| Although the facial nerve carries both afferent and efferent information, it
is usually the dysfunctional motor component that is brought to
the attention of the ophthalmologist. Clinically, dysfunction of the facial
nerve can be due to disorders of underactivity or overactivity; these
can be further localized anatomically to the supranuclear, nuclear, or
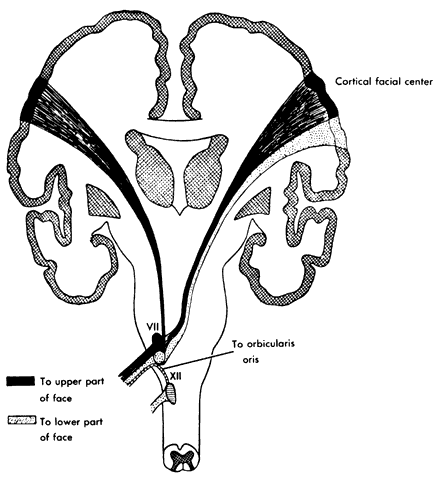
infranuclear segments. DISORDERS OF UNDERACTIVITY Supranuclear Lesions Motor function of the upper face derives from both hemispheres. As a result, supranuclear
lesions involving the face will spare the functions
of forehead wrinkling and eye closure. Rarely, infranuclear lesions have
been described with similar sparing of function, but the accompanying
lateralized neurologic findings involving the ipsilateral body usually
suggest an obvious supranuclear disturbance.12 In addition to the physical examination, computerized tomographic (CT) and
magnetic resonance imaging (MRI) scans allow prompt identification
of these lesions. Motor strip or corticobulbar tract lesions are characterized by paresis
of volitional movement, with preservation of facial tone and involuntary
expressions. These involuntary functions are subserved by extrapyramidal
input that originates in the basal ganglia and traverses extrapyramidal
pathways to the facial nucleus. When these fibers are damaged
in disease states involving the basal ganglia or brain stem (e.g., Parkinson's disease), volitional facial movements usually remain
normal, whereas little facial tone or emotional expression is displayed.3,5,15 The list of possible causes of supranuclear facial paresis is large. The
major categories of causes are vascular, infectious, demyelinating, and
tumorous. Nuclear Lesions (Including Those of the Intrapontine Fascicle) The facial nucleus lies in close proximity to the abducens nucleus and
the para-abducens (paramedian) reticular formation. Consequently, patients
with nuclear lesions often present with abnormalities of contiguous
structures. However, a dissociation between the facial nerve modalities
of somatic motor, visceral motor, and sensation can be seen at the
level of the pons. FOVILLE'S SYNDROME. Patients with Foville's syndrome display peripheral facial weakness, ipsilateral
conjugate horizontal gaze paresis, and contralateral hemiparesis. Lesions
associated with these clinical features extend from
the facial nucleus to the abducens nucleus and paramedian pontine reticular
formation dorsomedially, and into the corticospinal tracts ventrally.20 The paramedian reticular formation can be thought of as the conjugate
gaze center of the brain stem, which is why the gaze paresis is conjugate
rather than just an ipsilateral abducens palsy. MILLARD-GUBLER SYNDROME. In Millard-Gubler syndrome there is a combination of contralateral hemiparesis, abducens
palsy, and variable facial nerve palsy.20 Lesions associated with these clinical features lie more ventrally in
the brain stem than those of Foville's syndrome, and they cause an
infranuclear abducens palsy rather than a nuclear disruption. Ipsilateral
conjugate gaze paresis, as seen in Foville's syndrome, is absent. Although Foville's syndrome and Millard-Gubler syndrome are of historical
interest, they represent only signatures of brain stem involvement
in which there are varying degrees of nuclear and corticospinal tract
dysfunction. Clinically, if a patient has both peripheral facial weakness
and eye findings, the presence of a central lesion should be considered
and signs of contralateral hemiparesis should be inspected. Stroke
and infiltrating tumor are the two most common causes; sudden onset
suggests the former and subacute progression the latter. MÖBIUS' SYNDROME. Harlan, writing in 1881, described the first patient. In 1892, Möbius
collated a number of cases, bringing greater recognition to the syndrome
that now bears his name. Peripheral facial weakness and abduction
deficits are central features of this syndrome, and in the majority
of cases these findings are bilateral. A host of associated features
have been described, including other cranial nerve palsies, ptosis, musculoskeletal
abnormalities, cardiac anomalies, craniofacial defects, and
mental retardation.21 Nuclear agenesis of nerves VI and VII is considered the classic syndrome, but
investigators have found cases in which the abnormality was nuclear, neuropathic, or
myopathic.22,23 Furthermore, the etiologies postulated to date include congenital hypoplasias, vasculopathies, genetics, infections, neuronal degeneration, and
muscular dystrophy.24–26 Imaging studies in nuclear cases, when positive, have shown brain stem
atrophy and caudal pontine calcifications. At necropsy, the calcifications
in one patient correlated with necrotic areas.27 Treatment of the facial diplegia associated with this syndrome has been
successful. Treatment involves the use of muscle transplants that are
innervated by transposition of either cranial nerves V or XII.28 COMPLICATIONS OF AIDS. Supranuclear, nuclear, and infranuclear lesions can also be found in AIDS
patients. The meningeal space is a common site of infranuclear involvement. Depending
on the clinical location of disease, CT/MRI, cerebrospinal
fluid analysis, and serologic tests are very important ways of
differentiating possible causes. Consideration should be given to the
presence of toxoplasmosis, lymphoma (intraparenchymal and meningeal), neurosyphilis, tuberculosis, fungus (especially Cryptococcus), and viruses (HIV, cytomegalovirus, and progressive multifocal leukoencephalopathy). A study by Keane29 found that 50 of 2030 (2.5%) AIDS patients admitted to the hospital had
neuro-ophthalmologic pathology; of these 50 patients, 7 had a peripheral-type
facial paresis, all due to lesions of the pontine tegmentum. Infranuclear Lesions When facial weakness is progressive and of a peripheral nature, lesions
along the course of the facial nerve must be considered. By testing the
various facial nerve functions, these lesions can be clinically localized, and
specific neuroradiologic techniques can be used to define
further the extent of involvement and suggest a cause. Facial weakness
is bilateral in approximately 1% of cases.30 CEREBELLOPONTINE ANGLE LESIONS. As noted above, cranial nerves VII and VIII are enclosed in a common sheath
as they leave the brain stem in the cerebellopontine angle on their
way to the internal auditory canal. Thus, a combination of progressive
facial weakness, tinnitus, hearing loss, dizziness, and periorbital
dysesthesias (trigeminal nerve involvement) should alert the clinician
to a lesion in this area. Careful evaluation of facial function may
also uncover dysfunction of modalities carried by the nervus intermedius. Tumors of the cerebellopontine angle are generally of a benign histologic
character. The most common tumors are acoustic neuroma, meningioma, and
epidermal cyst. Extra-axial in location, these tumors grow slowly, which
is why dysequilibrium, rather than true vertigo, is most often
reported. The advent of MRI has made evaluation of the cerebellopontine
angle simple. Initial workup may also include an audiogram and brain
stem auditory evoked potentials.31 BELL'S PALSY (IDIOPATHIC FACIAL PALSY). Since Sir Charles Bell's classic descriptions of peripheral facial
weakness, a voluminous body of literature has developed on the possible
etiologies and treatment modalities of the idiopathic variety of facial
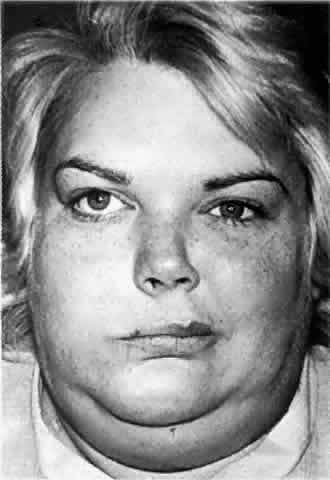
weakness.5,18,32–35 Clinical Features. Bell's palsy, by far the most common type of facial palsy, is a disease
that typically affects adults 20 to 40 years of age, but persons
of any age are susceptible. Men and women are equally affected. It is
characterized by an acute unilateral infranuclear facial nerve paresis (see Fig. 4). Maximal deficits are usually reached within 2 to 3 weeks, but the majority
are maximally affected by 2 to 5 days. In 50% of patients there
will be a complaint of retromastoid pain preceding, or concurrent with, the
onset of paresis.35 Many patients complain of either increased or decreased lacrimation; a
smaller group of patients report having a subjective feeling of numbness, despite
intact sensory testing. The explanation for a complaint of
increased lacrimation is not that there is true increased production,36 as demonstrated by normal Schirmer's test results. Rather, laxity
of the lower lid prevents normal flow of tears toward the lacrimal duct, and
the tears spill over. A subjective feeling of diminished taste
or perverted taste (parageusia) on the involved side is reported by 30% of patients. It is often surprising to find other cranial nerve signs, such as altered
facial sensation, corneal hypesthesia, or tongue deviation in a case
that is otherwise typical of Bell's palsy. Broadly defined, this
might not cause alarm if one considers Bell's palsy to be a viral
disease, and thus capable of causing a mononeuritis multiplex.36 If, however, one uses a more narrow definition, patients with more than
just facial nerve involvement probably should be diagnosed as having
idiopathic cranial polyneuritis.37 Bell's phenomenon, a normal upward deviation of the eye with attempted lid closure, is easily
seen when the orbicularis oculi muscles are paralyzed. Although the cause of Bell's palsy is unknown, an infectious process
probably accounts for the majority of cases; vascular and genetic causes
account for some cases. It is seen more commonly among diabetics, hypertensives, and
pregnant women. Few early pathologic examinations
have been done for obvious reasons. Liston38 examined the facial nerve 1 week after onset and found demyelination, axonal
changes, and inflammatory cells consistent with a viral cause. Other
investigators have not found a cellular infiltrate. Not all facial paresis is Bell's palsy. In fact, the differential
diagnosis is quite large.39,40 If any other features in the history or examination cannot be explained
by seventh cranial nerve dysfunction, alternative diagnoses should be
sought. Additional worrisome features include recurrence, bilaterality, subacute
progression, otologic findings, history of trauma, or central
nervous system dysfunction. Clinical Course. Overall, 70% to 80% of patients will have complete spontaneous recovery.19,36,39,41 The majority of the remaining patients will be left with partial paresis, and
less than 5% will have no recovery. Several early clinical features
predict which patients will have a worse outcome: total paralysis, dry
eye, dysacusis, and age greater than 60. The first sign of recovery
is gradual return of eyelid closure. Failure to achieve this within 8 to 12 weeks
after the onset of symptoms makes complete recovery unlikely. Electrodiagnostic studies have also been able to make similar predictions
as to poor prognosis. Poor outcome is predicted by diminished salivary
flow, absence of voluntary motor units on electromyography, decreased
compound action potential, and increased nerve excitability threshold.42 These studies are rarely done in the routine clinical setting because
of the lack of a clearly beneficial treatment modality that will change
the course of the disease. A few investigators, however, believe that
surgical decompression of the facial nerve in the worst cases is useful.43 Several other problems, both mechanical and cosmetic, can occur in patients
who don't obtain full recovery. Contracture of the previously involved
side may draw the mouth upward, increase the nasolabial fold, and
decrease the palpebral fissure. It may appear as if the unaffected side
is now paretic. Synkinesis, “crocodile tears,” and hemifacial
spasm are discussed below in the Disorders of Overactivity section. Treatment. Over the years, steroids have not invariably shown a benefit in the treatment
of Bell's palsy. Nonetheless, they are used frequently, and
summation of the data points toward a benefit, especially if used early
in the course.44 They may help prevent progression, hasten recovery, and prevent synkinesis. In
patients without a specific contraindication, a typical prednisone
course is 1 mg/kg/day for 5 days, followed by a tapered dosage for
an additional 5 days. As noted above, surgical decompression cannot
be recommended,45 although the future may define a special subgroup who will benefit. The mainstay of therapy remains close ophthalmologic observation for corneal
exposure and the use of ocular emollients in patients with a poor
or absent blink. Although ocular shields that moisturize the eye may
be quite helpful, patching or closing the eye with tape at night, unless
performed with meticulous care, adds little to the instillation of
emollients and may actually lead to pressure breakdown of the corneal
epithelium. If corneal breakdown appears likely, a soft contact lens or temporary tarsorrhaphy produced
either surgically or with botulinum toxin is indicated to provide ocular
protection. In patients who have experienced a poor recovery, muscle feedback training
and nerve transfers have been successful.46 IDIOPATHIC CRANIAL POLYNEURITIS. As the name suggests, multiple cranial nerve palsies are present at the
same time in this condition. The abducens nerve is most frequently affected, but
any combination is possible with the exception of the olfactory
nerve. In a retrospective review by Juncos and Beal,37 the facial nerve was found to be involved in 4 of 14 cases. Face or head
pain was almost invariable, and in one case this finding preceded any
nerve deficits by more than 3 months. The disease is self-limited, but
it tends to recur. Steroids are the mainstay of treatment. Idiopathic cranial polyneuritis should be distinguished from Guillain-Barré syndrome (see
Acute Idiopathic Polyneuritis section), carcinomatous
meningitis, and identifiable inflammatory or infectious causes. TRAUMATIC FACIAL NERVE PALSY. Trauma to the head may result in facial paresis, which can often be hard
to detect if accompanied by facial swelling. Cases of both immediate
and delayed paresis (up to days after the trauma) are seen. The natural
history is for both groups to do well, although the delayed group
probably fares a little better.47 CT scanning to look for temporal bone fractures should be performed. Longitudinal
fractures are more common than transverse and seem to have
a better prognosis. Although controversial, surgical exploration in cases
of immediate-onset total paralysis together with a transverse fracture
should be seriously considered.48 Electrodiagnostic studies can be helpful, as in some cases of Bell's
palsy.42 RAMSAY HUNT SYNDROME (GENICULATE HERPES, OTITIC HERPES). The association of facial paresis with herpetic eruptions along the ipsilateral
external auditory meatus constitutes the Ramsay Hunt syndrome49,50 (Fig. 5). Patients frequently give a history of a recent viral syndrome and auricular
pain that preceded the facial weakness and vesicular eruption. The
extent of the herpetic involvement may not be limited to the distribution
of the facial nerve. Other cranial nerve palsies in a mononeuritis
multiplex fashion can be seen. Vesicles can involve any aspect of
the ipsilateral face indicating trigeminal nerve involvement, and auditory
and vestibular symptoms are frequent. The pain associated with
this viral eruption is typically severe and often persists for weeks. 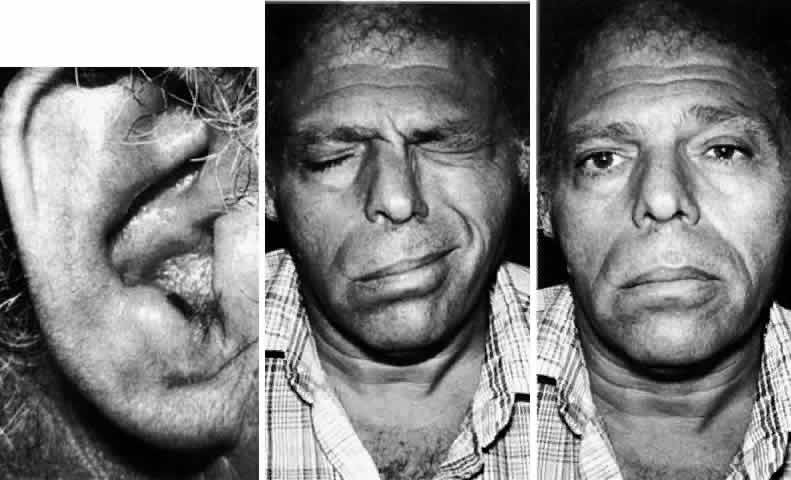 Fig. 5. Ramsay Hunt syndrome. Left. Healing herpetic vesicle in the external meatus. Middle. Right facial paresis, incomplete with residual orbicularis oculi closure. Right. Mild facial asymmetry noted at rest. Fig. 5. Ramsay Hunt syndrome. Left. Healing herpetic vesicle in the external meatus. Middle. Right facial paresis, incomplete with residual orbicularis oculi closure. Right. Mild facial asymmetry noted at rest.
|
The malignant nature of this condition is highlighted by reports showing
incomplete recovery in 40% to 70% of patients who received either no
treatment or only steroid treatment.51–54 The accuracy of these findings, however, is questionable because large
prospective trials have not been done. Also, a significant percentage
of patients develop a postherpetic neuralgia after resolution of the
skin lesions. Currently recommended treatment is a combination of steroids and acyclovir, although
a definitive prospective randomized trial has not been done.52 Several case studies53–55 have shown better results with acyclovir compared to the outcome of historical
controls. The dose, route, and length of treatment has not been
worked out, but a course of prednisone as used for Bell's palsy
cases is reasonable. Whether a dosage of 5 mg/kg intravenous acyclovir
every 8 hours for 5 to 7 days, followed by 800 mg acyclovir PO 5 times
a day for 7 days, should be given, or whether the whole course can
be given orally, is not clear. Zoster sine herpete is a condition in which varicella zoster titers rise, but
vesicles never develop.56 One approach would be to treat all patients with facial palsy and severe
auricular pain with acyclovir, so as not to miss treatment of this
condition. MELKERSSON-ROSENTHAL SYNDROME. MelkerssonRosenthal syndrome comprises a triad of recurrent infranuclear
facial paralysis, orofacial edema (predominately of the lips), and
lingua plicata.57,58 Not all three features need be present; only very rarely do they appear
in combination. Onset can be at any age. The swelling may be bilateral, despite
unilateral facial paresis, or it may occur independent of
the facial paresis, with the edema antedating the weakness by months to
years.35 Cheilitis granulomatosis is seen on lip biopsy and helps confirm the diagnosis. Several
causes have been put forth, such as inheritance,59 infection, autoimmunity, and allergic reaction.60,61 Treatment includes several drugs, such as clofazimine and steroids, as
well as the surgical reduction of granulomatous tissue.58 UVEOPAROTID FEVER (HEERFORDT'S DISEASE). As the name suggests, in its full presentation uveoparotid fever is characterized
by uveitis, parotitis, and mild pyrexia.62 The facial nerve is the most commonly involved cranial nerve in sarcoidosis,63 and it is affected in 50% of cases of uveoparotid fever. Although frequently
asymmetric in extent, the facial weakness can be bilateral.29,64 The site of facial nerve inflammation is often within its path through
the parotid gland, although it can be anywhere along its course, as demonstrated
by some patients with parageusia and reduced lacrimation.62,65 The cause is probably nerve infiltration with noncaseating granulomatous
material. The diagnosis is made on the basis of the appearance of bilateral, asymmetric, peripheral facial paresis accompanied by other systemic
signs or symptoms of sarcoidosis as well as a positive tissue
biopsy. An elevated serum angiotensin-converting enzyme (ACE) level can
be helpful. ACUTE IDIOPATHIC POLYNEURITIS ( GUILLAIN-BARRÉ SYNDROME). Although the typical presentation of Guillain-Barré syndrome is
an ascending paresis with depressed tendon reflexes, there are several
variants that involve the cranial nerves to a greater extent.66 In 1956, Fisher67 described the cases of three patients with ophthalmoplegia, ataxia, and
areflexia. Another less common variant is facial diplegia with distal
paresthesias. The acute or subacute onset of any combination of these
signs should alert the clinician to this potentially fatal disorder, since
respiratory and autonomic involvement may occur. Classically, motor
nerve conduction studies reveal slow responses, and cerebrospinal
fluid examination shows cytoalbuminologic dissociation. PROGRESSIVE HEMIFACIAL ATROPHY (PARRYROMBERG SYNDROME). Progressive hemifacial atrophy is clearly a syndrome, rather than a specific
disease. The unifying characteristic of all cases over the past 150 years
is acquired hemifacial atrophy, which must be distinguished
from congenital forms68 and bilateral lipodystrophy. Additional manifestations have included hyporeflexia, seizures, trigeminal anesthesia, and dementia as well as
the ophthalmologic manifestations of enophthalmos, lid atrophy, tonic
or irregular pupils, Horner's syndrome, Fuchs's heterochromic
cyclitis, retinal vascular abnormalities, scleral melting, extraocular
muscle imbalance and palsies, and bony defects of the inferior orbital
rim and floor.69–73 One case of prolonged follow-up for 43 years has been reported.74 Typically, patients present in the first or second decade of life, when
they notice that their face appears lopsided (Fig. 6). This early facial asymmetry progresses for 2 to 10 years to variably
involve the skin, subcutaneous tissue, muscle, cartilage, and bone. A
common coup de sabre deformity in the forehead (not shown in Fig. 6) represents the demarcation between normal and abnormal tissue. Rarely, the
ipsilateral body is also affected. 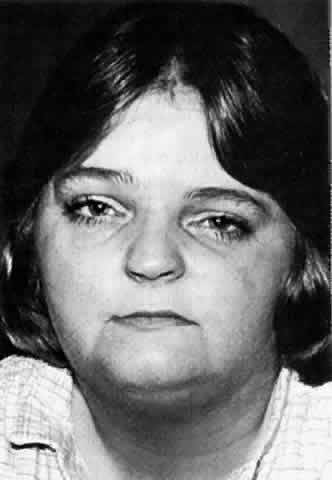 Fig. 6. Left hemifacial atrophy in a 30-year-old woman with mild to moderate atrophy
and an associated left abduction deficit (not shown). Fig. 6. Left hemifacial atrophy in a 30-year-old woman with mild to moderate atrophy
and an associated left abduction deficit (not shown).
|
Several explanations for the progressive atrophy have been put forth, and
all of them may be correct. These include trauma, sequelae of irradiation, infection, scleroderma, sympathetic dysfunction,75 lupus erythematosus,76 trigeminal neuropathy, and lymphocytic neurovasculitis.73,77–79 Recent microscopic and biochemical analyses of connective tissue are beginning
to find clues.77,79 MRI data were reported recently by Terstegge and associates.80 In cases in which MRI abnormalities were found, a high majority of these
abnormalities were ipsilateral to the hemifacial atrophy, supporting
the lymphocytic neurovasculitis theory. Cerebral hemiatrophy was the most common finding; gliosis, cortical calcification, and
meningeal enhancement were also seen. These findings
help explain why seizures, when present, are most often contralateral
to the facial atrophy. The overwhelming majority of MRI findings were
seen in patients with symptoms involving the central nervous system; most
asymptomatic patients had normal scans. Currently, treatment is limited to either chemotherapy, if an underlying autoimmune cause is found, or surgical reconstructive techniques after progression of the atrophy has halted.81 Surgical options include free flaps and lipofilling. DISORDERS OF OVERACTIVITY In any evaluation of unusual facial movements, the synkinetic movements
seen after aberrant regeneration and the reflex grimacing movements created
by trigeminal irritation must be considered first. Synkinetic Movements Synkinesis is defined as an unintentional movement following the initiation of volitional
movement. The term is most often applied to the mass movements
that follow incomplete recovery from Bell's palsy. (It also has
been used, perhaps inaccurately, to describe hemifacial spasms that can
occur spontaneously, without prior volitional movement.) Axonal compression or disruption along the course of the facial nerve may
lead to involuntary or synkinetic movements. Several theories have
been put forth, including facial nuclear reorganization, aberrant regeneration, ephaptic (false
synapse) transmission, and kindling. All explain
some components of involuntary facial movements, and electrophysiologic
studies provide support for each theory.82–86 No single theory, however, is able to explain all aspects of this complicated
subject. Only a simple overview will be provided here (please
see the appropriate references for more details). Facial nuclear reorganization refers to the process whereby deafferentation changes nuclear inhibitory
connections, thereby “unmasking” reflex movements.84 The complex nature of synkinesis is best explained by a nuclear origin. Aberrant regeneration refers to the resprouting of axons down incorrect myelin sheaths after
nerve disruption. Ephaptic transmission refers to the concept of an “artificial synapse” between contiguous
nerves, caused by the lateral spread of extra-axonal current
through the interstitium.82,83 Current spread may occur between efferent fibers, or from afferent to
efferent fibers, and the impulse may be transmitted in either direction
along the nerve. Ultimately, misdirected neural firing leads to the
synchronous contraction of unassociated muscle groups. Finally, kindling incorporates the concepts of both ephaptic transmission and nuclear reorganization, whereby
an antidromic impulse from the ephapse of the nerve
activates the facial nucleus, which then coordinates facial muscle
contraction.86 As noted in the section on Bell's palsy, several complications involving
the above mechanisms can occur after peripheral facial nerve disruption. Eye
closure when eating or smiling as well as elevation of the
corner of the mouth upon blinking are the most common forms of synkinesis, and
become apparent within weeks to months of the insult. Unilateral
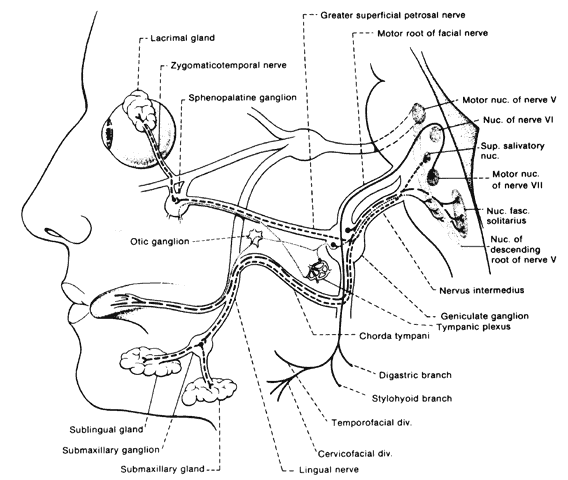
lacrimation while eating is a complication referred to as crocodile tears and is an example of aberrant regeneration. Fibers of the nervus intermedius
destined originally for the submandibular ganglion, become misdirected
to the lacrimal gland via the greater superficial petrosal nerve; this
suggests involvement of the facial nerve prior to the geniculate
ganglion.87 Hemifacial spasm is a rare, if at all existent, complication of facial
nerve palsy, and may be very hard to distinguish from the synkinetic movements
that follow facial nerve palsy.45 In addition to the history of previous palsy, close observation may help
with this distinction, since the synkinesias accompanying Bell's
palsy are invariably produced only after voluntary movement, whereas
hemifacial spasms can be self-perpetuating, as noted above.88 In the more mild forms of synkinesis, reassurance is the best treatment. When
the contractures are cosmetically upsetting, botulinum toxin is
the treatment of choice.89,90 Reflex Blepharospasm Irritation of any branch of the trigeminal nerve may lead to a bilateral
orbicularis oculi reflex spasm. This may be incorrectly labeled as essential
blepharospasm initially (see Essential Blepharospasm section), resulting
in potentially treatable underlying disorders going unrecognized. Patients
with anterior segment ocular inflammation or meningeal
inflammation (caused by subarachnoid hemorrhage or meningitis) may present
with referred pain in the first trigeminal division and reflex
blepharospasm. Eye irritation and photophobia may be initial complaints. Patients with reflex blepharospasm do, however, have volitional control
over their squinting. Because the lid closure is secondary rather than
primary, they prefer to keep their eyes closed; however, in the disorders
of overactivity to be discussed next, patients present with a primary
complaint of eyelid closure. Supranuclear Disorders HABIT SPASM OF THE FACE (NERVOUS TWITCH, FACIAL TIC). Although frequently confused clinically with blepharospasm or hemifacial
spasm, habit spasm of the face can be easily identified. The onset
typically occurs in childhood and is characterized by stereotypical, repetitive
facial movements that are reproducible and can be promptly inhibited
on command. Motor tics can occur as an isolated disorder, or
as a component of Tourette's syndrome. Treatment may be as simple
as reassurance, or it may require more aggressive drug therapy. FOCAL CORTICAL SEIZURES. Rarely, epileptiform discharges arising from the facial cortex of the
motor homunculus can manifest as gross clonic movements of the contralateral
face. These movements, when closely inspected, are seen to involve
contiguous cortical areas that serve a distribution beyond the facial
nerve. Postictally there can be a supranuclear type of paresis (e.g., Todd's paresis), representing exhaustion of the cortical tonic input. These
patients should be considered to have focal cortical disease, and
prompt neuroanatomic studies should be carried out to direct the
appropriate treatment course. ESSENTIAL BLEPHAROSPASM. Essential blepharospasm is a form of cranial dystonia limited to the orbicularis
oculi muscles. Age of onset is between 45 and 60 years and
is more prevalent in women than in men; excessive blinking is the usual
first symptom.91–93 This blinking gradually intensifies in character, insidiously becoming
a spasm of the eyelid that is not under volitional control (Fig. 7). Although involvement may appear unilateral in early stages or far into
the disease course, causing some diagnostic confusion with habit spasm
or hemifacial spasm, bilateral impairment is always found eventually. As
the disease progresses, the eye closure may become so frequent
and prolonged that the patient is functionally blind and may withdraw
from all social contact. 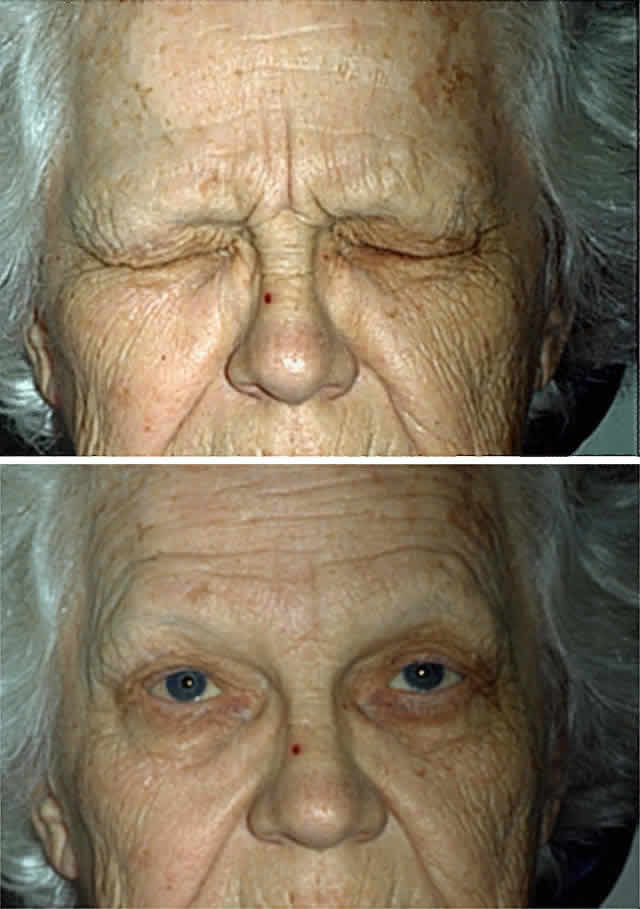 Fig. 7. Essential blepharospasm. A. Before treatment. B. After botulinum toxin injection of the orbicularis oculi muscles. Fig. 7. Essential blepharospasm. A. Before treatment. B. After botulinum toxin injection of the orbicularis oculi muscles.
|
Because essential blepharospasm and more widespread dystonic conditions
such as Meige's syndrome probably are part of a spectrum of the
same disease (cranial dystonia), clinical features, treatment, and pathophysiology will be discussed
together in the next section. MEIGE'S SYNDROME (BLEPHAROSPASM-OROMANDIBULAR DYSTONIA, OROFACIAL-CERVICAL
DYSTONIA, BRUEGHEL'S SYNDROME). Dystonic involvement of the lower cranial muscles (mouth retraction, jaw
opening or closing, facial grimacing), neck, vocal cords (spastic dysphonia), and
limbs is often referred to as Meige's syndrome.94 Also, in a study of 100 patients by Jankovic and Ford,95 a full 30% had tremor. Although the most frequent presenting sign is blepharospasm, it
is not uniformly present.95,96 Full expression of the syndrome often takes many years. Initial complaints in cranial dystonia are often dry eyes, ocular pain, and
photophobia. Exacerbating features include stress, bright lights, social
interaction, driving, reading, watching television, and wind. Relief
may be found in the morning or after rest, and by using sensory “ricks” such
as touching the side of the eye, singing, humming, yawning, sucking, chewing, drinking alcohol, extending the neck, and
wearing dark glasses.93,95,97 Cranial dystonia can be confused with tardive dyskinesia, which has a
more choreiform movement rather than the sustained postures of dystonia, although
overlap does occur. The presumed cause of cranial dystonia is an upset in the normal dopamine
balance in the basal ganglia and brain stem.98–100 The reason for differences in the extent of cranial and limb involvement
is not understood. Evidence for the dopamine hypothesis comes from
a complicated body of literature.93,95,97,101–103 These studies have reported on (1) different responses to pharmacologic
agents that exert their effect on the basal ganglia; (2) associated
conditions that affect the basal ganglia and can produce secondary dystonia, including
Wilson's disease, encephalitis lethargica, and levodopa
or neuroleptic use; (3) associated conditions that affect the
upper brain stem, including strokes and multiple sclerosis; and (4) pathologic
reports of cases in which abnormalities when evident have inconsistently
found gliosis and cell loss in the caudate, putamen, substantia
nigra, locus ceruleus, midbrain tectum, and dentate nuclei. Satisfactory treatment for blepharospasm is best accomplished with botulinum
A toxin injected into the muscles around the eye. Other muscles
of the face and neck can be injected, depending on the extent of dystonia, but
a satisfactory response is less likely than for pure essential
blepharospasm.90,104 The presynaptic nerve terminal is the site of action, where botulinum
toxin binds to acetylcholine-containing vesicles to prevent exocytosis. Muscle
paralysis is the result, with a peak effect at 5 to 7 days and
a duration of effect of between 10 and 16 weeks. Side effects occur
in approximately 25% of patients and include ptosis, lagophthalmos, entropion, epiphora, diplopia, bruising, and lower facial weakness.90 Changes at the neuromuscular junction have been reported by several investigators,105,106 but the importance of these changes and whether or not they are reversible
is debatable. Oral medications have become a second-line treatment, which is fortunate
considering that most patients respond incompletely or not at all to
this treatment. Several different medications have been tried, such as
tricyclic antidepressants, anticholinergics, neuroleptics (including
clozapine), dopamine depleters (reserpine, tetrabenazine), levodopa, cholinergics, clonazepam, baclofen, and lithium.92,95,97,107 One reason why such a varied number of drugs have been used in an attempt
to treat cranial dystonia is that this disorder was initially considered
a psychiatric illness. For the few patients who do not respond
to pharmacotherapy, surgical options include orbicularis myectomy, peripheral
facial nerve avulsion, and peripheral facial neurectomy.108 Nuclear Disorders FACIAL MYOKYMIA. The pathophysiology of facial myokymia needs to incorporate the variety
of conditions known to be associated with it, including brain stem tumors, pontine
tuberculoma, cerebellopontine angle tumors, carcinomatous
meningitis, sarcoidosis, syringobulbia, subarachnoid hemorrhage, multiple
sclerosis, Guillain-Barré syndrome, cysticercosis, timber
rattlesnake envenomation, hypoparathyroidism, and cardiopulmonary arrest.109–115 Unknown specific changes in the microenvironment of the motor neuron or
its axon due to edema, demyelination, toxins, ischemia, destruction, or
metabolic alterations are proposed.110,111,113–115 This may be due to direct damage or disruption of regulatory neurons. Pathologic
studies have documented cases of nuclear edema or damage, but
other studies have also documented cases of disease above or below
the facial nucleus without direct involvement.114,116 Myokymia is a continuous, undulating, involuntary movement of the facial muscles
involving predominantly the periocular and orbicularis oris musculature (Fig. 8). It is usually unilateral. This movement disorder is perceived by the
patient as a “nervous twitch” and rarely arises in isolation
of other neurologic signs or symptoms. Spastic paretic facial contracture
may accompany myokymia in intraparenchymal brain stem cases.111,117 In the absence of other obvious causes, the most likely causes are pontine
glioma in children less than 10 years old and multiple sclerosis
in persons older than 15. Facial myokymia should be differentiated from
the transient twitches of extreme fatigue, the rhythmic contractions
seen in hemifacial spasm, facial tics, focal seizures, and the synkinetic
movements that follow facial palsy. 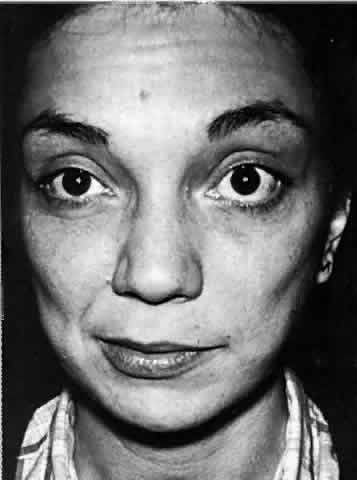 Fig. 8. A 30-year-old woman with right facial myokymia and a right internuclear
ophthalmoplegia as the initial presentation of demyelinating disease. Note
the “puckering” of the right cheek. Fig. 8. A 30-year-old woman with right facial myokymia and a right internuclear
ophthalmoplegia as the initial presentation of demyelinating disease. Note
the “puckering” of the right cheek.
|
Treatment of this condition should be aimed at the underlying pathology. Symptomatic
pharmacologic treatment options include carbamazepine, phenytoin, and
most recently, botulinum toxin.90 Infranuclear Disorders HEMIFACIAL SPASM. Rhythmic, intermittent, unilateral facial twitching, which begins insidiously
around the orbicularis oculi and spreads slowly over 1 to 5 years
to involve all the muscles of facial expression, is characteristic
of hemifacial spasm (Figs. 9 and 10). These bursts of clonic activity may last only seconds or eventually
may become tonic and continue for periods of minutes to hours. The prolonged, severe
contractions of facial musculature lead to an annoying, and
frequently socially disfiguring, grimacing appearance with partial
eyelid closure. Although this is usually painless, patients may complain
of mouth or neck pain during severe contractions as well as the occasional
sensation of oscillopsia and auditory discomfort during lid
and tensor tympani spasm.118 Often, voluntary movements such as smiling, eating, talking, or raising
the eyebrows may precipitate involuntary spasms. Spasms can be exacerbated
by stressful situations and may occur during sleep. Ultimately, progression
is the rule; however, the course is variable, some patients
having spontaneous exacerbations and remissions.93,97 Mild facial weakness can be seen, and because of the facial nerve's
proximity to cranial nerve VIII, hearing loss, tinnitus, and vertigo
may also occur.85,119  Fig. 9. A 50-year-old man with left hemifacial spasm. Note the marked involvement
of the orbicularis oris. Fig. 9. A 50-year-old man with left hemifacial spasm. Note the marked involvement
of the orbicularis oris.
|
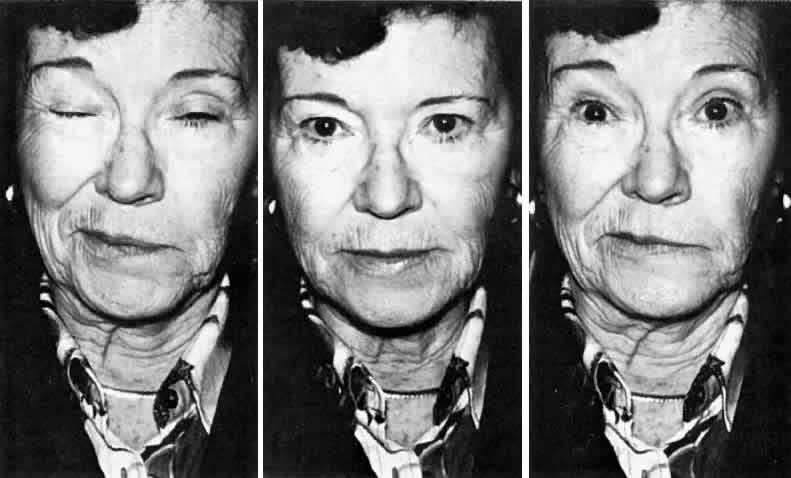 Fig. 10. A 52-year-old woman with right hemifacial spasm secondary to facial nerve
compression from a large ectatic vertebral artery. Right. Spasm beginning in the orbicularis oris. Middle. At rest. Left. Spasm spreading within seconds to involve the entire facial nerve distribution (including
the platysma). Fig. 10. A 52-year-old woman with right hemifacial spasm secondary to facial nerve
compression from a large ectatic vertebral artery. Right. Spasm beginning in the orbicularis oris. Middle. At rest. Left. Spasm spreading within seconds to involve the entire facial nerve distribution (including
the platysma).
|
The varied causes that can produce hemifacial spasm have in common the
ability to produce ephaptic transmission at some point along the nerve. By
far the most common site of disruption is at the root exit zone of
the facial nerve. In most cases, aberrant vascular loops of the basilar, posterior
inferior cerebellar, anterior inferior cerebellar, vertebral, or
internal auditory artery compress the facial nerve at its root (Fig. 11). Other known causes include cerebellopontine tumors, aneurysms, arteriovenous
malformations, and aberrant venous structures; in some reported
cases, the cause was undetermined.88,120,121 Only rarely is there an antecedent history of ipsilateral facial palsy. Tic convulsif is the term given to the combination of trigeminal neuralgia and hemifacial
spasm, and a common structural cause is often found. MRI and MR
angiography have greatly increased our ability to document the cause of
hemifacial spasm prior to surgery. 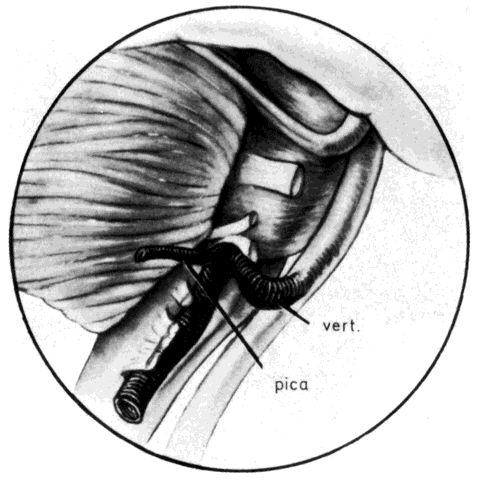 Fig. 11. View of the left brain stem showing vessel causing cross-compression of
the anterior caudal aspect of the root exit zone of the facial nerve. vert. = vertebral artery; pica = posterior inferior cerebellar artery. (Jannetta PJ: Hemifacial spasm. In Samii M, Jannetta PJ (eds): The Cranial
Nerves, p 486. New York, Springer-Verlag, 1981) Fig. 11. View of the left brain stem showing vessel causing cross-compression of
the anterior caudal aspect of the root exit zone of the facial nerve. vert. = vertebral artery; pica = posterior inferior cerebellar artery. (Jannetta PJ: Hemifacial spasm. In Samii M, Jannetta PJ (eds): The Cranial
Nerves, p 486. New York, Springer-Verlag, 1981)
|
Treatment. Surgical decompression of the facial nerve, in which a nonabsorbable sponge
is placed between the nerve and the offending artery, is the only
permanent treatment and has an 85% success rate.88,120,121 Occasionally a second operation is needed. Some authors have suggested
that this procedure can be beneficial even in the absence of an offending
vessel, suggesting that the real benefit comes from manipulation
of the nerve and subsequent fibrosis.122 Unfortunately the long-term complication rate is 16% and includes facial
palsy and hearing loss.121 Nonsurgical management is most successful with botulinum toxin, and given
its lack of permanent complications, one could argue that a therapeutic
course with this agent should be tried in all patients before surgery
is considered.90,123 Alternative drug trials, which are effective 25% of the time, include
carbamazepine, phenytoin, baclofen, clonazepam, and anticholinergics.85,93 |