pressure, mental retardation (13%), and seizures (11.5%).17,19 The most characteristic facial feature is midface retrusion, hypoplasia, and shallow orbits.29,30 Brachycephaly, in combination with hypoplasia of the frontal, sphenoid, ethmoid, and maxillary bones, is mostly responsible for these features.
|
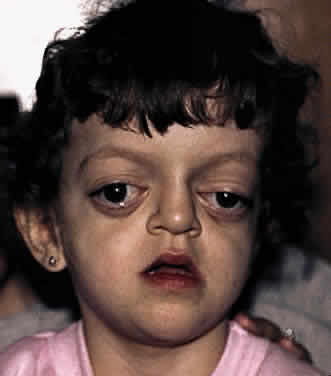
Hypertelorism and exophthalmos are universal features of all variants of Crouzon's syndrome. The exophthalmos is secondary to midfacial hypoplasia, resulting in shallow, widespread orbits. One theory states that premature suture closure results in increased intracranial pressure and preferential brain growth in the anterior and middle cranial fossae.24 Another, based on examination of affected fetuses and animal studies, suggests that cranial base changes with resultant stresses initiate osteogenic overstimulation and aberrant bony growth in the calvarium and face.13
Crouzon's syndrome can affect all ocular structures. Shallow orbits predispose to proptosis and exposure of the cornea and conjunctiva.16,22,31,32 Optic nerve atrophy and edema are vision-threatening complications and have been reported in as many as 80% of patients with this syndrome.33 However, in a series of 25 patients with Crouzon's syndrome, only 1 had optic nerve disease.32 The most plausible current explanation for optic nerve atrophy is that it is the result of a primary disturbance (hypoplasia or dysplasia) rather than the result of secondary factors (compression atrophy or chronic edema).32,33 Early surgical intervention has resulted in decreased morbidity, although the mechanisms involved in secondary optic nerve problems continue to occur.
Other structural abnormalities of the globe reported in patients with Crouzon's syndrome include aniridia, anisocoria, corectopia, blue sclera, cataract, ectopia lentis, glaucoma, chorioretinal colobomas, megalocornea, and microcornea.3,13,16,17,22,28,31
Strabismus is present in as many as 92% of patients with Crouzon's syndrome.32,34–39 V-pattern exotropia is the most common misalignment.32,34,39 The V-pattern and hypertropia on adduction usually are secondary to overaction of the inferior oblique muscles, which occurs as a result of an absent or ineffective ipsilateral superior oblique muscle.36–39 Morax has postulated that the widened, shallow orbits and anomalous muscle insertions change the actions of the muscles.37–39 The superior oblique seems to be sagittalized and less effective.33–39 In a series of 25 patients with Crouzon's syndrome, 48% had exotropia, 32% had esotropia, and 68% were hypertropic in some position of gaze.32
Patients with Crouzon's syndrome have large degrees of compound astigmatism, and many also are anisometropic.16,32 Adnexal abnormalities include ptosis, canthal dystopia, nasolacrimal duct obstruction, entropion, and ectropion.31,32,40–44
The advances made in the surgical treatment of Crouzon's syndrome have changed the pattern and prevalence of visual loss. Structural abnormalities of the globe (e.g., optic atrophy, corneal opacification), which previously were the main causes of vision loss, are less common. The main reason for vision loss is amblyopia. The cause of the amblyopia usually is multifactorial, with contributions from strabismus, uncorrected refractive errors, anisometropia, and ptosis.32
Apert's Syndrome
In 1896, Apert described this syndrome in a 15-month-old girl.45 He suggested the term acrocephalosyndactyly in a report of nine cases in 1906.46,47 The classification of this syndrome has since been modified and can be confused with other craniofacial syndromes with syndactyly48 (Table 1). This syndrome occurs with a frequency of 1:160,000 births with an autosomal dominant mode of inheritance. Spontaneous mutations have been associated with increased paternal age.46
TABLE 1. Craniosynostosis Syndromes Associated
| Syndactyly | Associated Syndrome |
| ACS, type I | Apert's syndrome |
| ACS, type II | Vogt's ACS |
| ACS, type III | Saethre-Chotzen syndrome |
| ACS, type IV | Waardenburg-type ACS |
| ACS, type V | Pfeiffer's syndrome |
| ACPS, type I | Noack's syndrome |
| ACPS, type II | Carpenter's syndrome |
| ACPS, type III | Sakati-Nyhan-Tiscale syndrome |
ACS, acrocephalosyndactyly; ACPS, acrocephalopolysyndactyly.
(McKusick VA: Mendellian Inheritance in Man: Catalogs of Autosomal Dominant, Autosomal Recessive, and X-Linked Phenotypes. Baltimore, The Johns Hopkins Press, 1974)
Clinically, the cranium, face, and extremities are involved. Although the cranial features vary, the coronal suture always is affected. The skull is oxycephalic with a flat occiput and steep forehead. The face has a hypoplastic midfacial region with a “parrot-beak” nose. Other features include low-set ears, hypertelorism, and exorbitism.45–47 More prominent features of Apert's syndrome are associated palatal and dental abnormalities, including a high-arched palate, clefting of the soft palate or a bifid uvula, a V-shaped maxillary arch with malocclusion, dental crowding, and supranumerary teeth.47,49 Ear anomalies include pinna defects, conductive hearing loss, and congenital otosclerosis45–47 (Fig. 4). Hydrocephalus and mental retardation also occur in these patients.50
|
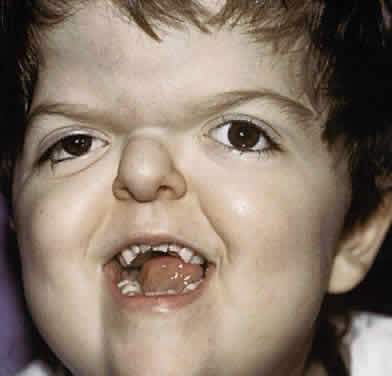
Ophthalmic manifestations are similar to those patients described with Crouzon's syndrome.31,32 Hypertelorism and proptosis are less severe than in Crouzon's syndrome. Structural abnormalities of the globe also occur in this syndrome and include keratoconus, congenital glaucoma, ectopia lentis, cataracts, exposure keratitis, and optic atrophy.30,31,44,45 Strabismus is common, occurring in 70% to 100% of patients.32,36,39 Exotropia with a V-pattern predominates, although V-pattern esotropia is common. Patients also can have nystagmus, dissociated vertical deviations, and incomitant vertical deviations. The structure of the extraocular muscles is abnormal both microscopically and grossly.38 Anomalous or missing muscles, especially the superior rectus and superior oblique, occur commonly in these patients.31,33,36–39,51 Refractive errors, in particular, high degrees of oblique astigmatism (from 60% to 100% of patients), are common, as is anisometropia.31,32,34
Adnexal abnormalities include ptosis, nasolacrimal duct obstruction, canthal dystopia, entropion, and ectropion.31,32,41–44 The high prevalence of visual loss (up to 57% of eyes) in this population of patients results from amblyopia for the same multifactorial reasons as in Crouzon's syndrome.32
The major associated systemic malformation is syndactyly of the hands and feet. This is most commonly a middigital hand mass involving the second, third, and fourth digits (Fig. 5). Other bony abnormalities include aplasia or ankylosis of the elbow, shoulder, or hip and cervical stenosis.13,46,47
|
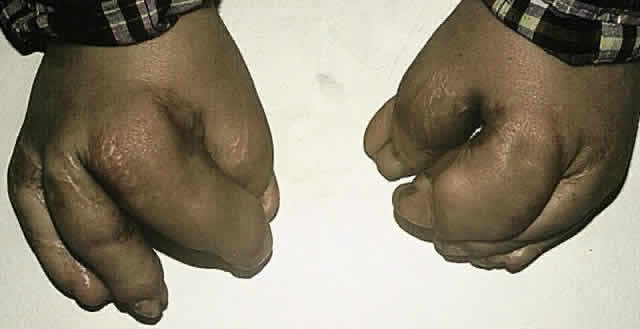
Other systemic associations include congenital heart disease, tracheoesophageal fistulas, hydrocephalus, and pyloric stenosis.13,46,47,51
Other Craniostenosis Syndromes
A variety of other craniostenoses (Table 2) share common features with Crouzon's and Apert's syndromes but occur less frequently. Pfeiffer's syndrome is autosomal dominant and has craniosynostosis with oxycephaly, maxillary hypoplasia, hypertelorism, proptosis, depressed nasal bridge, beaked nose, highly arched palate, crowded teeth, hydrocephalus, seizures, and, occasionally, mental retardation.13,52 Ophthalmic features are similar to Apert's and Crouzon's syndromes. Distinguishing features include broad, long thumbs and great toes.
TABLE 2. Clinical Characteristics of Craniofacial Synostoses
| Synostosis | Clinical Manifestations |
| Crouzon's syndrome | Coronal synostosis, shallow orbits, proptosis, hypertelorism, exotropia, exposure keratitis, luxation of globe, autosomal dominant |
| Apert's syndrome | Coronal synostosis, hypertelorism shalloworbits, strabismus, proptosis, syndactyly, autosomal dominant |
| Pfeiffer's syndrome | Coronal synostosis, hypertelorism, proptosis, strabismus, broad fingers and toes, auto-somal dominant |
| Saethre-Chotzen syndrome | Coronal synostosis, hypertelorism, ptosis, strabismus, nasolacrimal duct obstruction,optic atrophy, syndactyly, autosomal dominant |
| Carpenter's syndrome | All sutures synostotic, mental deficiency, short stature, obesity, syndactyly, hypertelorism, epithelial folds, medial canthal dystopia, corneal opacities, autosomal recessive |
| Kleeblattschädel anomaly | Variable: proptosis, hypertelorism, exposurekeratitis, iris coloboma, orbital hypoplasia, nasolacrimal duct obstruction (cloverleaf skull) |
| Christian syndrome | All sutures synostotic, hypertelorism, ophthalmoplegia, lateral canthal dystopia, microcephaly, cleft palate, arthrogryposis, autosomal recessive |
| Lowry's syndrome | Coronal synostosis, proptosis, strabismus, cleft palate, simian creases, autosomal recessive |
| Gorlin-Chaudhry-Moss syndrome | Coronal synostosis, hypertrichosis, lid colobomas, ptosis, microphthalmia, patent ductus arteriosus, autosomal recessive. |
| Baller-Gerold syndrome | Coronal synostosis, hypertelorism, epicanthal folds, radial aplasia, absent carpal bones, autosomal recessive |
Saethre-Chotzen syndrome is characterized by a more variable cranial vault with coronal synostosis, plagiocephaly, scaphocephaly, and oxycephaly. Other features include low-set frontal hairline, facial asymmetry, variable brachydactyly and syndactyly, low-set ears, hearing loss, highly arched palate, and dental abnormalities. Intelligence usually is normal, but mental retardation has been reported.11,13,22
Ophthalmic manifestations of Saethre-Chotzen syndrome differ slightly from Apert's or Crouzon's syndrome because of less severe maxillary and midfacial hypoplasia. Hypertelorism and exorbitism are less common. Ptosis is much more common in this syndrome, as well as strabismus and nasolacrimal dysfunction. Amblyopia and optic atrophy again contribute to visual loss in these patients.32
Patients with plagiocephaly exhibit a cranial asymmetry caused by a premature closure of the coronal sutures in one half of the skull. The deformities may be subtle. The skull is flattened in the frontal region on the affected side, often with a compensatory expansion on the opposite side. There is retrusion of the orbital arch on the affected side with a raised eyebrow. The maxillary bone and ear may be affected, with abnormal dental apposition and a downward and forward displaced ear.
Up to 68% of patients with plagiocephaly have ophthalmic involvement. This includes strabismus, cranial nerve palsies, optic neuropathy, ptosis, canthal dystopia, and nasolacrimal duct dysfunction.52,53 Although esotropia is common in these patients, an unusually high prevalence of clinical underaction of the superior oblique muscle ipsilateral to the stenotic suture is present. This manifests with a head tilt and hypertropia on the affected side, which increases on contralateral gaze and ipsilateral head tilt. Often there is underaction of the ipsilateral superior oblique and overaction of the ipsilateral inferior oblique muscle.54 This is postulated to result from a “sagittalization” of the superior oblique because of the orbital deformity.39,44,54 This condition is treated in the same manner as a superior oblique palsy. If the superior oblique muscle is found to be redundant or “loose,” a “tuck” of the tendon is necessary.
Carpenter's syndrome, first described in 1901, is an autosomal recessive craniosynostosis involving many sutures with preaxial polysyndactyly of the feet and variable brachydactyly and syndactyly of the hands. Mental retardation is uniformly present. Congenital heart disease, short stature, obesity, and lower limb abnormalities are other systemic associations. Hypertelorism, epicanthal folds, and telecanthus are reported ocular abnormalities.19,55–57 Other less common craniostenosis syndromes are described in Table 2.
CLEFTING SYNDROMES
The most familiar form of clefting syndromes is isolated cleft lip and palate syndromes. These clefts occur early in development before the 17-mm crown-to-rump length (17 to 20 weeks' of gestation).58 Interruptions in fusion of the developing facial processes during embryonic development possibly are responsible for these clefting syndromes.58,59 There are five facial processes: a frontonasal, two maxillary, and two mandibular. There may be insufficient outgrowth of the facial processes, poor cellular adhesion resulting in an abnormal epithelial plate, or abnormal cell degeneration of the epithelial plate. The defects of bone, musculature, and soft tissues result from poor fusion of the facial swellings. Failure of mesodermal penetrators with subsequent epithelial dehiscence in the developing facial process as it reaches the midline also is a postulated mechanism of soft tissue defects.7,9,13,58,59
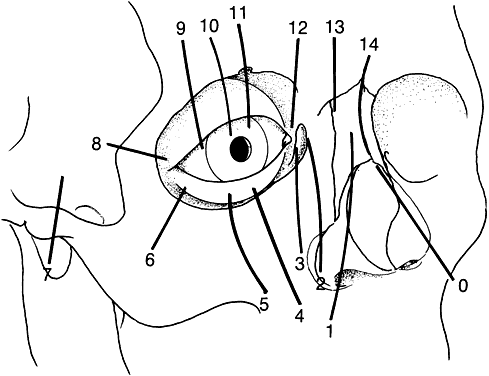
Tessier has revolutionized the classification of clinical clefting syndromes.59 This system assigns each major cranial, orbital, maxillary, and mandibular cleft a number from 0 to 14 in a clocklike circle, beginning at 0 in the lower facial midline and running clockwise on the right side of the face and counterclockwise of the left, and ending at 14 in the upper facial midline (Fig. 6). This system represents an architectural system rather than a true pathognomonic description of specific clinical syndromes.
|
Heredity appears to play a limited role in the etiology of clefting syndromes, except for Treacher Collins syndrome (mandibulofacial dysostosis). Some factors seem to be implicated in the development of craniofacial clefts and include radiation, influenza A2 virus, toxoplasmosis, chemotherapeutic agents, anticonvulsant agents, steroids, and tranquilizers.53
The amniotic band syndrome is a related, nonhereditary syndrome that can lead to facial clefts, probably secondary to pressure necrosis. Amniotic bands result from a rupture in the amniotic membrane with formation of tissue cords that can wrap themselves around limbs and digits and, if swallowed, can tighten across or around the face, cranium, or mandible distorting or “cutting” the developing craniofacial structures.1,5,53
Fries and Katowitz suggest that orbital clefts can be grouped into four regional categories that emphasize the associated ophthalmic abnormalities53:
- Median facial clefts (Tessier cleft numbers 0, 1, 13, 14), which primarily
present with hypertelorism
- Medial canthal and nasolacrimal system clefts (Tessier cleft numbers 2, 3, 4, 11, 12, 13), which
primarily present with medial canthal dystopia
and nasolacrimal duct abnormalities
- Central eyelid clefts (Tessier cleft numbers 4, 5, 6, 9, 10, 11), which
primarily present with colobomatous defects
- Lateral facial clefts (Tessier cleft numbers 6, 7, 8), which primarily
present with lateral canthal dystopia
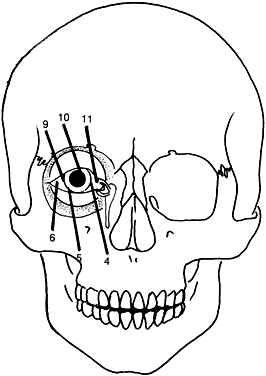
Although these clefts may occur as independent malformations, combinations of clefts frequently occur. Commonly observed pairings include 0–14, 4–10, and the 5–9 clefts (see Fig. 6). Area 1 (midline) clefts usually have orbital hypertelorism as the major ophthalmic manifestation. This also includes a broadened nasal root; bifid anterior cranium and midline facial cleft involving the nose, lip, and sometimes the palate; a lack of formation of the nasal tip; and a widow's peak (Fig. 7). Area 2 (medial canthal/nasolacrimal) clefts produce variable degrees of hypertelorism, medial canthal dystopia, nasolacrimal abnormalities, lid colobomas, and occasional displaced eyebrows and frontal and forehead dysplasia (Fig. 8). Area 3 (eyelid) clefts (see Fig. 8) produce defects that involve the lid and orbit, and, depending on the severity, can be associated with significant structural consequences. In addition to the eyelid deformities, underlying soft tissue and bony absence may permit the orbital contents to be prolapsed into the maxillary sinus. The globe usually is microphthalmic, and true anophthalmos can be present. The lateral lower lid may be affected to varying degrees, often without an oral component. The skin defect is mild and associated with malar hypoplasia and a pseudocoloboma. This lid deformity is manifested by a bowing of the lateral lid structures, usually associated with lateral canthal dystopia, and contrasts with a true coloboma, which has an actual defect in the lid margin (see Fig. 8). Area 4 (lateral canthal clefts) are rarely isolated and more often are associated with distinguishing features categorizing them into a syndrome such as Treacher Collins syndrome, Goldenhar's syndrome, or facial microsomia. When they are isolated, they can be responsible for lateral canthal dystopia; ear deformities; auricular tags; and maxillary, mandibular, parotid, tongue, and soft palate hypoplasia (Fig. 9).
|
|
|

Goldenhar's Syndrome (Oculoaurovertebral Dysplasia)
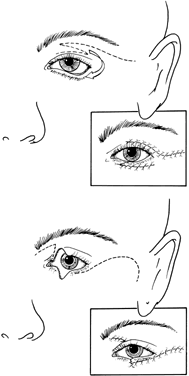
Goldenhar's syndrome first was described by Van Duyse in 1882, with additional cases and differentiation from the other first and second branchial arch syndromes reported by Goldenhar in 1952.13,31,53,60,61 The clinical features include corneoscleral dermoids, subconjunctival anterior dermoids or lipodermoids, upper eyelid colobomas, unilateral aplasia or hypoplasia of the mandibular ramus, small size or abnormal shape of one or both ears, preauricular skin tags, and vertebral anomalies13 (Fig. 10).
|
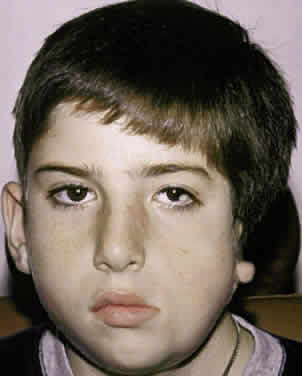
Ocular abnormalities have been well documented, and epibulbar choristomas are the most consistent finding (32% to 92%).53,60 Lid colobomas frequently are lateral and appear in association with epibulbar dermoids, but they also can occur nasally.60,61 Ptosis, nasolacrimal duct dysfunction, iris colobomas, and lid skin tags are less frequent but also have been reported.60,61 Strabismus is noted in 10% to 19% of patients and includes esotropia, exotropia, Duane's syndrome, and sixth nerve palsy.31,53,60,61 Microphthalmos with associated intraocular malformations can occur in severe cases.53,60,61
Associated systemic features include microcephaly, hydrocephalus, plagiocephaly, intracranial dermoids, congenital heart disease, and kidney malformations.8
The etiology of Goldenhar's syndrome is unknown, but the clinical findings suggest aberrant development of the first and second branchial arches similar to Treacher Collins syndrome (see later) and the facial microsomias.23–25,58 An early cell induction abnormality originated by some environmental, vascular, or topographic insult also has been postulated.53 An animal model of stapedial artery hemorrhage with resultant maxillary and mandibular hypoplasia has been proposed to explain some forms of the first and second branchial arch syndromes.62
Treacher Collins Syndrome
Treacher Collins syndrome is the only clefting syndrome with a definite hereditary pattern. It has autosomal dominant transmission with variable penetrance. Treacher Collins syndrome also is known as mandibulofacial dysostosis or zygoauromandibular dysplasia.63–67 This bilateral anomaly represents a cluster of malformations produced by zygomatic temporoaural and mandibular dysplasia. Zwahlen in 1944 and immediately afterward Franceschetti and Zwahlen (1944) confirmed and elaborated on the original reports of Treacher and Collins.63–67
Mandibulofacial dysostosis is characterized by retrusion of the malar region, obliteration of the frontonasal angle, and a receding chin. Protrusion of the nose and maxilla may produce an enophthalmic appearance. The inferior-lateral angle of the orbit is defective, and the superolateral orbit is displaced caudally, giving the orbit an egg-shaped appearance. The orbital contents appear displaced into the deficiency created by the malar hypoplasia63–67 (Fig. 11).
|
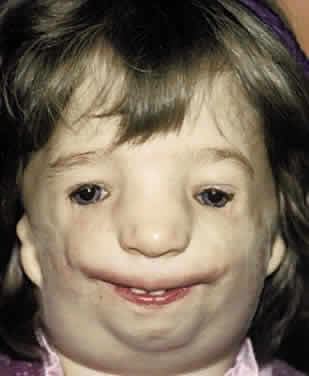
Ocular abnormalities include pseudocolobomas and true colobomas of the lids, especially inferolaterally; canthal dystopia; orbital lipodermoids; limbal dermoids; and, occasionally, microphthalmos and anophthalmos. Strabismus is less common than in the syndromes described earlier but has been reported in several forms including esotropia, exotropia, Duane's syndrome, and cranial nerve palsies.31,63–67 Less frequently, nasolacrimal dysfunction and structural abnormalities of the globe can be present.
An infinite variety of ear abnormalities can be present. Respiration can be affected by choanal atresia and by lingual obstruction from mandibular retrusion.13,63
Facial Microsomia
Facial microsomia is one of the more frequent craniofacial malformations occurring with an estimated frequency of 1:4000 live births.68,69 Only rarely have familial cases been reported. A male predominance has been suggested, which was confirmed by our report of 49 patients with facial microsomias, 73% of whom were male.70 The deformity varies in extent and degree, at times showing a predominance of ear or jaw findings. A characteristic finding is hypoplasia of the mandible on the affected side or sides. The ear deformity ranges from complete absence to a normal ear with additional “ear tags.” The muscles of mastication, subcutaneous tissue, and facial muscles often are hypoplastic. The temporal bone, middle ear, mastoid process, base of the skull, and craniofacial skeleton may be involved and hypoplastic (Fig. 12). A classification scheme has been proposed.67 The hemifacial form accounts for 90% to 94% of facial microsomias (Table 3).
|
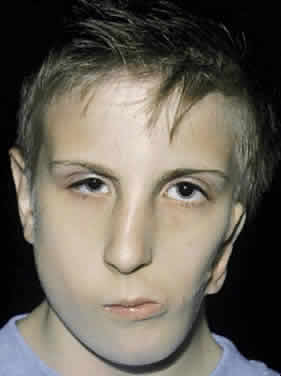
TABLE 3. Classification of Hemifacial Microsomia
| Type | Clinical Manifestations |
| I | Complete facial skeleton, infact intramuscular joint, asymmetric mandible |
| II | Aplasia of the condylar head, hypoplastic ascending ramus, ear deformities |
| III | Type II defects and hypoplastic zygomatic arch, rudimentary glenoid fossa |
| IV | Type III defects and hypoplastic lateral and inferior orbital rims |
| V | Type IV defects and orbital hypoplasia and dystopia, flattened temporal fossa |
Ocular findings in patients with facial microsomia are varied. In a study of 49 patients, ocular or adnexal findings were present in 67% of patients. Sixteen percent of patients had visual loss, all secondary to unilateral amblyopia.70 This resulted from various combinations of uncorrected refractive errors (27%), anisometropia (8%), strabismus (22%), nystagmus (2 of 49 patients), and ptosis (12%). Forty-one percent of patients had associated lid or adnexal abnormalities, most commonly dacryostenosis and ptosis. As in other first and second branchial arch syndromes, incomitant strabismus was common. Of 11 patients with strabismus, 3 had Duane's syndrome, 2 had sixth nerve palsies, and 1 had bilateral superior oblique palsies.70
The absence of more severe systemic features (vertebral, renal, cerebral, multiple cranial nerves) justifies the separation of facial microsomias from Treacher Collins and Goldenhar's syndromes. The etiology may be explained by the animal model of stapedial artery hemorrhage discussed earlier.53,62,63,70
HYPERTELORISM
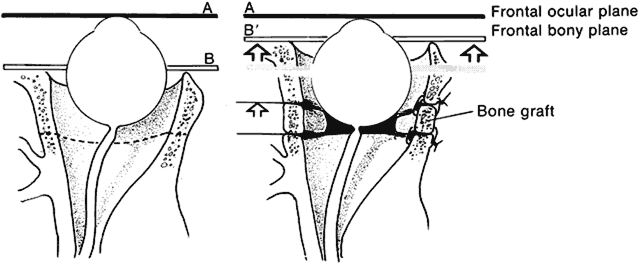
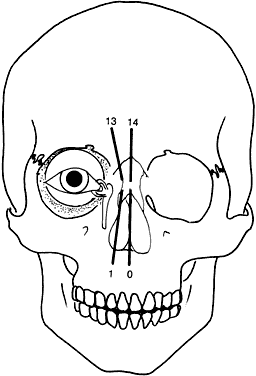
Converse and colleagues stressed the importance of differentiating between hypertelorism and teleorbitism.3 They defined teleorbitism as a congenital condition in which the interorbital distance increases from the apex to the orbital rim (true lateralization of the orbits)1,3,13 (Fig. 13). Hypertelorism, however, shows no true lateralization of the orbits but rather an increase only in the interorbital width. Hypertelorism is differentiated from telecanthus, which refers to a lateral displacement of the soft tissue. Telecanthus can occur without underlying hypertelorism, whereas hypertelorism always has associated telecanthus. Sometimes, a low nasal bridge may be another source of a false impression of hypertelorism. Measurement of the inner canthal distance and outer canthal distance may be used. Hypertelorism is present when the inner canthal distance is increased and the outer canthal distance is normal. Teleorbitism exists when both distances are increased.
|
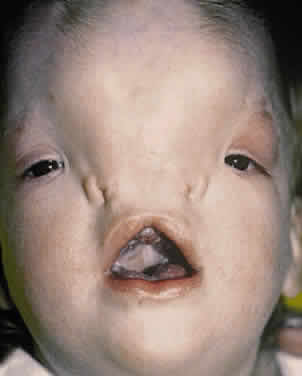
Hypertelorism and telecanthus may present as isolated anomalies but more often are associated with other malformations.1,3,13,71 Teleorbitism is seen most commonly in patients with craniofacial synostosis.72 Other causes of teleorbitism include encephaloceles, pneumatoceles, meningoencephaloceles, and frontonasal dysplasia.72 In teleorbitism, the orbital volumes may be symmetric or asymmetric, and it is more commonly associated with a syndrome.
Greig's syndrome, or primary orbital hypertelorism in combination with other skeletal abnormalities, was the first form of primary hypertelorism to be described.13 This is a rare syndrome (1:100,000). Cohen's syndrome, or craniofrontonasal dysplasia, is more common. Teleorbitism, internasal dysplasia, and maxillary arching are the main characteristics and may be associated with premature coronal synostosis inherited as an autosomal dominant trait.12,13,71
Teleorbitism and hypertelorism often are associated with other anomalies. This includes the brain (agenesis of the corpus callosum, V-shaped separation of the lateral ventricles), the forehead (widow's peak, meningoencephaloceles), the eyebrow (dystopia, distortion), the eye (anophthalmia, microphthalmia, colobomata, strabismus, ptosis, refractive errors, and amblyopia), and the nose (internasal or nasal dysplasia).4,26,31,42,44,53,73