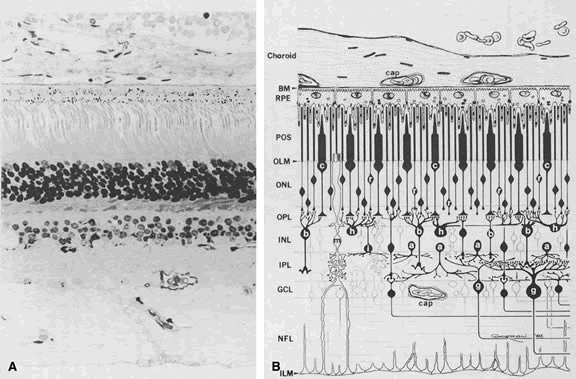
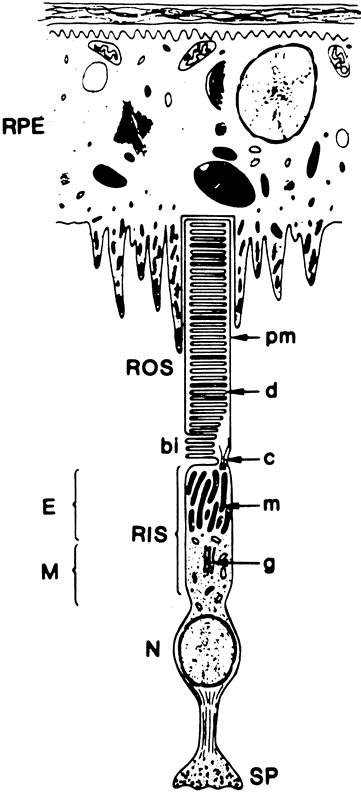
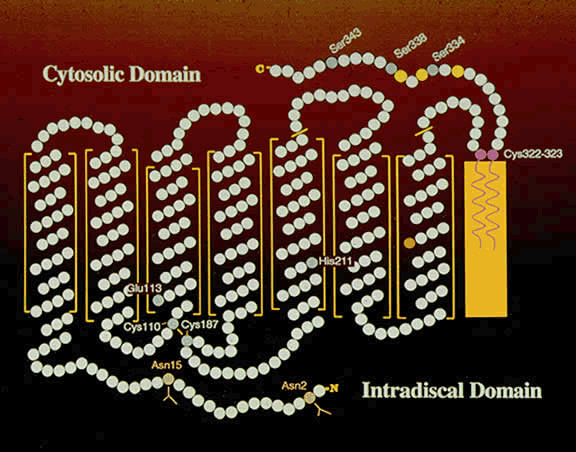
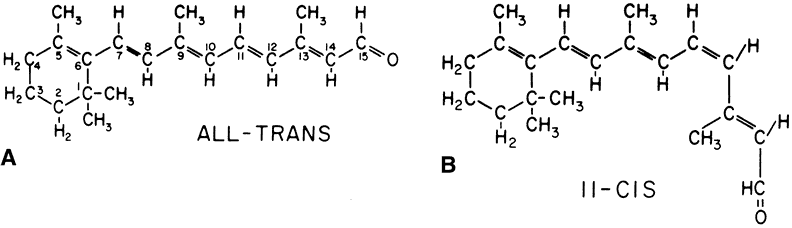
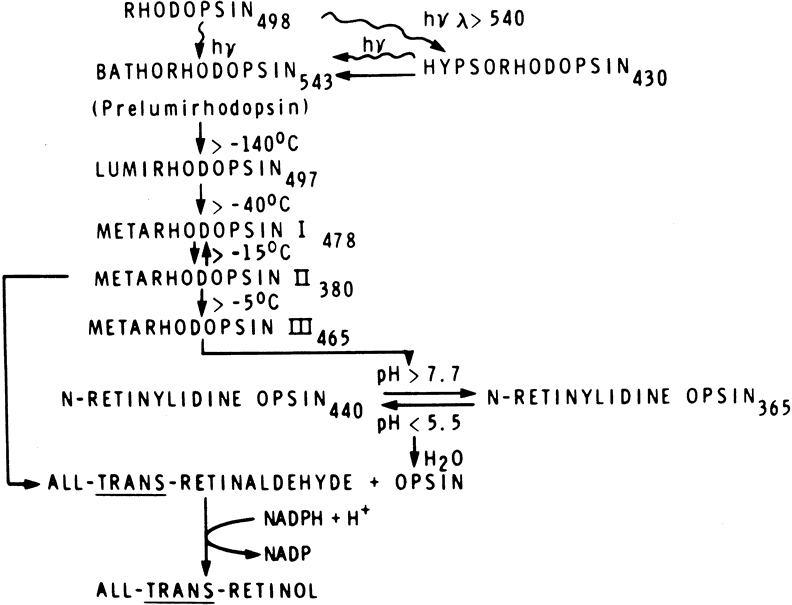
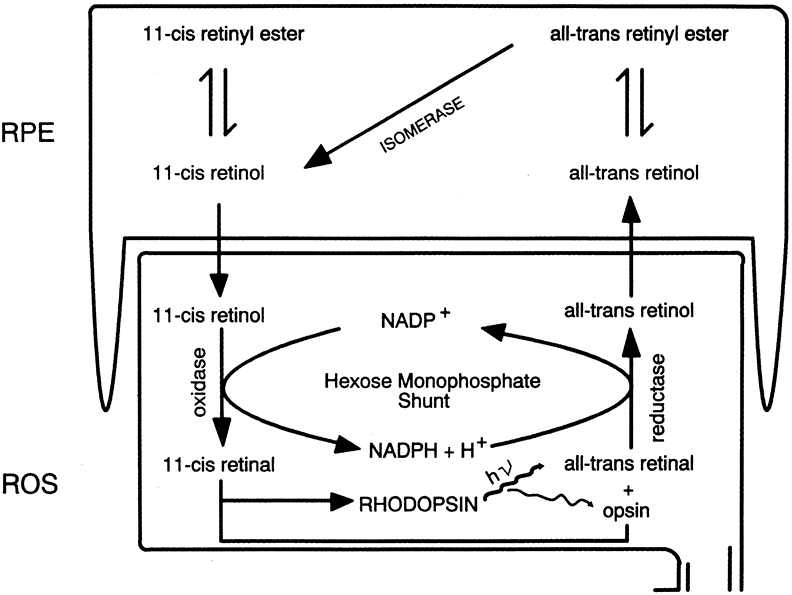
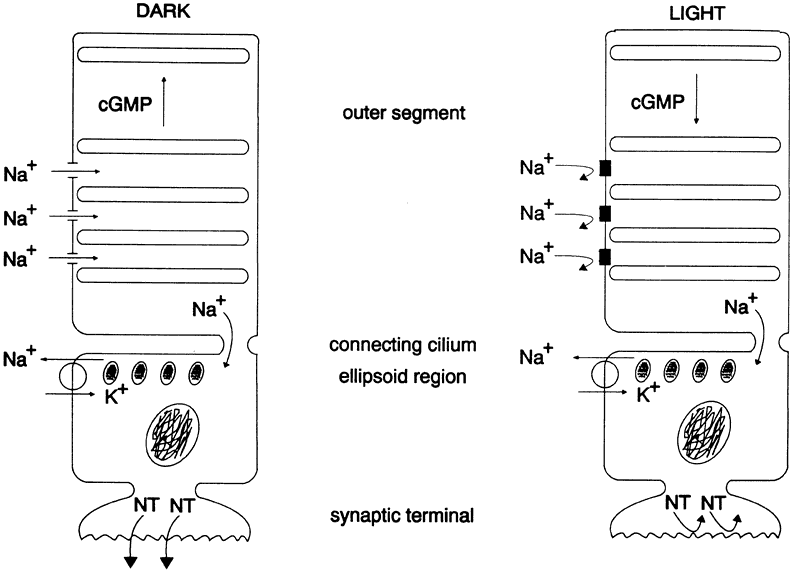
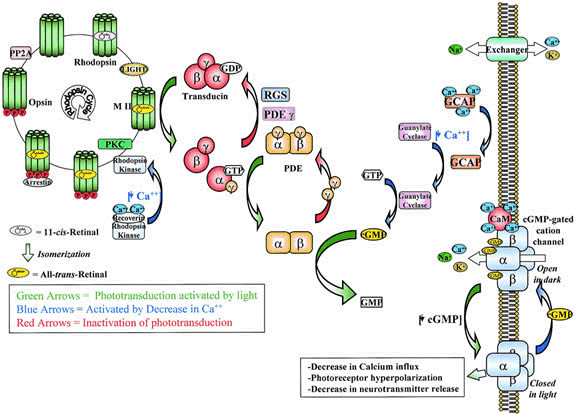
1. Andrews LD, Cohen AI. Freeze-fracture evidence for the presence of cholesterol
in particle-free patches of basal disks and the plasma membrane
of retinal rod outer segments of mice and frogs. J Biol Chem 1979;81:215 2. Boesze-Battaglia K, Fliesler SJ, Albert AD. Relationship of cholesterol
content to spatial distribution and age of disc membranes in retinal
rod outer segments. J Biol Chem 1990;265:18867 3. Fliesler SJ, Anderson RE. Chemistry and metabolism of lipids in the vertebrate
retina. In Holman RT (ed). Progress in Lipid Research. Elmsford, New
York: Pergamon Press, 1983:79–131 4. Findlay JBC, Pappin DJC, Eliopoulos EE. The primary structure, chemistry
and molecular modeling of rhodopsin. In Osbourne NN, Chader GJ, (eds). Progress
in Retinal Research. Elmsford, New York: Pergamon Press, 1988:63–87 5. Ohguro H, Johnson RS, Ericsson LH et al. Control of rhodopsin multiple
phosphorylation. Biochemistry 1994;33:1023 6. Ohguro H, Van Hooser JP, Milam AH et al. Rhodopsin phosphorylation and
dephosphorylation in vivo. J Biol Chem 1995;270:14259 7. St. Jules RS, Smith SB, O'Brien PJ. The localization and timing of post-translational
modifications of rat rhodopsin. Exp Eye Res 1990;51:427 8. Hargrave PA, McDowell JH. Rhodopsin and phototransduction: A model system
for G protein-linked receptors. FASEB J 1992;6:2323 9. Moench SJ, Moreland J, Stewart DH et al. Fluorescence studies of the location
and membrane accessibility of the palmitoylation sites of rhodopsin. Biochemistry 1994;33:5791 10. O'Dowd BF, Hnatowich M, Caron MG et al. Palmitoylation of the human β2-adrenergic receptor. J Biol Chem 1989;264:7564 11. Connell GJ, Molday RS. Molecular cloning, primary structure, and orientation
of the vertebrate photoreceptor cell protein peripherin in the rod
outer segment disk membrane. Biochemistry 1990;29:4691 12. Molday RS, Molday LL. Differences in the protein composition of bovine
retinal rod outer segment disk and plasma membranes isolated by a ricin-gold-dextran
density perturbation method. J Cell Biol 1987;105:2589 13. Boesze-Battaglia K, Albert AD. Fatty acid composition of bovine rod outer
segment plasma membrane. Exp Eye Res 1989;49:699 14. Nathans J, Thomas D, Hogness DS. Molecular genetics of human color vision: The
genes encoding blue, green, and red pigments. Science 1986;232:193 15. Nathans J, Piantanida TP, Eddy RL et al. Molecular genetics of inherited
variations in human color vision. Science 1986;232:203 16. Nathans J. The evolution and physiology of human color vision: Insights
from molecular genetic studies of visual pigments. Neuron 1999;24:299 17. Downes SM, Holder GE, Fitzke FW et al. Autosomal dominant cone and cone-rod
dystrophy with mutations in the guanylate cyclase activator 1A gene-encoding
guanylate cyclase activating protein-1. Arch Ophthalmol 2001;119:96 18. Dudley PA, Anderson RE. Phospholipids transfer protein from bovine retina
with high activity towards retinal rod disc membranes. FEBS Letters 1978;95:57 19. Besharse JC, Pfenninger KH. Membrane assembly in retinal photoreceptors, part
I. Freeze-fracture analysis in cytoplasmic vesicles in relationship
to disc assembly. J Cell Biol 1980;87:451 20. Steinberg RH, Fisher SK, Anderson DH. Disc morphogenesis in vertebrate
photoreceptors. J Comp Neurol 1980;190:501 21. Deretic D. Rab proteins and post-Golgi trafficking of rhodopsin in photoreceptor
cells. Electrophoresis 1997;18:2537 22. Fliesler SJ, Basinger SF. Monensin stimulates glycerolipid incorporation
into rod outer segment membranes. J Biol Chem 1987;262:17516 23. Young RW. The daily rhythm of daily shedding and degradation of rod and
cone outer segment membranes in the chick retina. Invest Ophthalmol Vis
Sci 1978;17:105 24. Tierstein PS, Goldman AI, O'Brien PJ. Evidence for both local and central
regulation of rat rod outer segment disc shedding. Invest Ophthalmol
Vis Sci 1980;19:1268 25. Hollyfield JG, Basinger SF. Photoreceptor shedding can be initiated within
the eye. Nature 1978;274:794 26. Blest AD. The rapid synthesis and destruction of photoreceptor membrane
by a dinopid spider: A daily cycle. Proc R Soc Lond 1978;200:463 27. Bok D, Hall MO. The role of the pigment epithelium in the etiology of inherited
retinal dystrophy in the rat. J Cell Biol 1971;49:664 28. Bernstein PS, Law WC, Rando RR. Isomerization of all-trans-retinoids to 11-cis-retinoids
in vitro. Proc Natl Acad Sci U S A 1987;84:1849 29. Chader GJ. Interphotoreceptor retinoid-binding protein (IRBP): A model
protein for molecular biological and clinically relevant studies. Friedenwald
lecture. Invest Ophthalmol Vis Sci 1989;30:7 30. Ho MT, Massey JB, Pownall HJ et al. Mechanism of vitamin A movement between
rod outer segments, interphotoreceptor retinoid-binding protein, and
liposomes. J Biol Chem 1989;264:928 31. Rando RR. Membrane phospholipids as an energy source in the operation of
the visual cycle. Biochemistry 1991;30:595 32. Fung BK-K. Transducin: Structure, function and role in phototransduction. In
Osborne NN, Chader GJ (eds). Progress in Retinal Research. Elmsford, New
York: Pergamon Press, 1987:151–177 33. Sears S, Erickson A, Hendrickson A. The spatial and temporal expression
of outer segment proteins during development of Macaca monkey cones. Invest
Ophthalmol Vis Sci 2000;41:971 34. Hargrave PA. Rhodopsin structure, function, and topography the Friedenwald
lecture. Invest Ophthalmol Vis Sci 2001;42:3 35. Sallese M, Iacovelli L, Cumashi A et al. Regulation of G protein-coupled
receptor kinase subtypes by calcium sensor proteins. Biochim Biophys
Acta 2000;1498:112 36. Mendez A, Burns ME, Roca A et al. Rapid and reproducible deactivation of
rhodopsin requires multiple phosphorylation sites. Neuron 2000;28:153 37. Bruel C, Cha K, Niu L et al. Rhodopsin kinase: Two mAbs binding near the
carboxyl terminus cause time-dependent inactivation. Proc Natl Acad
Sci U S A 2000;97:3010 38. Bruel C, Cha K, Reeves PJ et al. Rhodopsin kinase: Expression in mammalian
cells and a two-step purification. Proc Natl Acad Sci U S A 2000;97:3004 39. Chen CK, Hurley JB. Purification of rhodopsin kinase by recoverin affinity
chromatography. Methods Enzymol 2000;315:404 40. Johnson WC, Palczewski K, Gorczyca WA et al. Calcium binding to recoverin: Implications
for secondary structure and membrane association. Biochim
Biophys Acta 1997;1342:164 41. Zhou J, Moroi K, Nishiyama M et al. Characterization of RGS5 in regulation
of G protein-coupled receptor signaling. Life Sci 2001;68:1457 42. Lyubarsky AL, Naarendorp F, Zhang X et al. RGS9-1 is required for normal
inactivation of mouse cone phototransduction. Mol Vis 2001;7:71 43. He W, Lu L, Zhang X et al. Modules in the photoreceptor RGS9-1.Gbeta 5L
GTPase-accelerating protein complex control effector coupling, GTPase
acceleration, protein folding, and stability. J Biol Chem 2000;275:37093 44. Chen CK, Burns ME, He W et al. Slowed recovery of rod photoresponse in
mice lacking the GTPase accelerating protein RGS9-1. Nature 2000;403:557 45. Koch KW, Stryer L. Highly cooperative feedback control of retinal rod guanylate
cyclase by calcium ions. Nature 1988;334:64 46. Gorczyca WA. Role of calcium ions in vertebrate phototransduction. Pol
J Pharmacol 1999;51:167 47. Duda T, Goraczniak R, Surgucheva I et al. Calcium modulation of bovine
photoreceptor guanylate cyclase. Biochemistry 1996;35:8478 48. Gorczyca WA, Polans AS, Surgucheva IG et al. Guanylyl cyclase activating
protein. A calcium-sensitive regulator of phototransduction. J Biol
Chem 1995;270:22029 49. Dryja TP, McGee TL, Reichel E et al. A point mutation of the rhodopsin
gene in one form of retinitis pigmentosa. Nature 1990;343:364 50. Dryja TP, McGee TL, Hahn LB et al. Mutations within the rhodopsin gene
in patients with autosomal dominant retinitis pigmentosa. N Engl J Med 1990;323:1302 51. Inglehearn CF, Bashir R, Lester DH et al. A 3-bp deletion in the rhodopsin
gene in a family with autosomal dominant retinitis pigmentosa. Am
J Hum Genet 1991;48:26 52. Chen J, Makino CL, Peachey NS et al. Mechanism of rhodopsin inactivation
in vivo as revealed by COOH-terminal truncation mutant. Science 1995;267:374 53. Gryczan C, Kusvak JR, Novak L et al. A transgenic mouse model for autosomal
dominant retinitis pigmentosa caused by a three base pair deletion
in codon 255/256 of the opsin gene. Invest Ophthalmol Vis Sci 1995;36:S423 54. Naash MI, Hollyfield JG, Al-Ubaidi MR et al. Simulation of human autosomal
dominant retinitis pigmentosa in transgenic mice expressing a mutated
murine opsin gene. Proc Natl Acad Sci U S A 1993;90:5499 55. Naash MI, Al-Ubaidi MR, Hollyfield JG et al. Simulation of autosomal dominant
retinitis pigmentosa in transgenic mice. In Hollyfield JG, Anderson
RE, LaVail MM (eds). Retinal Degeneration: Clinical and Laboratory
Applications. New York: Plenum, 1993:201–210 56. Olsson JE, Gordon JW, Pawlyk BS et al. Transgenic mice with a rhodopsin
mutation (Pro23His): A mouse model of autosomal dominant retinitis pigmentosa. Neuron 1992;9:815 57. Sung C-H, Makino C, Baylor D et al. A rhodopsin gene mutation responsible
for autosomal dominant retinitis pigmentosa results in a protein that
is defective in localization to the photoreceptor outer segment. J
Neurosci 1994;14:5818 58. Zozulya SA, Gurevich VV, Zvyaga TA et al. Functional expression in vitro
of bovine visual rhodopsin. Protein Eng 1990;3:453 59. Berson EL, Rosner B, Sandberg MA et al. Ocular findings in patients with
autosomal dominant retinitis pigmentosa and rhodopsin, proline-347-leucine. Am
J Ophthalmol 1991;111:614 60. Jacobson SG, Kemp CM, Sung CH et al. Retinal function and rhodopsin levels
in autosomal dominant retinitis pigmentosa with rhodopsin mutations. Am
J Ophthalmol 1991;112:256 61. Apfelstedt-Sylla E, Kunisch M, Horn M et al. Ocular findings in a family
with autosomal dominant retinitis pigmentosa and a frameshift mutation
altering the carboxyl terminal sequence of rhodopsin. Br J Ophthalmol 1993;77:495 62. Allikmets R, Shroyer NF, Singh N et al. Mutation of the Stargardt disease
gene (ABCR) in age-related macular degeneration. Science 1997;277:1805 63. Sun H, Smallwood PM, Nathans J. Biochemical defects in ABCR protein variants
associated with human retinopathies. Nat Genet 2000;26:242 64. Allikmets R. Further evidence for an association of ABCR alleles with age-related
macular degeneration. The International ABCR Screening Consortium. Am
J Hum Genet 2000;67:487 65. Rivera A, White K, Stohr H et al. A comprehensive survey of sequence variation
in the ABCA4 (ABCR) gene in Stargardt disease and age-related
macular degeneration. Am J Hum Genet 2000;67:800 66. Fuse N, Suzuki T, Wada Y et al. Molecular genetic analysis of ABCR gene
in Japanese dry form age- related macular degeneration. Jpn J Ophthalmol 2000;44:245 67. Souied EH, Ducroq D, Rozet JM et al. ABCR gene analysis in familial exudative
age-related macular degeneration. Invest Ophthalmol Vis Sci 2000;41:244 68. De La Paz MA, Guy VK, Abou-Donia S et al. Analysis of the Stargardt disease
gene (ABCR) in age-related macular degeneration. Ophthalmology 1999;106:1531 69. Simonelli F, Testa F, de Crecchio G et al. New ABCR mutations and clinical
phenotype in Italian patients with Stargardt disease. Invest Ophthalmol
Vis Sci 2000;41:892 70. Lewis RA, Shroyer NF, Singh N et al. Genotype/phenotype analysis of a photoreceptor-specific
ATP-binding cassette transporter gene, ABCR, in
Stargardt disease. Am J Hum Genet 1999;64:422 71. Kramer F, White K, Pauleikhoff D et al. Mutations in the VMD2 gene are
associated with juvenile-onset vitelliform macular dystrophy (Best disease) and
adult vitelliform macular dystrophy but not age-related macular
degeneration. Eur J Hum Genet 2000;8:286 72. Caldwell GM, Kakuk LE, Griesinger IB et al. Bestrophin gene mutations in
patients with Best vitelliform macular dystrophy. Genomics 1999;58:98 73. Marquardt A, Stohr H, Passmore LA et al. Mutations in a novel gene, VMD2, encoding
a protein of unknown properties cause juvenile-onset vitelliform
macular dystrophy (Best's disease). Hum Mol Genet 1998;7:1517 74. Lotery AJ, Munier FL, Fishman GA et al. Allelic variation in the VMD2 gene
in best disease and age-related macular degeneration. Invest Ophthalmol
Vis Sci 2000;41:1291 |