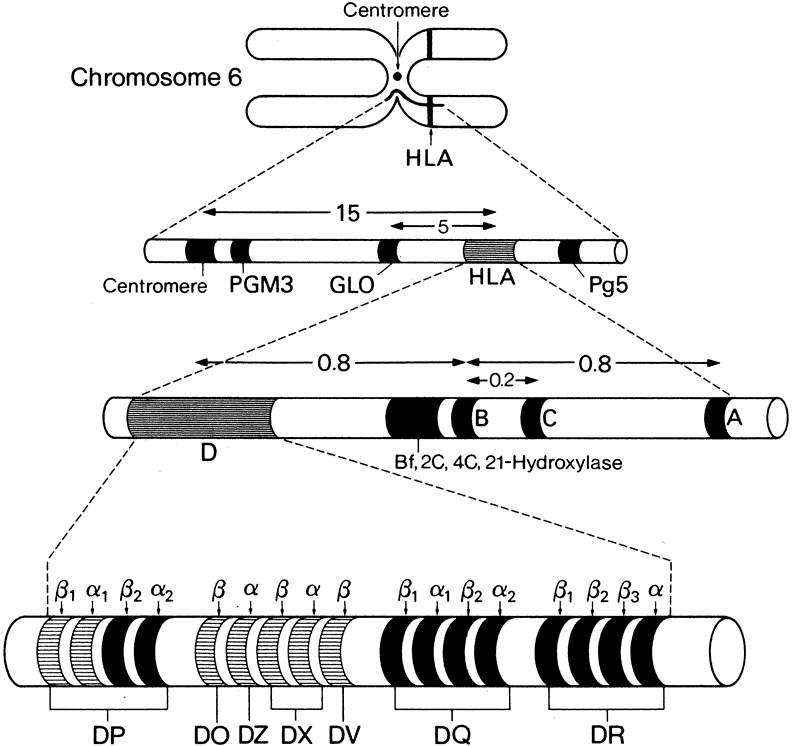
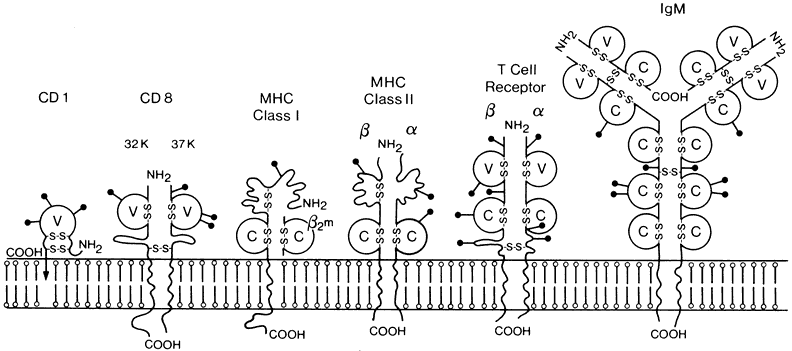
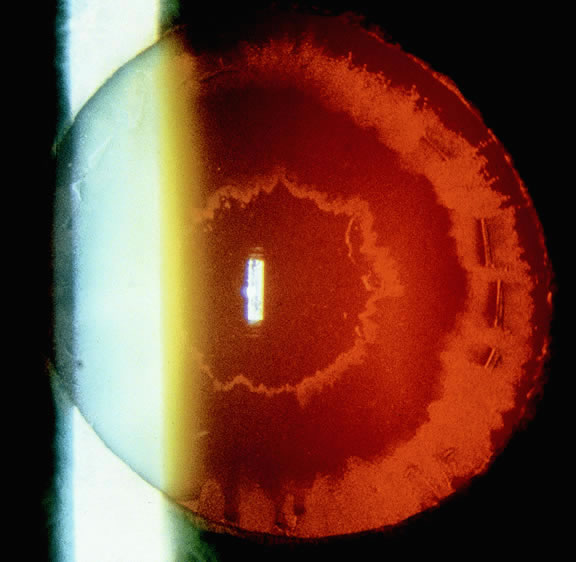
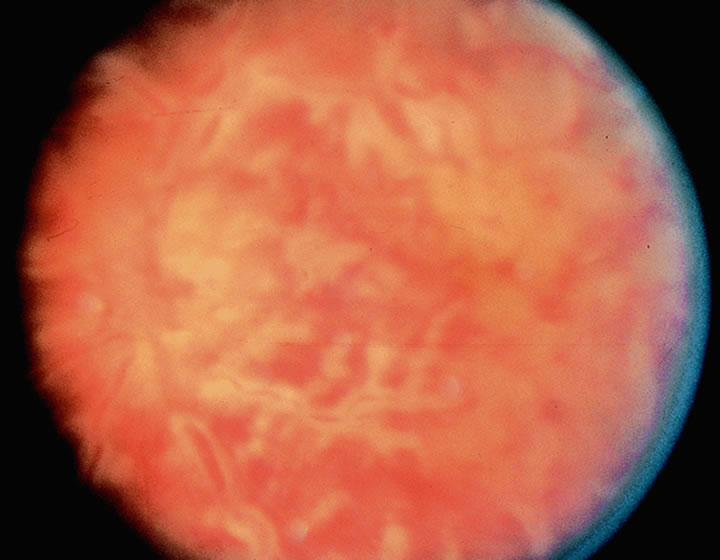
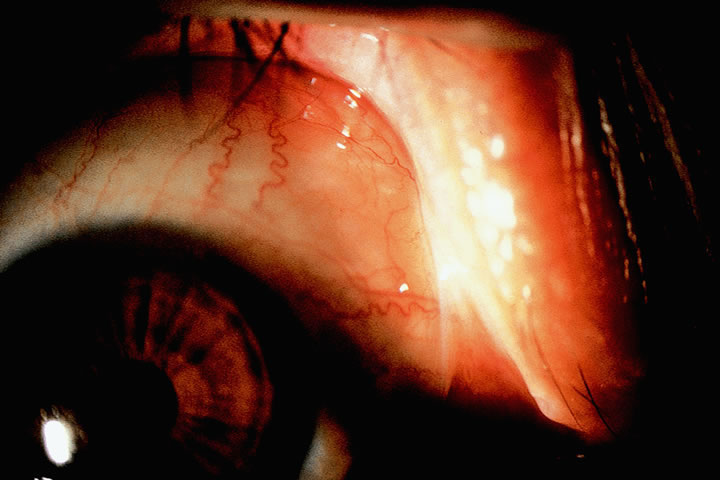
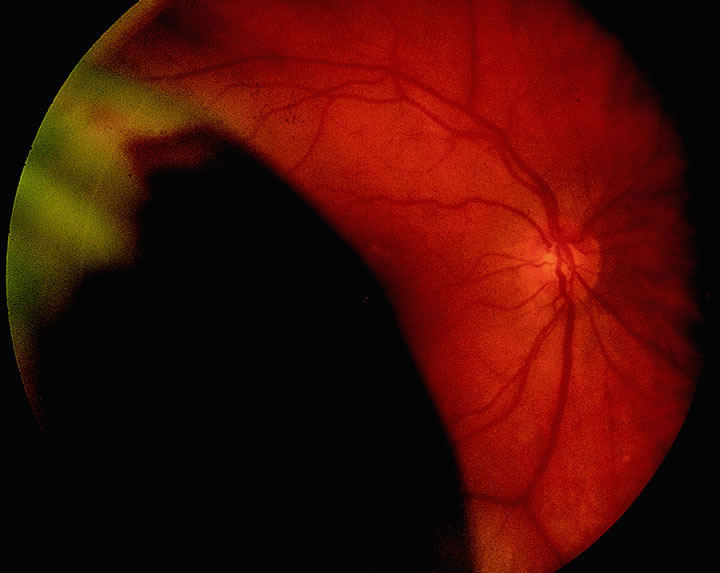
1. Lechler R, Warrens AN. HLA in Health and Disease. 2nd ed. San Diego, CA: Academic, 2000:xvii, 472 2. Bach FH. Immunogenetics of HLA. Wien Klin Wochenschr 1991;103:141 3. Taylor CJ, Dyer PA. Histocompatibility antigens. Eye 1995;9:173 4. Wordsworth P. HLA nomenclature: A user-friendly system? Ann Rheum Dis 1994;53:153 5. Zmijewski CM. HLA and disease. Crit Rev Clin Lab Sci 1984;20:285 6. Trowsdale J, Campbell RD. Complexity in the major histocompatibility complex. Eur
J Immunogenet 1992;19:45 7. Bodmer WF. HLA: What's in a name? A commentary on HLA nomenclature
development over the years. Tissue Antigens 1997;49:293 8. Bodmer JG, Marsh SG, Albert ED et al. Nomenclature for factors of the HLA
system, 1994. Eur J Immunogenet 1994;21:485 9. Kasahara M. Major Histocompatibility Complex: Evolution, Structure, and
Function. Tokyo; New York: Springer, 2000:xxiii, 561 10. Holmes N, Parham P. Exon shuffling in vivo can generate novel HLA Class
I molecules. EMBO J 1985;4:2849 11. Schreuder GM, Hurley CK, Marsh SG et al. The HLA dictionary 1999: A summary
of HLA-A, -B, -C, -DRB1/3/4/5, -DQB1 alleles and their association
with serologically defined HLA-A, -B, -C, -DR and -DQ antigens. Tissue
Antigens 1999;54:409 12. Wujciak T, Opelz G. Evaluation of HLA matching for CREG antigens in Europe. Transplantation 1999;68:1097 13. Reveille, JD. HLA-B27 and the seronegative spondyloarthropathies. Am J
Med Sci 1998;316:239 14. Reveille JD, Ball EJ, Khan MA. HLA-B27 and genetic predisposing factors
in spondyloarthropathies. Curr Opin Rheumatol 2001;13:265 15. Carrington M. Recombination within the human MHC. Immunol Rev 1999;167:245 16. Alfonso C, Karlsson L. Nonclassical MHC Class II molecules. Annu Rev Immunol 2000;18:113 17. Fischer GF. Molecular genetics of HLA. Vox Sang 2000;78(Suppl 2):261 18. Davidson JA, Kippax RL, Dyer PA. A study of HLA-A, B, DR and Bf bearing
haplotypes derived from 304 families resident in the north west of England. J
Immunogenet 1988;15:227 19. Mittal KK. Immunobiology of the human major histocompatibility complex: Association
of HLA antigens with disease. Acta Anthropogenet 1984;8:245 20. Schreuder GM, Hurley CK, Marsh SG et al. HL-A antigen in Sjögren's
syndrome. Ann Ophthalmol 1980;12:1084 21. Marga M, Denisova A, Sochnev A et al. Two HLA DRB 1 alleles confer independent
genetic susceptibility to Graves disease: Relevance of cross-population
studies. Am J Med Genet 2001;102:188 22. Philippou G, Krimitzas A, Kaltsas G et al. HLA DQA1*0501 and DRB1*0301 antigens
do not independently convey susceptibility to Graves' disease. J
Endocrinol Invest 2001;24:88 23. Browning MJ, McMichael AJ. HLA and MHC: Genes, Molecules, and Function. Human
Molecular Genetics Series. Oxford, UK; Herndon, VA: BIOS Scientific, 1996:xvii, 438 24. Hood L, Campbell JH, Elgin SC. The organization, expression, and evolution
of antibody genes and other multigene families. Annu Rev Genet 1975;9:305 25. Huang SK, Yi M, Kumai M et al. Immunogenetic aspects of IgE-mediated responses. Adv
Exp Med Biol 1994;347:11 26. Le Bouteiller P, Blaschitz A. The functionality of HLA-G is emerging. Immunol
Rev 1999;167:233 27. Nelson CA, Fremont DH. Structural principles of MHC Class II antigen presentation. Rev
Immunogenet 1999;1:47 28. O'Callaghan CA, Bell JI. Structure and function of the human MHC Class
Ib molecules HLA-E, HLA-F and HLA-G. Immunol Rev 1998;163:129 29. Natarajan K, Li H, Mariuzza RA et al. MHC Class I molecules, structure
and function. Rev Immunogenet 1999;1:32 30. Le Bouteiller P. HLA Class I chromosomal region, genes, and products: Facts
and questions. Crit Rev Immunol 1994;14:89 31. Volpi EV, Chevret E, Jones T et al. Large-scale chromatin organization
of the major histocompatibility complex and other regions of human chromosome 6 and
its response to interferon in interphase nuclei. J Cell
Sci 2000;113:1565 32. Morita N, Munkhbat B, Gansuvd B et al. Effect of HLA-A and -DPB1 matching
in corneal transplantation. Transplant Proc 1998;30:3491 33. Urban RG, Chicz RM. MHC molecules: Expression, Assembly, and Function. Medical
intelligence unit. New York; Austin: Chapman & Hall; RG Landes, 1996:298 34. Woolfrey AE, Nepom GT. Differential transcription elements direct expression
of HLA-DQ genes. Clin Immunol Immunopathol 1995;74:119 35. Sonderstrup G, McDevitt HO. DR, DQ, and you: MHC alleles and autoimmunity. J
Clin Invest 2001;107:795 36. Truedsson L, Alper CA, Awdeh ZL et al. Characterization of type I complement
C2 deficiency MHC haplotypes. Strong conservation of the complotype/HLA-B-region
and absence of disease association due to linked Class
II genes. J Immunol 1993:151:5856 37. Treseler PA, Foulks GN, Sanfilippo F. The expression of HLA antigens by
cells in the human cornea. Am J Ophthalmol 1984;98:763 38. Carpenter CB. HLA Class I DNA typing in organ transplantation. Tissue Antigens 1997;50:322 39. Niederkorn JY, Chiang EY, Ungchusri T et al. Expression of a nonclassical
MHC Class Ib molecule in the eye. Transplantation 1999;68:1790 40. Batchelor JR, Casey TA, Werb A et al. HLA matching and corneal grafting. Lancet 1976;1:551 41. Nelson KA. Human leukocyte antigen system and the immune response to it. Clin
Orthop 1996;326:35 42. Streilein JW, Dana MR, Ksander BR. Immunity causing blindness: Five different
paths to herpes stromal keratitis. Immunol Today 1997;18:443 43. Sompayrac L. How the Immune System Works. Malden, MA: Blackwell Science, 1999:vii, 111 44. Davey MP, Rosenbaum JT. The human leukocyte antigen complex and chronic
ocular inflammatory disorders. Am J Ophthalmol 2000;129:235 45. Abi-Hanna D, Wakefield D, Watkins S. HLA antigens in ocular tissues. I. In
vivo expression in human eyes. Transplantation 1988;45:610 46. Steptoe RJ, Holt PG, McMenamin PG. Origin and steady-state turnover of
major histocompatibility complex Class II-positive dendritic cells and
resident-tissue macrophages in the iris of the rat eye. J Neuroimmunol 1996;68:67 47. Steptoe RJ, Holt PG, McMenamin PG. Functional studies of major histocompatibility
Class II-positive dendritic cells and resident tissue macrophages
isolated from the rat iris. Immunology 1995;85:630 48. Diaz-Araya CM, Madigan MC, Provis JM et al. Immunohistochemical and topographic
studies of dendritic cells and macrophages in human fetal cornea. Invest
Ophthalmol Vis Sci 1995;36:644 49. McCluskey J, Peh CA. The human leucocyte antigens and clinical medicine: An
overview. Rev Immunogenet 1999;1:3 50. Fernandez N, Butcher G. MHC: A Practical Approach. Practical approach series. Oxford; New
York: IRL Press at Oxford University Press, 1997:v, 180 51. Whitton JL. Antigen Presentation. Current Topics in Microbiology and Immunology, 232. Berlin; New
York: Springer, 1998:vi, 244. 52. Singh N, Agrawal S, Rastogi AK. Infectious diseases and immunity: Special
reference to major histocompatibility complex. Emerg Infect Dis 1997;3:41 53. Viret C, Janeway CA Jr. MHC and T cell development. Rev Immunogenet 1999;1:91 54. Begovich AB, Erlich HA. HLA typing for bone marrow transplantation. New
polymerase chain reaction-based methods. JAMA 1995;273:586 55. Wernet P, Kogler G, Enczmam J et al. Rapid method for successful HLA Class
I and II typing from cadaveric blood for direct matching in cornea
transplantation. Graefes Arch Clin Exp Ophthalmol 1998;236:507 56. Bray RA. Flow cytometry in human leukocyte antigen testing. Semin Hematol 2001;38:194 57. Erlich HA, Opelz G, Hansen J. HLA DNA typing and transplantation. Immunity 2001;14:347 58. Odum N. Cellular typing for HLA-DF: Bulk-expansion of primed lymphocyte
typing (PLT) cells. Technical and immunogenetic aspects. Dan Med Bull 1989;36:551 59. Munkhbat B, Hagihara M, Sato T et al. HLA Class II DNA typing using ocular
tissue and its usefulness in corneal transplantation. Transplant Proc 1996;28:1257 60. Blake E, Mihalovich J, Higuchi R et al. Polymerase chain reaction (PCR) amplification
and human leukocyte antigen (HLA)-DQ alpha oligonucleotide
typing on biological evidence samples: Casework experience. J Forensic
Sci 1992;37:700 61. Baumgartner I, Huber-Spitzy V, Grabner G et al. HLA typing from human donor
eyes. Graefes Arch Clin Exp Ophthalmol 1989;227:541 62. Zavazava N, Halene M, Westphal E et al. Expression of MHC Class I and II
molecules by cadaver retinal pigment epithelium cells: Optimization
of post-mortem HLA typing. Clin Exp Immunol 1991;84:163 63. Kapasi K, Inman RD. ME1 epitope of HLA-B27 confers Class I-mediated modulation
of gram- negative bacterial invasion. J Immunol 1994;153:833 64. Liblau R, Gautam AM. HLA, molecular mimicry and multiple sclerosis. Rev
Immunogenet 2000;2:95 65. Ebringer A, Ahmadi K, Fielder M et al. Molecular mimicry: The geographical
distribution of immune responses to Klebsiella in ankylosing spondylitis
and its relevance to therapy. Clin Rheumatol 1996;15(Suppl 1):57 66. Schwimmbeck PL, Yu DT, Oldstone MB. Autoantibodies to HLA B27 in the sera
of HLA B27 patients with ankylosing spondylitis and Reiter's syndrome. Molecular
mimicry with Klebsiella pneumoniae as potential mechanism of autoimmune disease. J Exp Med 1987;166:173 67. Yu DT, Choo SY, Schaack T. Molecular mimicry in HLA-B27-related arthritis. Ann
Intern Med 1989;111:581 68. Bittencourt PL, Goldberg AC, Cancado EL et al. Different HLA profiles confer
susceptibility to autoimmune hepatitis type 1 and 2. Am J Gastroenterol 1998;93:1394 69. Thomson G. HLA disease associations: Models for the study of complex human
genetic disorders. Crit Rev Clin Lab Sci 1995;32:183 70. Huhtinen M, Karma A. HLA-B27 typing in the categorisation of uveitis in
a HLA-B27 rich population. Br J Ophthalmol 2000;84:413 71. Kijlstra A. The value of laboratory testing in uveitis. Eye 1990;4:732 72. Allen RL, Bowness P, McMichael A. The role of HLA-B27 in spondyloarthritis. Immunogenetics 1999;50:220 73. Criteria for diagnosis of Behçet's disease. International Study
Group for Behçet's Disease. Lancet 1990;335:1078 74. Zierhut M, Feltkamp B, Forrester J et al. Immunology of the eye and the
joint. Immunol Today 1994;15:249 75. Dodds EM, Lowder CY, Meisler DM. Posterior segment inflammation in HLA-B27+ acute
anterior uveitis: Clinical characteristics. Ocul Immunol
Inflamm 1999;7:85 76. Saari KM, Hakli J, Solja J et al. HLA-B27 frequency and MLC reactions in
acute anterior uveitis. Albrecht Von Graefes Arch Klin Exp Ophthalmol 1981;216:23 77. Feltkamp TE. HLA and uveitis. Int Ophthalmol 1990;14:327 78. Shinzato M, Yamamoto J, Hirata CE et al. Eye disease in a patient with
rheumatoid arthritis. Postgrad Med J 1999;75:676 79. Lyons JL, Rosenbaum JT. Uveitis associated with inflammatory bowel disease
compared with uveitis associated with spondyloarthropathy. Arch Ophthalmol 1997;115:61 80. Schlosstein L, Terasaki PI, Bluestone R et al. High association of an HL-A
antigen, W27, with ankylosing spondylitis. N Engl J Med 1973;288:704 81. Jabs DA, Rosenbaum JT, Foster CS et al. Guidelines for the use of immunosuppressive
drugs in patients with ocular inflammatory disorders: Recommendations
of an expert panel. Am J Ophthalmol 2000;130:492 82. Baarsma GS, Priem HA, Kijlstra A. Association of birdshot retinochoroidopathy
and HLA-A29 antigen. Curr Eye Res 1990;9(Suppl):63 83. Meredith TA, Smith RE, Duquesnoy RJ. Association of HLA-DRw2 antigen with
presumed ocular histoplasmosis. Am J Ophthalmol 1980;89:70 84. Spaide RF, Skerry JE, Yannuzzi LA et al. Lack of the HLA-DR2 specificity
in multifocal choroiditis and panuveitis. Br J Ophthalmol 1990;74:536 85. Davis JL, Mittal KK, Freidlin V et al. HLA associations and ancestry in
Vogt-Koyanagi-Harada disease and sympathetic ophthalmia. Ophthalmology 1990;97:1137 86. Richard G, Dieckhues B, Junemann G. [Immunological examinations in
Vogt-Koyanagi-Harada disease (author's transl)]. Albrecht Von
Graefes Arch Klin Exp Ophthalmol 1981;217:325 87. Islam SM, Numaga J, Fujino Y et al. HLA Class II genes in Vogt-Koyanagi-Harada
disease. Invest Ophthalmol Vis Sci 1994;35:3890 88. Rathinam SR, Nampcrumalsamy P, Nozik RA et al. Vogt-Koyanagi-Harada syndrome
after cutaneous injury. Ophthalmology 1999;106:635 89. Goldberg AC, Yamamoto JH, Chiarella JM et al. HLA-DRB1*0405 is the predominant
allele in Brazilian patients with Vogt-Koyanagi-Harada disease. Hum
Immunol 1998:59:183 90. Reynard M, Shulman IA, Azen SP et al. Histocompatibility antigens in sympathetic
ophthalmia. Am J Ophthalmol 1983;95:216 91. Shindo Y, Ohno S, Usui M et al. Immunogenetic study of sympathetic ophthalmia. Tissue
Antigens 1997;49:111 92. Biswas J, Raizada S. Immunopathology of uveitis. Indian J Lepr 1998;70:11 93. Nussenblatt RB. Proctor Lecture. Experimental autoimmune uveitis: Mechanisms
of disease and clinical therapeutic indications. Invest Ophthalmol
Vis Sci 1991;32:3131 94. Malinovsky VE. Glaucomatocyclitic crisis. J Am Optom Assoc 1983;54:1069 95. Hirose S, Ohno S, Matsuda H. HLA-Bw54 and glaucomatocyclitic crisis. Arch
Ophthalmol 1985;103:1837 96. Olivius E, Polland W. Histocompatibility (HLA) antigens in capsular glaucoma
and simplex glaucoma. Acta Ophthalmol Copenh 1980;58:406 97. Slagsvold JE, Nordhagen R. The HLA system in primary open angle glaucoma
and in patients with pseudoexfoliation of the lens capsule (exfoliation
or fibrillopathia epitheliocapsularis). Acta Ophthalmol Copenh 1980;58:188 98. FitzSimon JS, Mulvihill A, Kennedy S et al. Association of HLA type with
pseudoexfoliation of the lens capsule. Br J Ophthalmol 1996;80:402 99. Ohno S, Kimura SJ, O'Connor GR et al. HLA antigens and uveitis. Br J Ophthalmol 1977;61:62 100. Kilmartin DJ, Finch A, Acheson RW. Primary association of HLA-B51 with
Behçet's disease in Ireland. Br J Ophthalmol 1997;81:649 101. Yazici H, Kimura SJ, O'Connor GR et al. HLA antigens in Behçet's
disease: A reappraisal by a comparative study of Turkish and British
patients. Ann Rheum Dis 1980;39:344 102. Mizuki M, Ohno S, Ando H et al. Major histocompatibility complex Class
II alleles in Kazak and Han populations in the Silk Route of northwestern
China. Tissue Antigens 1997:50:527 103. Gul A, Uyar FA, Inanc M et al. Lack of association of HLA-B*51 with a severe
disease course in Behçet's disease. Rheumatology Oxford 2001;40:668 104. Kotter I, Gunaydin I, Stubiger N et al. Comparative analysis of the association
of HLA-B*51 suballeles with Behçet's disease in patients
of German and Turkish origin. Tissue Antigens 2001;58:166 105. Murray KJ, Moroldo MB, Donnelly P et al. Age-specific effects of juvenile
rheumatoid arthritis-associated HLA alleles. Arthritis Rheum 1999;42:1843 106. Cerna M, Vavrincova P, Havelka S. HLA and juvenile rheumatoid arthritis. Acta
Univ Carol Med 1994;40:69 107. Gasset AR, Richman AV, Frias JL. HLA antigens and keratoconus. Ann Ophthalmol 1977;9:767 108. Ihalainen A. Clinical and epidemiological features of keratoconus genetic
and external factors in the pathogenesis of the disease. Acta Ophthalmol 1986;178(Suppl):1 109. Gorskova EN, Sevost'ianov EN. [Associations of HLA Class I haplotype
antigens with various patterns of keratoconus]. Vestn Oftalmol 1997;113:31 110. Cogan DG, Sullivan WR Jr. Immunologic study of nonsyphilitic interstitial
keratitis with vestibuloauditory symptoms. Am J Ophthalmol 1975;80:491 111. Kaiser-Kupfer MI, Del Valle LA, Mittal KK et al. HLA and Cogan's syndrome. N
Engl J Med 1978;298:1094 112. Taylor CJ, Smith SI, Morgan CH et al. HLA and Mooren's ulceration. Br
J Ophthalmol 2000;84:72 113. Wang Z, Chen J, Zheng H. Changes in local immune functions in Mooren's
ulcer. Yan Ke Xue Bao 1996;12:33 114. Chan LS, Hammerberg C, Cooper KD. Significantly increased occurrence of
HLA-DQB1*0301 allele in patients with ocular cicatricial pemphigoid. J
Invest Dermatol 1997;108:129 115. Power WJ, Saidman SL, Zhang DS et al. HLA typing in patients with ocular
manifestations of Stevens-Johnson syndrome. Ophthalmology 1996;103:1406 116. Mondino BJ, Brown SI, Biglan AW. HLA antigens in Stevens-Johnson syndrome
with ocular involvement. Arch Ophthalmol 1982;100:1453 117. Darrell, RW. Thygeson's superficial punctate keratitis: Natural history
and association with HLA DR3. Trans Am Ophthalmol Soc 1981;79:486 118. Hida M, Kuwahara K. [Cataract and HLA antigen (author's transl)]. Nippon
Ganka Gakkai Zasshi 1978;82:751 119. Saari KM. HLA antigens in ophthalmology. Acta Ophthalmol 1984;165(Suppl):18 120. Ladas ID. Histocompatibility (HLA) antigens and eye diseases other than
uveitis. Surv Ophthalmol 1983;27:233 121. Ritch R, Podos SM, Henley W et al. Lack of association of histocompatibility
antigens with primary open-angle glaucoma. Arch Ophthalmol 1978;96:2204 122. Salmon JF, Martell R. The role of ethnicity in primary angle-closure glaucoma. S
Afr Med J 1994;84:623 123. Shin DH, Sugar HS. HLA in pigment dispersion and glaucoma. Tissue Antigens 1982;19:301 124. Kaiser-Kupfer MI, Mittal KK. The HLA and ABO antigens in pigment dispersion
syndrome. Am J Ophthalmol 1978;85:368 125. Bertrams J, Schildberg P, Hopping W et al. HL-A antigens in retinoblastoma. Tissue
Antigens 1973;3:78 126. Roberts DF, Duggan-Keen M, Aherne GE et al. Immunogenetic studies in retinoblastoma. Br
J Ophthalmol 1986;70:686 127. Osborne A, Tschickardt M, Blanck G. Retinoblastoma protein expression facilitates
chromatin remodeling at the HLA-DRA promoter. Nucleic Acids
Res 1997;25:5095 128. Biswas J, Mukesh BN, Narain S et al. Profiling of human leukocyte antigens
in Eales' disease. Int Ophthalmol 1997;21:277 129. Bertrams J, Spitznas M, Rommelfanger M. Missing evidence for HLA antigen
association with Eales' disease, chorioretinitis, central serous retinopathy, and
malignant choroidal melanoma. Invest Ophthalmol Vis Sci 1978;17:918 130. Metzelaar-Blok JA, Jager MJ, Moghaddam PH et al. Frequent loss of heterozygosity
on chromosome 6p in uveal melanoma. Hum Immunol 1999;60:962 131. Ericsson C, Seregard S, Bartolazzi A et al. Association of HLA Class I
and Class II antigen expression and mortality in uveal melanoma. Invest
Ophthalmol Vis Sci 2001;42:2153 132. Proenca R, Carvalho M, Proenca D et al. HLA antigens and lymphocytes in
proliferative vitreoretinopathy. Graefes Arch Clin Exp Ophthalmol 1994;232:25 133. Ohno S. HLA antigens in ophthalmology. Acta Ophthalmol 1984;165(Suppl):7 134. Bertrams J, Dewald G, Spitznas M et al. HLA-A, B, C, DR, Bf, and C2 alleles
in insulin-dependent diabetes mellitus with proliferative retinopathy. Immunobiology 1980;158:113 135. Agardh D, Gaur LK, Agardh E et al. HLA-DQB1*0201/0302 is associated with
severe retinopathy in patients with IDDM. Diabetologia 1996;39:1313 136. Gonzalez-Gay MA. Genetic epidemiology. Giant cell arteritis and polymyalgia
rheumatica. Arthritis Res 2001;3:154 137. Rauzy O, Fort M, Nourhashemi F et al. Relation between HLA DRB1 alleles
and corticosteroid resistance in giant cell arteritis. Ann Rheum Dis 1998;57:380 138. Salvarani C, Boiardi L, Mantovani V et al. HLA-DRB1, DQA1, and DQB1 alleles
associated with giant cell arteritis in northern Italy. J Rheumatol 1999;26:2395 139. Hunt PJ, Marshall SE, Weetman AP et al. Histocompatibility leucocyte antigens
and closely linked immunomodulatory genes in autoimmune thyroid
disease. Clin Endocrinol Oxf 2001;55:491 140. Streilein JW, Yamada J, Dana MR et al. Anterior chamber-associated immune
deviation, ocular immune privilege, and orthotopic corneal allografts. Transplant
Proc 1999;31:1472 141. Creemers PC, Kahn D, Hill JC. HLA-A and -B alleles in cornea donors as
risk factors for graft rejection. Transpl Immunol 1999;7:15 142. Katami M. Corneal transplantation—immunologically privileged status. Eye 1991;5:528 143. Sonoda Y, Streilein JW. Orthotopic corneal transplantation in mice—evidence
that the immunogenetic rules of rejection do not apply. Transplantation 1992;54:694 144. Goulmy E, Pool J, Van Lochem E et al. The role of human minor histocompatibility
antigens in graft failure: A mini-review. Eye 1995;9:180 145. Boisjoly HM, Roy R, Dube I et al. HLA-A,B and DR matching in corneal transplantation. Ophthalmology 1986;93:1290 146. Beekhuis WH. Current clinician's opinions on risk factors in corneal
grafting. Results of a survey among surgeons in the eurotransplant
area. Cornea 1995;14:39 147. Stulting RD, Waring GO, 3rd, Bridges WZ et al. Effect of donor epithelium
on corneal transplant survival. Ophthalmology 1988;95:803 148. The collaborative corneal transplantation studies (CCTS). Effectiveness
of histocompatibility matching in high-risk corneal transplantation. The
Collaborative Corneal Transplantation Studies Research Group. Arch
Ophthalmol 1992;110:1392 149. Volker-Dieben HJ, Claas FH, Schreuder GM et al. Beneficial effect of HLA-DR
matching on the survival of corneal allografts. Transplantation 2000;70:640 150. Holland EJ. Epithelial transplantation for the management of severe ocular
surface disease. Trans Am Ophthalmol Soc 1996;94:677 151. Donnelly JJ, Orlin SE, Wei ZG et al. Class II alloantigen induced on corneal
endothelium. Role in corneal allograft rejection. Invest Ophthalmol
Vis Sci 1990;31:1315 152. McBride BW, McGill JI, Smith JL. MHC Class I and Class II antigen expression
in normal human corneas and in corneas from cases of herpetic keratitis. Immunology 1988;65:583 153. Pepose JS, Gardner KM, Nestor MS et al. Detection of HLA Class I and II
antigens in rejected human corneal allografts. Ophthalmology 1985;92:1480 154. Hopkins KA, Maguire MG, Fink NE et al. Reproducibility of HLA-A, B, and
DR typing using peripheral blood samples: Results of retyping in the
collaborative corneal transplantation studies. Collaborative Corneal Transplantation
Studies Group (corrected). Hum Immunol 1992;33:122 155. Vail A, Gore SM, Bradley BA et al. Influence of donor and histocompatibility
factors on corneal graft outcome. Transplantation 1994;58:1210 |