OCULAR IMMUNOLOGIC PRIVILEGE
Ocular immunologic privilege is conferred by multiple contributory factors, including the following: (1) the blood-ocular barrier; (2) the absence of intraocular lymphatic drainage; (3) intraocular APCs; (4) immunosuppressive ocular fluids13; and (5) Fas ligand (Fas l)-mediated apoptosis. Ocular immune privilege likely represents an evolutionary adaptation to downregulate immunogenic ocular inflammation, with its potential threat to vulnerable intraocular tissues, in order to thereby preserve vision. This exists, however, in delicate counterbalance with the threat of unchecked infection due to inadequate immune responses.
The blood-ocular barrier comprises the zonulae occludentes of the nonpigmented ciliary epithelium and RPE cells, as well as the nonfenestrated vascular endothelium of retinal and iris blood vessels. Disruption of vascular tight junctions may increase vascular permeability, promoting chronicity and recurrence of inflammation as well as other consequences, such as cystoid macular edema. The presence of uveitis or systemic immune responses to ocular antigens generally indicates a breach of the blood-ocular barrier. The choroid, by virtue of its high rate of blood flow and its anatomy, is particularly susceptible to blood-borne diseases. It may function as a repository for immune cells, including antibody-producing lymphocytes with immunologic memory.14 The local humoral response to an intraocular antigen is not entirely directed against this antigen, but also activates other antibody-producing cells that have migrated to the choroid. For example, in recurrent allergic uveitis due to degranulation of choroidal mastocytes, the triggering antigen may vary with each recurrence. The vitreous may serve as a long-term depot for foreign antigens.
Because of the absence of intraocular lymphatic drainage, intraocular antigens are presented to the immune system via the bloodstream, and not via lymphatics.15,16 This produces an atypical immune response in which the spleen, rather than the local lymphatics, functions as the primary lymphoid organ. In contrast, the response to antigens presented to external ocular tissues or soluble antigens placed in areas with lymphatic drainage elicit typical complete immune responses with both antibody and cell-mediated components.
Another unusual factor in ocular immune privilege is the presence of marrow-derived MHC class II+ APCs within the uveal tract, which have been implicated in the induction of deviant immune responses, such as ACAID (see later discussion).13 Antigens must be presented in association with MHC class II (HLA-DR) molecules in order for recognition by T cells to occur. In the normal uninflamed eye, MHC class II expression is conspicuously deficient in certain structures, such as corneal endothelium, trabecular meshwork, anterior iris layer, vascular endothelium, RPE, and retinal cells. Thus, the normal eye is generally a poor site in which to elicit an immune response. However, both blood-borne (ED1+ ) and resident ocular (ED2+ ) macrophages, as well as MHC class II+ dendritic cells, have been identified in the eye. Iris macrophages may contribute to the development of ACAID. Furthermore, dendritic cells, which are found throughout the uveal tract and in close apposition to the RPE, have been shown to be effective presenters of retinal antigens to naive T cells, and are able to induce proliferative responses to both S-Ag and interphotoreceptor retinoid binding protein.3
The local ocular factors found in aqueous humor also inhibit intraocular immune responses. Human aqueous humor is able to suppress tumor cell growth and mitogen-stimulated T-cell proliferation.17 Normal iris and ciliary body epithelial cells produce TGF-β2,18,19 an immunosuppressive cytokine with multiple functions, including negative regulation of cell growth in the immune system.20 Other immunoinhibitory ocular fluid components include alpha-melanocyte-stimulating hormone, vasoactive intestinal peptide, calcitonin gene-related peptide and anticomplement activity (not yet defined). In addition, low levels of cortisol-binding globulin permit higher effective cortisol concentrations.13 The end result is a reduction in antigen-driven T-cell activation. There is little influence on systemic immune responses or local response to extraocular antigen. Soluble and membrane-bound inhibitors of complement activation and fixation have also been described.21,22
An important mechanism involved in ocular immune privilege is Fas l-mediated apoptosis. Fas l expression within the eye has been localized to the corneal epithelium and endothelium, iris, ciliary body, and retina. By means of a Fas-Fas l interaction, eyes expressing Fas l are able to delete Fas+ T cells by apoptosis.23,24 Fas- eyes are unable to delete Fas- or Fas+ cells. Thus ocular Fas l expression permits direct destruction of invading activated cells with the potential to destroy vision.
UNIQUE ORGAN-SPECIFIC ANTIGENS SEQUESTERED WITHIN THE EYE
Several sequestered antigens able to induce uveitis reside within the eye, which lends support to the concept of an autoimmune basis for uveitis, as proposed by Elschnig25 in 1910. Most of these antigens are of retinal origin, and the retina is more effective in producing uveitis than uveal tissue26; such antigens include S-Ag, interphotoreceptor retinoid binding protein, and rhodopsin. Systemic sensitization to retinal antigens causes inflammation of the uvea and the aqueous and vitreous humors, as well as retinal destruction.27 Injection of S-Ag at a remote site causes bilateral immune-mediated uveitis.28–31 This observation has been manipulated to produce the experimental model of EAU, which is associated with the production of autoreactive T cells and antibodies, and resembles sympathetic ophthalmia in humans. Lens antigens are also sequestered during embryologic development of immune self-tolerance. Later release of crystallin proteins leads to recognition of antigens as foreign rather than self, and elicits lens-induced uveitis.32,33 Many patients with uveitis or cataracts demonstrate sensitization to lens antigens, whereas many patients with choroiditis or retinitis are sensitized to retinal antigens.34
ANTERIOR CHAMBER-ASSOCIATED IMMUNE DEVIATION
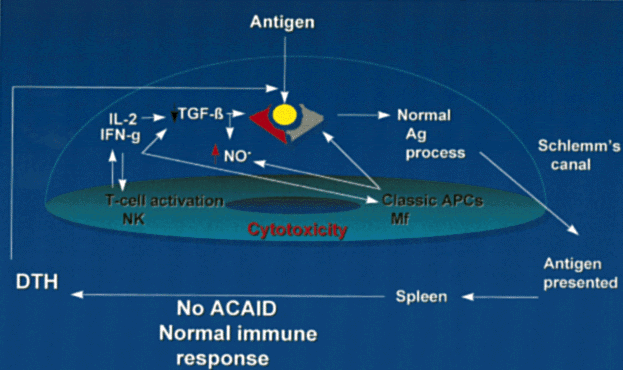
Inoculation of antigens or alloantigens into the anterior chamber, vitreous, or subretinal space elicits a deviant systemic immune response. A selective, transient depression of cell-mediated immunity (CMI), particularly delayed-type hypersensitivity (DTH), is observed, while the humoral response mediated by B cells that produce complement fixing antibodies is preserved.35 This phenomenon is known as anterior chamber-associated immune deviation (ACAID). Antigens reported to induce ACAID include lymph node cell suspensions,36,37 neoplastic cells,38,39 hapten-derived splenocytes,40 and soluble antigens.41
An intact oculosplenic axis enabling splenic stimulation while completely bypassing local lymphatic antigen processing is essential for the development of ACAID. A splenectomy performed before or within 6 days of intracameral antigen stimulation will prevent ACAID.42 The route of antigen administration is significant, because the absence of intraocular lymphatics permits the requisite bypass of lymphatics to produce ACAID with intraocular antigen stimulation. In contrast, subconjunctivally administered antigen is drained via lymphatics to regional lymph nodes for conventional processing and elicits conventional immune responses.43
Bone marrow-derived cells within the iris and ciliary body stroma serve a critical function in the induction of ACAID. These cells are unable to present antigen to T cells and also inhibit antigen-driven T-cell activation.44 Introduction of donor Ia+ APCs into the anterior chamber can subvert ocular immune privilege and elicit instead a normal systemic alloimmune response.45 Similarly, under chronic pathologic conditions or when HLA-DR+ APCs develop within the eye, local DTH can occur.
The mechanisms of ACAID have been the focus of much study and are summarized in Figure 1. Intraocular APCs, such as iris and ciliary body dendritic cells/macrophages (F4/80+ cells), are influenced by local immunoregulatory factors, particularly TGF-β, to acquire ACAID-inducing potential. APCs capture antigens entering the eye and direct these to the spleen, where deviant processing occurs.13 A good humoral response ensues, along with specific cytotoxic cells directed against the inciting antigen.46 Priming of DTH effectors does not occur.47 ACAID is dominant to conventional immunity. If the same antigen is injected subconjunctivally and into the anterior chamber (theoretically permitting conventional or deviant antigen processing and, therefore, conventional immune responses or ACAID to develop), ACAID develops rather than conventional immune responses because ACAID is dominant.
|
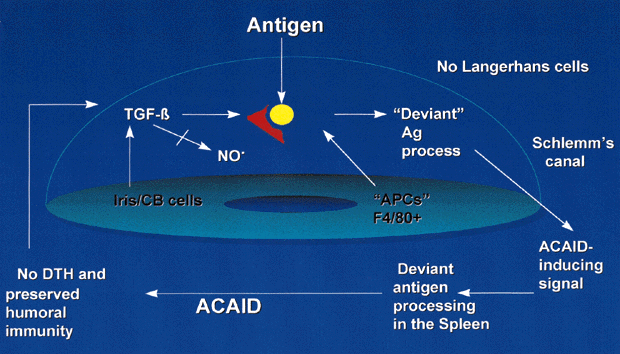
Although DTH is an effective means of eliminating pathogens, it is also potentially very destructive to surrounding tissues because of the intense lymphokine-induced secondary inflammation. Suppression of intraocular DTH by ACAID may have evolved as a means of preserving vision while permitting some degree of local immunity,35 producing what Streilein48 termed a “dangerous compromise” between the eye and the immune system.