IMMUNOPATHOLOGY
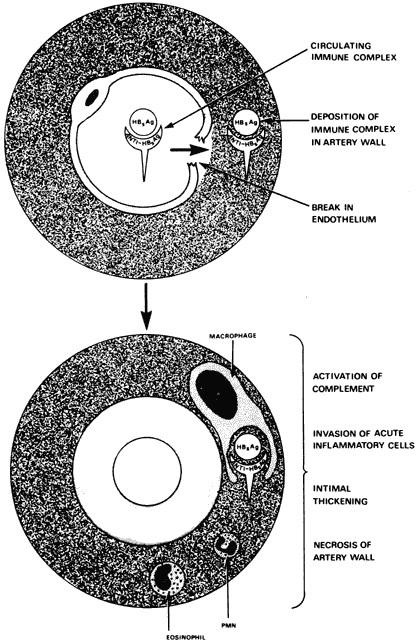
The antigenic stimulus that initiates joint inflammation in rheumatoid arthritis still is a mystery. Certain microorganisms have been implicated, but their causal roles still are uncertain. Viruses, particularly the slow viruses, may play a role, although no virus particles have been certainly identified. The results of attempts to isolate Mycoplasma and rubella virus from the joints of patients with rheumatoid arthritis have been inconclusive.12 During the prodromal stage of hepatitis, hepatitis B virus can form immune complexes and produce a syndrome resembling serum sickness with polyarthralgia and vasculitis. This virus seems to be the first virus known to produce a chronic rheumatic disorder in humans, and, like many other infectious agents, it can give rise to rheumatoid factor in serum. The clinical syndrome, however, bears no resemblance to either rheumatoid arthritis or systemic lupus erythematosus (SLE).
Other possible infectious causes of rheumatoid arthritis have been investigated. Rheumatoid synovial cells show a diminished sensitivity to infection with either Newcastle disease virus or rubella virus. Mycoplasma antibodies have been isolated from patients with rheumatoid arthritis, especially from those with longstanding disease, and recently a slow-growing infectious agent with some of the properties of Mycoplasma has been isolated from the synovial fluid of such patients.13 In other studies, however, investigators have failed to find any evidence of previous Mycoplasma infection in patients with rheumatoid arthritis.14
A variety of immunologic abnormalities have been found in patients with rheumatoid arthritis, and there now is considerable evidence that the disease is caused by an autoimmune process. Antibodies against IgG are formed in the patient's blood and synovial fluid. Immune complexes that are formed and deposited in the joints and other tissues activate the complement system through the classic and alternate pathways.15 Activation of the complement system results in a number of inflammatory phenomena, including chemotaxis of leukocytes, histamine release, and cell lysis. Enzymes released by the synovial leukocytes produce inflammatory changes in the joints and the destruction of normal structures. The inflammatory response is amplified by the various Immoral amplification systems.
The Immoral immune system appears to be highly active and important in the pathogenesis of rheumatoid arthritis (Fig. 1). The number of synovial B lymphocytes, which are precursors of antibody-producing plasma cells, often is abnormally high. More than 50% of the synovial plasma cells produce IgG rheumatoid factor, an antibody directed against other IgG molecules.10 Immune complexes may be found within plasma cells of the synovial membrane, a finding unparalleled in any other immunopathologic disorder. Although a greater-than-normal number of peripheral B cells usually are found, the number is hard to estimate because of the antilymphocyte antibodies present.16 When these antibodies are removed, the number of peripheral B cells may in fact be reduced.
|
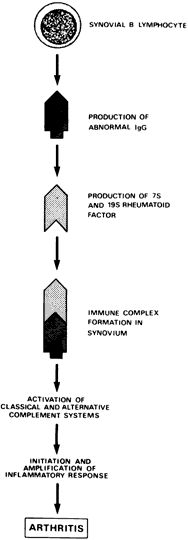
The following antibodies also have been identified in the sera of patients with rheumatoid arthritis: (1) antibodies to double-stranded DNA; (2) antibodies to human native and denatured collagen; and (3) antinuclear antibodies (ANAs).17,18 Recently, it has been shown that IgG molecules in the sera of patients with rheumatoid arthritis have a conformational anomaly in the hinge region.19 This altered IgG may be recognized as abnormal by B-lymphocyte receptors, leading to an Immoral autoimmune response directed against IgG.
The following defects in cellular immunity also have been associated with rheumatoid arthritis:
- When tested with multiple skin-test antigens, 20% of affected patients
are anergic.20
- Although some investigators have reported abnormally low levels of peripheral
blood T lymphocytes during active disease, others have found the
levels slightly elevated. In synovial fluid from actively inflamed joints, they
have been consistently high.21,22
- Heat-aggregated IgG and, to a lesser extent, native IgG inhibit the migration
of rheumatoid leukocytes.
This T-cell response to IgG antigens and the fact that soluble mediators of lymphocytes contribute to the inflammatory changes that take place in the rheumatoid joint strongly suggest that there is a cell-mediated immune component in rheumatoid arthritis.
A third population of lymphocytes, lacking conventional B- and T-cell markers, may be important in the pathogenesis of rheumatoid disease. This population, known as “null cells,” may include the so-called killer lymphocytes (K cells) that are cytotoxic to IgG-coated target cells.22 Null cells may be responsible for the formation of “rheumatoid rosettes,” which are formed by the interaction of lymphocytes and IgG-coated indicator erythrocytes. In addition, peripheral blood leukocytes collected from patients with rheumatoid arthritis may be cytotoxic for synovial cells.23
Antigen preparations of uvea-retina, synovial membrane, and articular cartilage inhibit the migration of leukocytes obtained from patients with rheumatoid arthritis.24 In ankylosing spondylitis, inhibition is induced only by synovial membrane antigens. Lymphocytes from blood and synovial fluid of patients with rheumatoid arthritis also show a markedly diminished blastogenic response to phytohemagglutinin and pokeweed mitogen.25
The reason for depressed cellular immunity in rheumatoid arthritis is unknown. It may result from a preoccupation of the host's immune mechanism with cell-mediated immune reactions related to the pathogenesis of the disease, or it may be related to a systemic viral infection. Alternatively, depressed cellular immunity may be caused by immune complex formation or by rheumatoid arthritis therapy. The HLA-Dw4 allele occurs with high frequency and DQw7 influences severity.26