GENERAL FEATURES
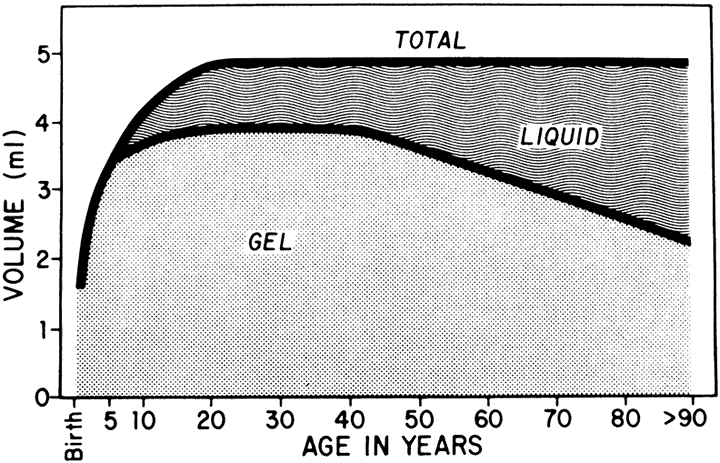
The vitreous of the normal human eye weighs approximately 4 g and occupies a volume of almost 4 ml. The precise weight and volume vary with the age and the size of the eye. The vitreous body is spherical with a depression in the anterior surface, the patellar fossa, corresponding to the posterior surface of the crystalline lens. Larsen1 ultrasonically measured the axial length of the vitreous body in 926 children. He noted that the mean value in newborns was 10.48 mm in boys and 10.22 mm in girls. A sex difference of 0.23 to 0.38 mm greater mean length was noted in males at birth and persisted throughout the growth period. By the age of 13 years, when the axial growth of the vitreous body is essentially completed, the average length was 16.09 mm in males and 15.59 mm in females. Larsen further noted that the relationship between vitreous length and refraction was already established by 1 year of age.
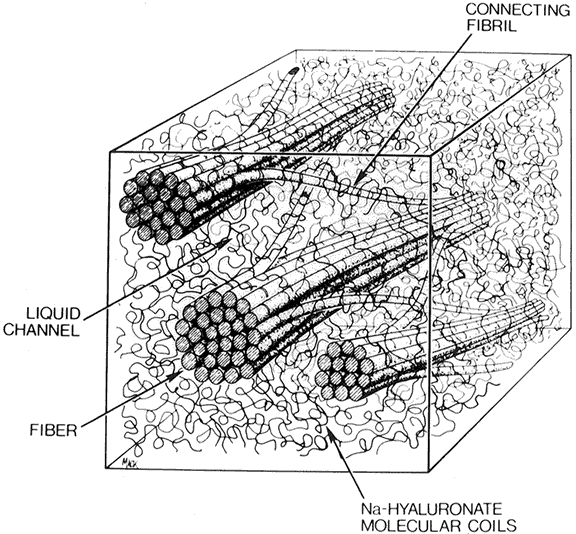
The vitreous body can be divided into two zones or regions. The more peripheral zone, the cortical vitreous, encases the medullary vitreous. The cortical vitreous consists of a relatively more condensed, fibrillar vitreous. Although the cortical vitreous represents only 2% of the total vitreous volume, it is the metabolic center of the vitreous body, because it contains the hyalocytes.2,3 Another connective tissue cell, the fibrocyte, is also found in the cortical vitreous. Most of the vitreous body, the medullary vitreous, is essentially a cell-free mixture of collagens and hyaluronic acid (HA) existing either in a gel or a liquid state depending on the age, refraction, and condition of the eye.
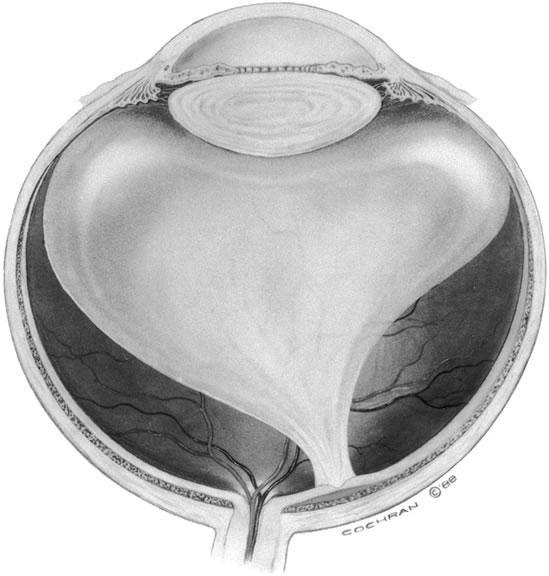
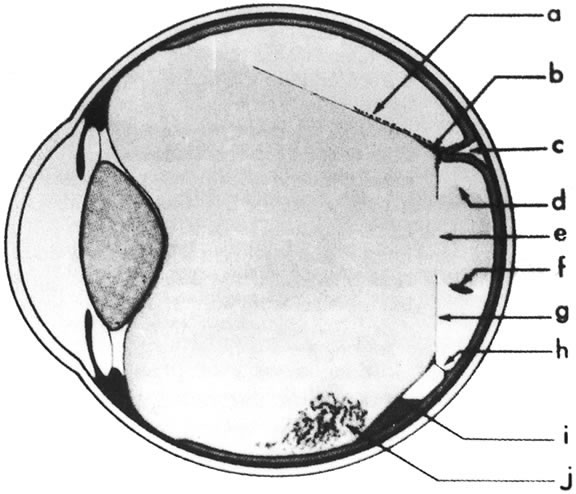
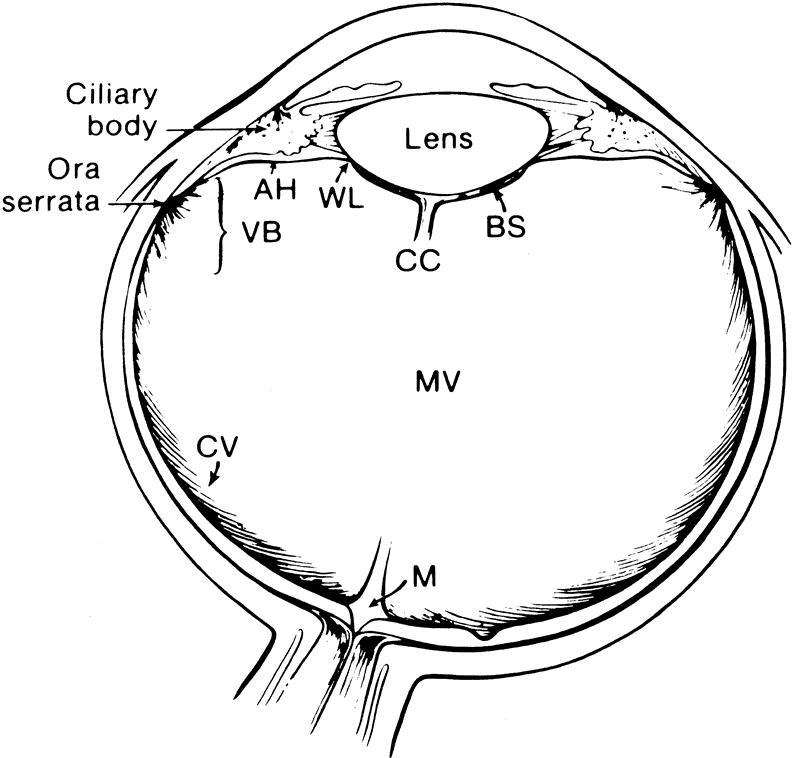
The vitreous body interfaces with a number of ocular structures through the vitreous cortex (Fig. 1). The vitreous cortex extends anteriorly from the vitreous base to form the anterior vitreous cortex and posteriorly to form the posterior vitreous cortex. Recently, the clinical importance of vitreous cortex has become increasingly apparent. The vitreous cortex has been implicated as a primary factor in a variety of vitreoretinal disorders, including retinal breaks, proliferative vitreoretinopathy (PVR), anterior hyaloidal fibrovascular proliferation, macular holes, and epiretinal membranes.
|
ANTERIOR VITREOUS CORTEX
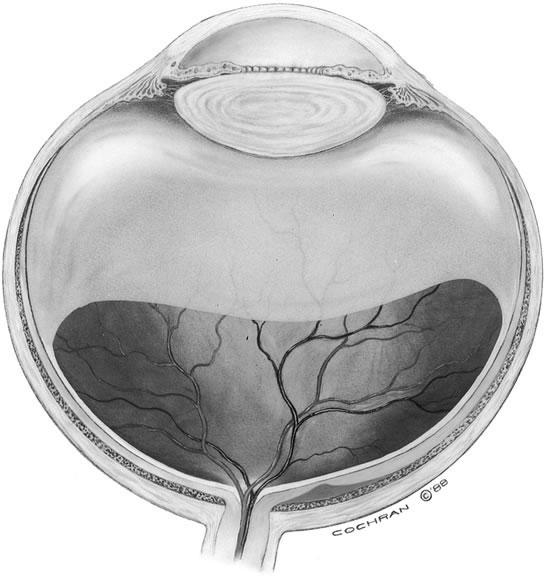
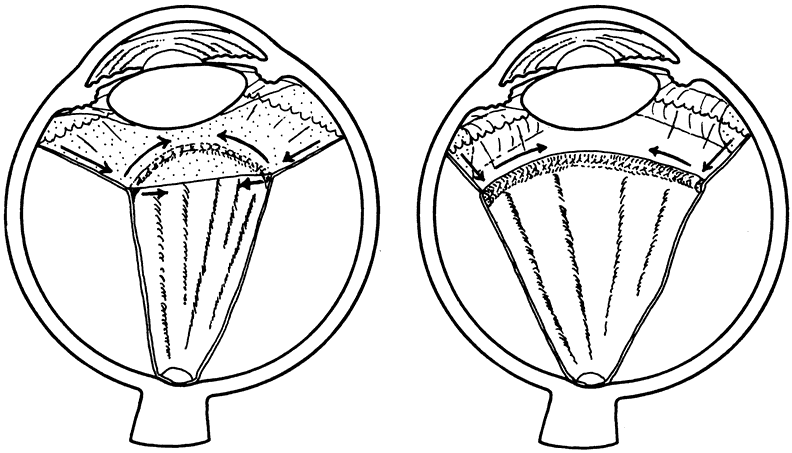
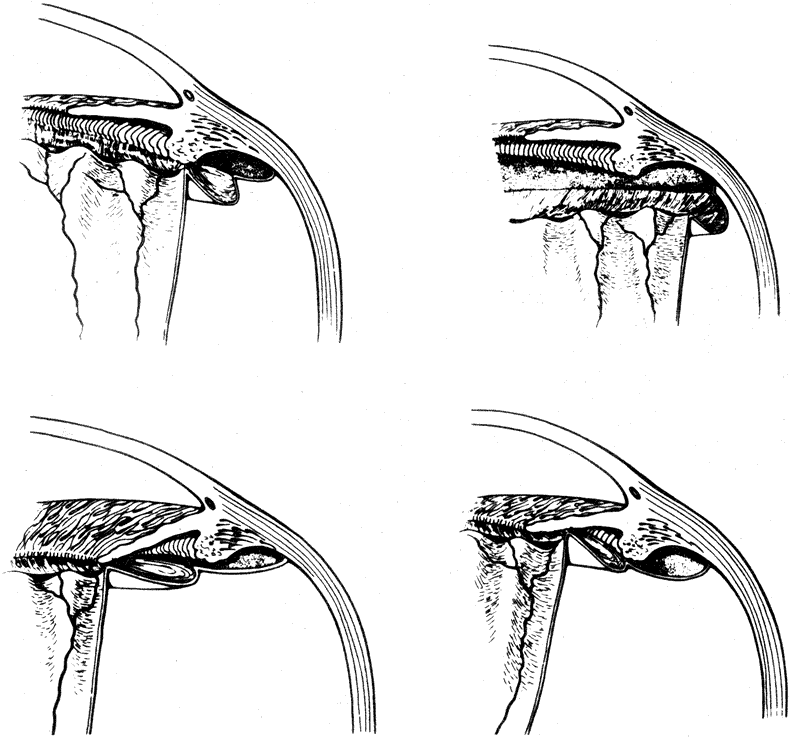
The anterior vitreous cortex or anterior hyaloid is the anterior surface layer or condensation of the vitreous body. There is no specialized membrane that constitutes the anterior vitreous cortex, but instead there is a greater density of collagen fibrils. Microscopically, the framework of collagen fibrils that run throughout the vitreous body ends in an interwoven network to form the anterior vitreous cortex. The superficial fibrils tend to run parallel to the surface in this region. The smooth surface and membrane-like appearance are due to the lamellar distribution of the cortical fibers and the associated highly polymerized mucoproteins.4,5 Anatomically, the anterior hyaloid forms the posterior limits of the posterior chamber. This portion of the vitreous cortex functions in the physiologic communication between the vitreous cavity and the aqueous humor. The anterior surface of the vitreous body separates from the pars plana approximately 1.5 mm anterior to the ora serrata. It extends medially to contact the lens posterior to the lens equator. Thus, the anterior hyaloid is in contact with the ciliary processes and the lens zonules, as well as the posterior lens capsule. The vitreous attaches to the lens capsule in a ring-like manner, forming the hyaloideocapsular ligament of Wieger. This ligament is believed by some to be synonymous with the attachment ring of the posterior zonular fibers.4 The circular area of attachment measures approximately 9 mm in diameter and is especially firm in younger persons or after intraocular inflammation.5 In this circular area the anterior hyaloid is thickened. Central to the attachment of Wieger's ligament (also known as Egger's line), the vitreous lens attachment is less pronounced and appears to be due to surface tension. This central area contains a potential space within the 9-mm ring known as Berger's space, or the patellar fossa. The anterior hyaloid then turns posteriorly to form the anterior portion of Cloquet's canal in the midportion of Berger's space. Cloquet's canal represents the remnants of the primary vitreous and can sometimes be seen with the slit lamp. It arises from the optic disc in a funnel-shaped manner, in the area of Martegiani, and extends forward to the posterior lens surface. The canal is 1 to 2 mm in width and has a down turn in the central vitreous cavity. The area of contact with the posterior lens capsule can at times be identified by a tag of embryonic tissue, known as a Mittendorf dot, located slightly nasal to the posterior pole of the lens. Similarly, a remnant of the posterior primary vitreous can occasionally be identified on the optic disc. This remnant, representing the embryonic point of exit of the hyaloid vascular system from the optic nerve head, is known as Bergmeister's papilla. The walls of Cloquet's canal are formed by a vitreous condensation rather than a true membrane (see Fig. 1).
POSTERIOR VITREOUS CORTEX
The mechanical relationship between the vitreous and the retina is mediated by the posterior vitreous cortex, which is also called the posterior hyaloid. The posterior vitreous cortex consists of relatively densely packed type II collagen fibrils arranged tangentially to the retina. The retinal basal lamina is the basement membrane of the Müller's cells that comprise the internal limiting membrane (ILM) of the retina.6,7 Ultrastructurally, the ILM consists of three layers.8 Adjacent to the end feet of the Müller's cells is the lamina rara interna. The lamina rara externa is contiguous with the vitreous cortex. In between these layers is the lamina densa. Collagen fibers of cortical vitreous are tangential to the lamina rara externa. The ILM is composed of primarily type IV collagen but also contains fibronectin, laminin, and type I collagen.9 The morphology of the ILM varies topographically in the retina. The ILM, and in particular the lamina densa, is thin in the retinal periphery and becomes increasingly thicker and irregular in the posterior retina.6,8
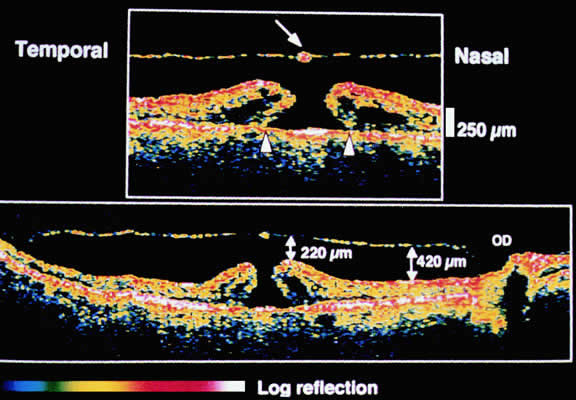
The ILM thickens from 50 nm at the vitreous base to 300 nm at the equator to 1900 nm posteriorly. In the foveal region the ILM thins to 10 to 20 nm.10
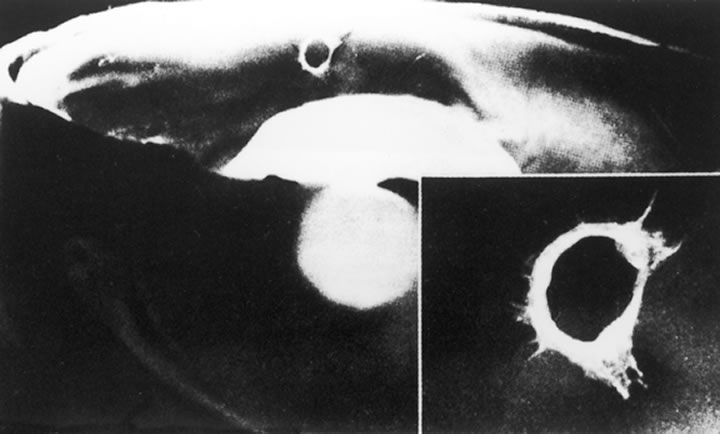
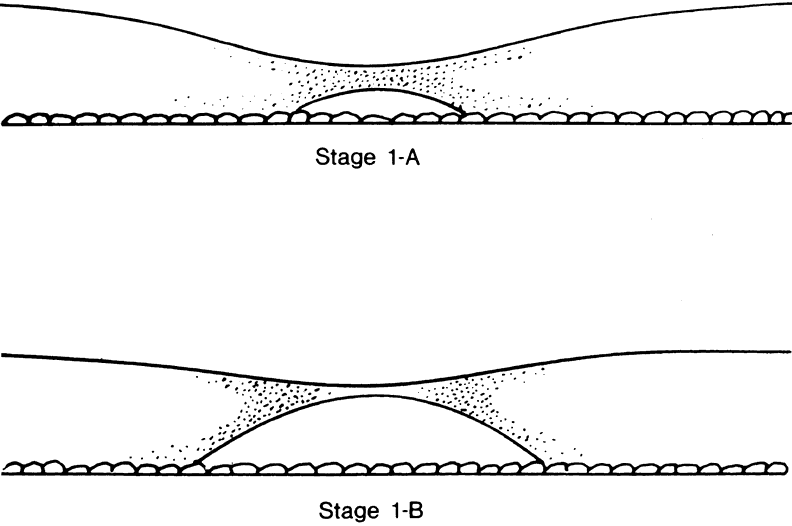
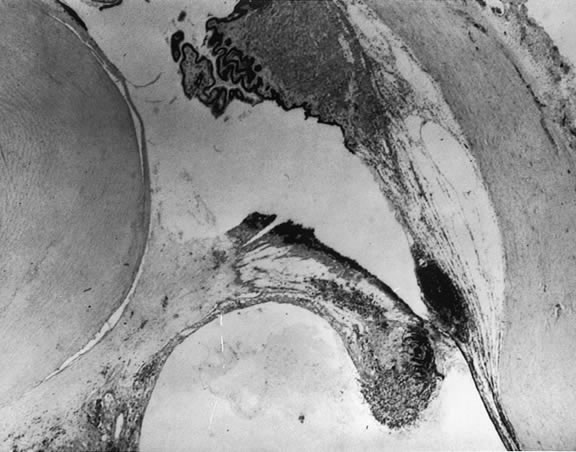
The nature of the adhesion between the vitreous cortex and the ILM is enigmatic. This adhesion is strongest at the optic nerve, the macula, the vitreous base, and retinal vessels. Foos6 demonstrated attachment plaques between Müller's cell cytoplasm and the ILM in the basal and equatorial retina (Fig. 2). Vitreous traction mediated by vitreous fibrils may contribute to these adhesions. These attachment plaques are not present posterior to the equator except where the ILM is thinned in the fovea. This anatomic variation in the fovea region may play a role in the pathogenesis of some of the vitreomacular disorders.
|

The ILM also thins over major blood vessels. Vitreous strands extend through pores in the ILM to surround the vessels11 and have been termed vitreoretinovascular bands.12 These bands may explain the strong adhesion between the vitreous and retinal vessels.
Despite the presence of these anatomic interfaces between the vitreous cortex and ILM, the adhesion of these structures is still poorly understood. Biochemical investigations suggest that vitreoretinal adhesion involve molecular adhesion mediated by glycoproteins such as fibronectin, laminin, and other glycoconjugates. The ILM consists of type IV collagen, laminin, fibronectin, type I collagen, and as yet uncharacterized glycoproteins. It is believed these substances mediate vitreoretinal adhesion.13
The ILM undergoes age-related changes that are anatomic, biochemical, and functional. Anatomically, the ILM thickens with age.6,8 Biochemically, the distribution of fibronectin and laminin changes with age. In young eyes, anti-fibronectin antibody binding has a homogeneous distribution. In older eyes, the posterior ILM shows a segmented bilaminar distribution of both antifibronectin and antilaminin antibodies.14 An age-dependent distribution of other ILM glycoconjugates has also been demonstrated.13 Variations in the distribution of these and other glycoproteins may play a role in conditions that are characterized by alterations in vitreoretinal adhesion.15
VITREOUS BASE
The vitreous base is a three-dimensional zone centered on the ora serrata where the vitreous is the most adherent to the retina and pars plana epithelium. It extends approximately 1.5 mm anteriorly to the ora serrata. Nasally, it extends 3.0 mm posterior to the ora and 1.8 mm posterior to the ora temporally. The functional base of the vitreous extends several millimeters into the vitreous body in this region. As the eye ages, the firm attachment of the vitreous base may extend posteriorly for several millimeters. This may lead to localized areas of enhanced vitreoretinal traction and result in retinal tears.
In the vitreous base, collagen fibers are relatively coarse, are numerous, and insert perpendicularly to the retina and pars plana. These fibers consist of collagen fibrils with diameters of 10.8 to 12.4 nm.16 The microscopic detail of the vitreous fibers attachment to the vitreous base varies from anterior to posterior. Anterior to the ora the fibers are less dense than posterior to the ora. Electron microscopic studies by Gartner17 demonstrate that the vitreous fibers have complex interdigitations with the reticular fibrillar materials of the basement membrane of the nonpigmented ciliary epithelium but do not pass between the cells. The anteriormost fibers splay out anteriorly to form the anterior loop of the vitreous base, which is important in the pathogenesis of anterior PVR.18 Posterior to the ora, bundles of vitreous fibrils attach to the ILM. Cords of vitreous collagen insert into gaps between the neuroglia. Gloor and Daicker19 likened this arrangement to Velcro and suggested that this may explain the strong vitreoretinal adhesion of the vitreous base. Cellular elements are also present in the vitreous base. Fibroblast-like cells are present anterior to the ora, and macrophage-like cells are posterior to the ora.17 These cells may play a role in the hypocellular gel contraction that characterizes anterior PVR.
VITREOUS CELLS
The vitreous normally contains relatively few cells compared with other connective tissues. Most of these are found within the thin layer of cortical vitreous. These few cells account for the metabolic activity of the vitreous body, which as a result is among the lowest in metabolic activity in the body. The cells are variously called vitreous cells, vitreous cortex cells, vitreocytes, or, most commonly, hyalocytes. In the physiologic state, the vitreous body appears to have a mixed population of cells. Morphologically, most are hyalocytes, whereas approximately 10% are similar to fibroblasts.
Hyalocytes are found throughout the vitreous cortex, but they appear to be concentrated in the basal region.3 These cells are more numerous in the fetal vitreous than in older eyes, and their exact origin and function are open to question. Some investigators believe hyalocytes are remnants of the primary vitreous.20 Based on developmental studies in animals and humans, hyalocytes arise from either monocytes originating from mesenchymal cells in the optic cup or embryonic fissure or from blood elements in the vasa hyaloidea propria. These cells migrate through the vitreous cavity and accumulate in the cortical vitreous as hyalocytes by the fifth month of gestation. Following birth, there is no further migration of cells to the vitreous cortex and existing hyalocytes do not normally proliferate. As a result, ocular growth results in increasing vitreous cortex surface area with a corresponding decrease in hyalocyte density. Gloor21 contests the previously mentioned hypothesis and believes hyalocytes are continuously derived from blood monocytes through life. The hyalocytes remain in the vitreous cortex for about a week and are then replaced by blood borne monocytes to maintain a constant cell population.
The hyalocytes appear as flattened, 10 to 15 μm in diameter, spindle-shaped cells when found over the retina. Whereas, in the region of the vitreous base, the cells are larger, rounder, and at times star-shaped with prominent nuclei.21,22 The hyalocytes are active metabolically with numerous lysosomal granules, phagosomes, and Golgi complexes. Hyalocytes synthesize vitreous glycosaminoglycans (GAGs) including HA and hexosamine.23 The highest density of hyalocytes is in the region of the vitreous base followed by the posterior pole, with the lowest density at the equator.
In addition to the hyalocytes and fibroblasts, there are cells found in the vitreous that appear to be in varying stages of degeneration.24 These cells are believed to represent the involutional remnants of the original cell population of the embryonic vitreous and are seen more often in young eyes. By electron microscopy, in the region of the ora serrata, cells are seen that appear to be damaged retinal cells that have been sloughed into the vitreous base.17
There is a mechanism for the elimination of foreign and degenerated substances from the vitreous that is found partly in the region of the optic disc. When India ink is injected into the anterior portion of the vitreous in a rabbit eye and the enucleated eye examined at varying intervals, it is noted the ink particles are eliminated from the vitreous by way of the optic nerve.25 Later the particles are removed by phagocytic cells that appear to enter the vitreous body from the region of the optic nerve and ciliary body. Surgical treatment may also affect the cellular composition of the vitreous. Cryopexy induces a macrophage influx into the vitreous cavity.26 This may play a role in the clearing of vitreous opacities.