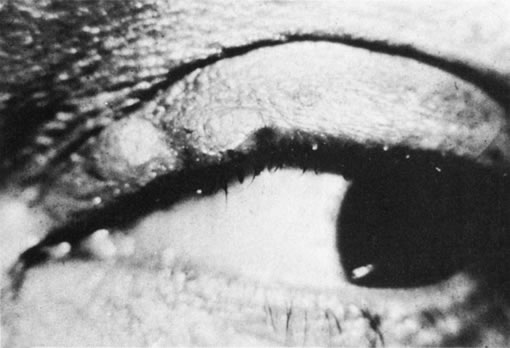
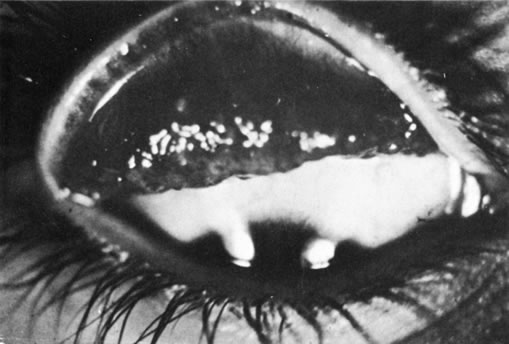
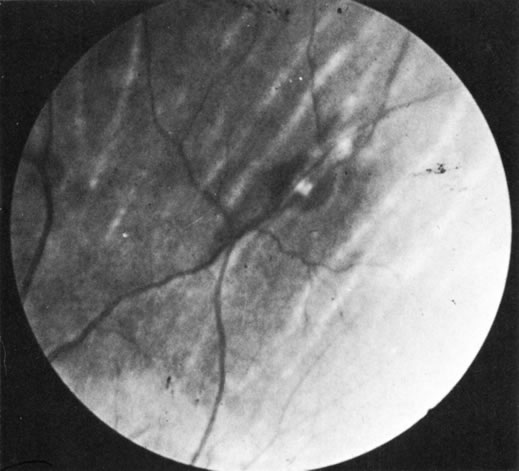
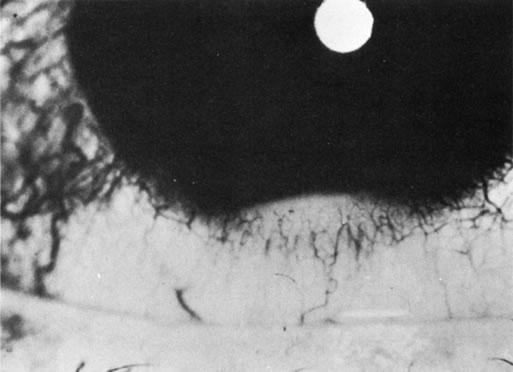
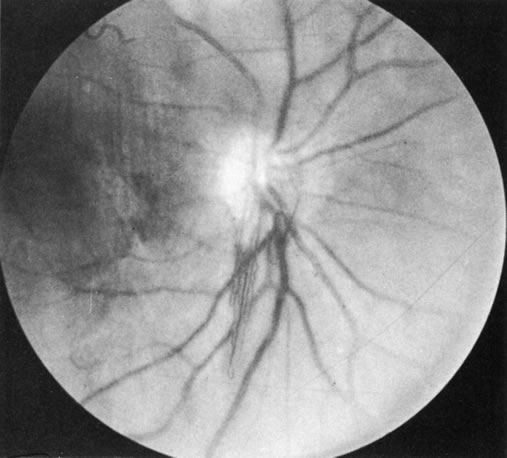
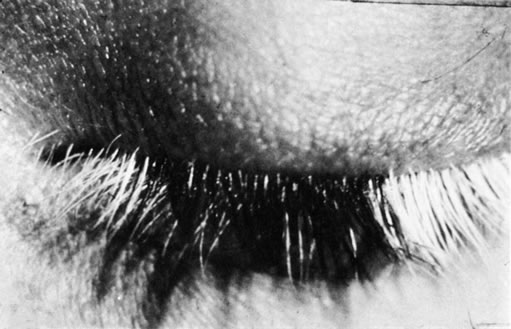
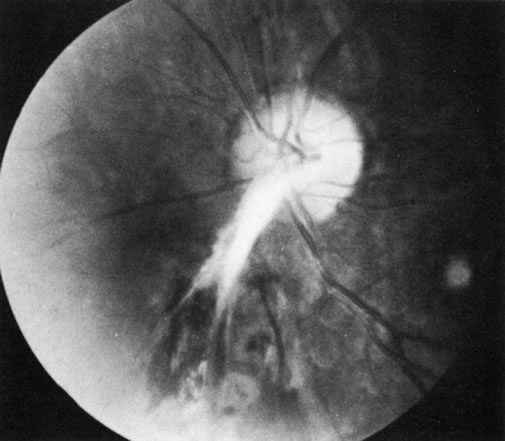
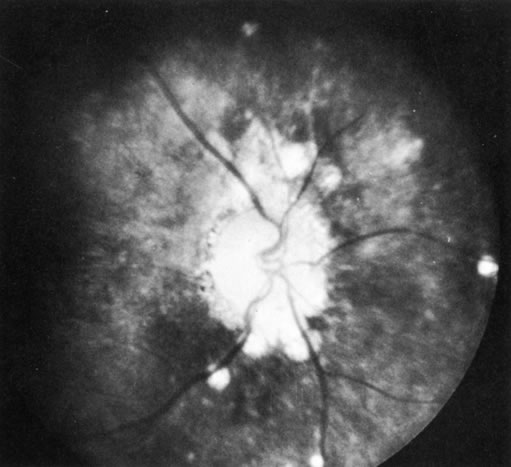
1. Morgan SJ, Williams JH, Walls AF et al: Mast cell number and staining characteristics in the normal and allergic
conjunctiva. J Allergy Clin Immunol; 87:111, 1991. 2. Irani AM, Butrus S, Tabbara KF et al: Human conjunctival mast cell: Distribution of MCT and MCTC in vernal and
giant papillary conjunctivitis. J Allergy Clin Immunol 86:34, 1990. 3. Graziano FM, Stahl JL, Cook EB et al: Conjunctival mast cells in ocular allergic disease. Allergy Asthma Proc 3:121, 2001. 4. Terr AI: Inflammation. In: Parslow TC, Stites DP, Terr AI, Imboden JB (eds): Medical Immunology, pp 189–203. New York: Lange, 2001. 5. Gleich GJ: Mechanisms of eosinophil-associated inflammation. J Allergy Clin Immunol 105:651, 2000. 6. Buckley CF, Cohen AB: Nasal mucosal hyperpermeability to macromolecules in atopic rhinitis and
extrinsic asthma. J Allergy Clin Immunol 55:213, 1975. 7. Kaufmann HS, Hobbs JR: Immunoglobulin deficiencies in an atopic population. Lancet 2:1061, 1970. 8. Hubscher TT: Immune and biochemical mechanisms in the allergic disease of the upper
respiratory tract: Role of antibodies, target cells, mediators, and eosinophils. Ann Allergy 38:83, 1977. 9. Cooke RA, Veer VA: Human sensitization. J Immunol 1:201, 1916. 10. Marsh DG, Bias WB, Hsu SH et al: Association of an HLA-7 cross-reacting group with a specific
reaginic antibody response in allergic man. Science 179:691, 1973. 11. Kisimoto T, Ishizaka K: Regulation of antibody response in vitro: 6. Carrier-specific helper
cells for IgG and IgE antibody response. J Immunol 111:720, 1973. 12. Theodore FH, Lewson AS: Bilateral iritis complicating serum sickness. Arch Ophthalmol 21:828, 1939. 13. Erffmeyer JE: Serum sickness. Ann Allergy 56:105–109, 1986. 14. Hoover RE: Nongranulomatous uveitis: A complication of serum sickness. Am J Ophthalmol 41:534, 1956. 15. Wong VG, Anderson RR, McMaster PRB: Endogenous immune uveitis. Arch Ophthalmol 85:93, 1971. 16. James DG, Neville E, Walker A: Immunology of sarcoidosis. Am J Med 59:388, 1975. 17. Kataria VP: Sarcoidosis: An overview. Clin Notes Respir Dis 14:2, 1975. 18. Mitchell DN, Scadding JG: Sarcoidosis. Am Rev Respir Dis 110:774, 1974. 19. Steplewski Z, Israel HL: The search for viruses in sarcoidosis. Proceedings of the Seventh International
Conference on Sarcoidosis. Ann N Y Acad Sci 278:260, 1976. 20. Mueller-Quernheim J: Sarcoidosis: Immunopathogenetic concepts and their application. Eur Respir J 12:716, 1998. 21. Cummiskey JM, McLaughlin H, Keelan P: T and B lymphocytes in sarcoidosis: A clinical correlation. Thorax 31:665, 1976. 22. Ramachandar K, Douglas DS, Siltzbach LE: Peripheral blood lymphocyte subpopulations in sarcoidosis. Cell Immunol 16:442, 1975. 23. Hirschorn K, Schriebman RR, Bach FH et al: In vitro studies of lymphocytes from patients with sarcoidosis and lymphoproliferative
disease. Lancet 2:842, 1964. 24. Becker FW, Krul P, Deicher H et al: The leukocyte migration test in sarcoidosis. Lancet 1:120, 1972. 25. Byrne EB, Evans AS, Fouts DW et al: A seroepidemiological study of Epstein-Barr virus and other viral
antigens in sarcoidosis. Am J Epidemiol 97:355, 1973. 26. Wahren B, Carlens E, Espmark A et al: Antibodies to various herpes viruses in sera from patients with sarcoidosis. J Natl Cancer Inst 47:747, 1971. 27. Sands JH, Palmer PP, Maycock RL: Evidence for serological hyperreactivity in sarcoidosis. Am J Med 19:401, 1955. 28. Goldstein RA, Israel HA, Janicki BW et al: Serum immunoglobulin D levels in sarcoidosis. Proceedings of the Sixth
International Conference on Sarcoidosis. Tokyo: Tokyo University Press, 196, 1974. 29. Buckley CE, Trayer HR: Serum IgD concentrations in sarcoidosis and tuberculosis. Clin Exp Immunol 10:257, 1972. 30. Von Bergmann K-C, Zausmmell I, Lachmann B: IgE konzentratione im serum von patienten mit sarkoidose und lungentuberkulose. Dtsch Geisindheitsw 27:1774, 1972. 31. Buckley CE, Nagaya H, Sieker HO: Altered immunologic activity in sarcoidosis. Ann Intern Med 64:508, 1966. 32. Agnello V, Winchester RJ, Kunkel HG: Precipitin reaction of the C1q component of complement with aggregated
gamma-globulin and immune complexes in gel diffusion. Immunology 19:909, 1970. 33. Hedfors E, Norberg R: Evidence for circulating immune complexes in sarcoidosis. Clin Exp Immunol 16:493, 1974. 34. Behçet H: Uber die rezidivierende, Aphthose, durch ein Virus verursachte Geschwure
am Mund, am Auge und an den Genitalien. Dermatol Wochenschr 105:1152, 1937. 35. Noyan B, Gursky G, Aktiu E: Inoculation of cerebrospinal fluid of a neuro-Behçet patient
into mice. Acta Neuropathol (Berl) 12:195, 1969. 36. Sezer N: Further investigations on the virus of Behçet's disease. Am J Ophthalmol 41:41, 1956. 37. Evans AD, Pallis CA, Spillane JD: Involvement of the nervous system in Behçet's syndrome: Report
of three cases and isolation of virus. Lancet 2:349, 1957. 38. Dowling GB: Behçet's disease. Proc R Soc Med 54:101, 1961. 39. Sigel N, Larson R: Behçet's syndrome: A case with benign pericarditis and recurrent
neurologic involvement treated with adrenal steroids. Arch Intern Med 115:203, 1965. 40. Oshima Y, Shimizu T, Yokohari et al: Clinical studies on Behçet's syndrome. Ann Rheum Dis 22:36, 1963. 41. Shimizu T, Katsuta Y, Oshima Y: Immunological studies on Behçet's syndrome. Ann Rheum Dis 24:494, 1965. 42. Cooper DA, Penny R: Behçet's syndrome: Clinical, immunological, and therapeutic
evaluation of 17 patients. Aust N Z J Med 4:585, 1974. 43. Lehner T: Behçet's syndrome and autoimmunity. Br Med J 1:465, 1967. 44. O'Duffey JD, Carney JA: Behçet's disease: Report of 10 cases, 3 with new manifestations. Ann Intern Med 75:561, 1971. 45. Haim S, Sobel JD, Friedman-Birnbaum R et al: Histological and direct immunofluorescence study of cutaneous hyperactivity
in Behçet's disease. Br J Dermatol 95:631, 1976. 46. Lehner T: Immunological aspects of recurrent oral ulcers. Lancet 2:80, 1964. 47. Cooper D, Penny R, Fiddes P: Autologous-plasma sensitization in Behçet's disease. Lancet 1:910, 1971. 48. Kogure M, Shimada K, Hara HG: Complement titer in patients with Behçet's disease. Acta Soc Ophthalmol Jpn 75:1260, 1971. 49. Kawachi-Takahashi S, Takahashi M, Kogure M et al:. Evaluation of C9 level associated with Behçet's disease. Jpn J Exp Med 44:488, 1974. 50. Lehner T: Stimulation of lymphocyte transformation by tissue homogenate in recurrent
oral ulceration. Immunology 13:159, 1967. 51. Dolby AE: Recurrent aphthous ulceration: Effect of sera and peripheral blood lymphocytes
upon oral epithelial tissue culture cells. Immunology 17:709, 1967. 52. Rogers RS, Sams WM, Shorter EG: Lymphocytotoxicity in recurrent aphthous stomatitis: Lymphotoxicity for
oral epithelial cells in recurrent aphthous stomatitis and Behçet's
syndrome. Arch Dermatol 109:361, 1974. 53. Coe JE, Feldman JD, Lee S: Immunologic competence of thoracic duct cells: I. Delayed hypersensitivity. J Exp Med 123:267, 1966. 54. Lehner T: Characterization of mucosal antibodies in recurrent aphthous ulceration
and Behçet's syndrome. Arch Oral Biol 14:843, 1969. 55. Lehner T: Pathology of recurrent oral ulceration and oral ulceration in Behçet's
syndrome: Light, electron, and fluorescence microscopy. J Pathol 97:481,1969. 56. Marquardt JL, Snyderman R, Oppenheim JJ: Depression of lymphocyte transformation and exacerbation of Behçet's
syndrome by ingestion of English walnuts. Cell Immunol 9:263, 1973. 57. Ohno S, Nakayama E, Suguira S et al: Specific histocompatibility antigens associated with Behçet's
disease. Am J Ophthalmol; 80:636, 1975. 58. O'Duffy JD, Taswell HF, Elvebeck LR: HLA antigens in Behçet's disease. J Rheumatol 3:1, 1976. 59. Goolamli SK, Comaish JS, Hassanyeh F: Familial Bench's syndrome. Br J Dermatol 95:637, 1976. 60. Meador R, Ehrlich G, Von Feldt JM: Behçet's disease: Immunopathologic and therapeutic aspects. Curr Rheumatol Rep 4:47, 2002. 61. Takahashi M: Clinical and experimental studies on the nature of idiopathic, bilateral, severe
uveitis. Acta Soc Ophthalmol Jpn 34:506, 1930. 62. Friedenwald JS, McKee EM: A filter-pasing agent as a cause in endophthalmitis. Am J Ophthalmol 21:723, 1938. 63. Remky H: Beitrag zur Kenntnis schwerer Foremen der Uveoencephalitis. Klin Monatsbl Augenheilkd 123:166, 1953. 64. Morris RW, Schlaegel TF: Virus-like inclusion bodies in subretinal fluid in uveoencephalitis. Am J Ophthalmol 58:940, 1964. 65. Wilczek M, Feltynowski A: Study of sympathetic ophthalmia by electron microscopy. Klin Oczna 25:11, 1956. 66. Rao NA: Mechanisms of inflammatory response in sympathetic ophthalmia and VKH syndrome. Eye 11:213, 1997. 67. Pivetti-Pezzi P, Accorinti M, Colabelli-Gisoldi RA et al: Vogt-Koyanagi-Harada disease and HLA type in Italian patients. Am J Ophthalmol 122:889, 1996. 68. Gocho K, Kondo I, Yamaki K: Identification of autoreactive T cells in Vogt-Koyanagi-Harada
disease. Invest Ophthalmol Vis Sci 42:2004, 2001. 69. Kahan A, Sztanojevits A, Szabados T et al: Pigment-autoagression in der Pathogenese des Vogt-Koyanagi-Harada
syndrome. Graefes Arch Ophthalmol 167:246, 1964. 70. McPherson SD, Woods AC: The significance of the intracutaneous test for hypersensitivity to uveal
pigment. Am J Ophthalmol 31:35, 1948. 71. Hammer H: Lymphocyte transformation test in sympathetic ophthalmitis and Vogt-Koyanagi-Harada
syndrome. Br J Ophthalmol 55:850, 1971. 72. Hammer H: Cellular hypersensitivity to uveal pigment confirmed by leukocyte migration
tests in sympathetic ophthalmitis and the Vogt-Koyanagi-Harada
syndrome. Br J Ophthalmol 58:773, 1974. 73. Sanefuji M: Cell-mediated immunity in uveitis: II. Leukocyte migration inhibition
test in Harada disease. Acta Soc Ophthalmol Jpn 78:306, 1974. 74. Char DH, Brunn J, West W: Thymus-derived lymphocytes in the Vogt-Koyanagi-Harada
syndrome. Invest Ophthalmol 16:179, 1977. 75. Tagawa Y, Sugiura S, Yakura H et al: HLA and Vogt-Koyanagi-Harada syndrome. N Engl J Med 295:173, 1976. 76. Ohno S, Char DH, Kimura SJ et al: HLA and Vogt-Koyanagi-Harada syndrome. N Engl J Med 295:788, 1976. 77. Perry HD, Font RL: Clinical and histopathologic observations in severe Vogt-Koyanagi-Harada
syndrome. Am J Ophthalmol 83:242, 1977. 78. Chan CC, Palestine AG, Kuwabara T et al: Immunohistologic studies of Vogt-Koyanagi-Harada syndrome. Am J Ophthalmol 105:607, 1988. 79. Inomata L, Sakamoto T: Immunohistochemical studies of Vogt-Koyanagi-Harada disease
with sunset sky fundus. Curr Eye Res 924, 1990. 80. Mitchell DN, Rees RJW: Agent transmissible from Crohn's disease tissue. Lancet 2:168, 1970. 81. Cave DR, Mitchell DN, Kane SP et al: Further animal evidence of a transmissible agent in Crohn's disease. Lancet 192:1120, 1973. 82. Gitnick GL, Rosen VJ: Electron microscopic studies of viral agents in Crohn's disease. Lancet 2:217, 1976. 83. Parent K, Mitchell P: Cultivation of viral agents from Crohn's disease. Lancet 2:576, 1976. 84. Anderson AFR: Gastrointestinal manifestations in food allergy. Med J Rec 122:271, 1925. 85. Binder V, Hvidberg E: Histamine content of rectal mucosa in ulcerative colitis. Gut 8:24, 1967. 86. Broberger O, Perlmann P: Autoantibodies in human ulcerative colitis. J Exp Med 110:657, 1959. 87. Weinstein L: Bacterial aspects of ulcerative colitis. Gastroenterology 40:323, 1961. 88. Kraft SC, Kirsner JB: The immunology of ulcerative colitis and Crohn's disease: Clinical
and humoral aspects. In: Kirsner JB, Shorter RG (eds): Inflammatory Bowel Disease, pp 72–73. Philadelphia: Lea & Febiger, 1975. 89. Ballard J, Shiner M: Evidence of cytotoxicity in ulcerative colitis from immunofluorescent staining
of the rectal mucosa. Lancet 1:1014, 1974. 90. Jones JV, Housley J, Ashurst PM et al: Development of delayed hypersensitivity to dinitrochlorobenzene in patients
with Crohn's disease. Gut 10:52, 1969. 91. Meyers S, Sachar DB, Taub RN et al: Anergy to dinitrochlorobenzene and depression of T-lymphocytes in
Crohn's disease and ulcerative colitis. Gut 17:911, 1976. 92. Whorwell PJ, Wright R: Immunological aspects of inflammatory bowel disease. Clin Gastroenterol 5:303, 1976. 93. Perlmann P, Broberger O: In vitro studies of ulcerative colitis: Cytotoxic action of white blood
cells from patients on human fetal colon cells. J Exp Med 117:717, 1963. 94. Shorter EG, Cardoza M, Spencer RJ et al: Further studies of in vitro cytotoxicity of lymphocytes from patients with
ulcerative and granulomatous colitis for allogeneic colonic epithelial
cells including the effects of colectomy. Gastroenterology 56:304, 1969. 95. Watson DW, Shorter EG: The immunology of ulcerative colitis and Crohn's disease: Cell-mediated
immunity responses. In: Kirsner JB, Shorter EG (eds): Inflammatory Bowel Disease, p 61. Philadelphia: Lea & Febiger, 1975. 96. Asquith P, Mackintosh P, Stokes PL et al: Histocompatibility antigens in patients with inflammatory bowel disease. Lancet 1:113, 1974. 97. Nahir M, Gideoni O, Eidelman S et al: HLA antigens in ulcerative colitis. Lancet 1:573, 1976. 98. Mallas EG, Mackintosh P, Asquith P et al: Histocompatibility antigens in inflammatory bowel disease. Gut 17:906, 1976. 99. Brophy S, Pavy S, Lewis P et al: Inflammatory eye, skin, and bowel disease in spondyloarthritis: Genetic, phenotypic, and
environmental factors. J Rheumatol 28:2667, 2001. 100. Lyons JL, Rosenbaum JT: Uveitis associated with inflammatory bowel disease compared with uveitis
associated with spondyloarthropathy. Arch Ophthalmol 115:61, 1997. |