|
AMPHOTERICIN B
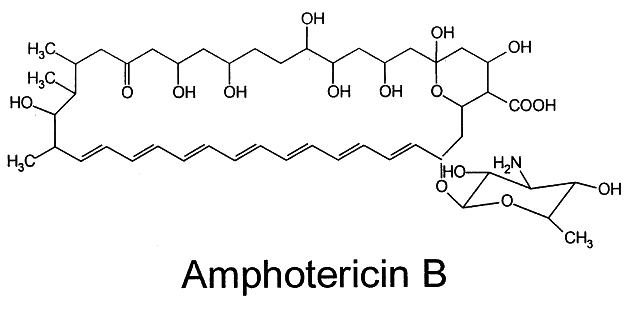
Amphotericin B (Fig. 2), produced by Streptomyces nodosus,1 was the first polyene shown to be effective in treating systemic mycoses.7 It is poorly soluble in water and has a tendency to degrade when exposed to light.8 In vivo, the rapid tissue binding of amphotericin B limits the actual amount of bioactive drug. In an attempt to increase solubility and drug bioavailability while limiting toxicity, several approaches have been tried, including methyl esterification and combination with liposomes and other vehicular compounds. Methyl esterification appeared promising initially because of enhanced solubility9; however, severe toxicity led to abandonment of this approach.10 Amphotericin B lipid complexes such as ABLC (Liposome Co., Princeton, NJ), Amphocil (Sequus Pharmaceuticals, Menlo Park, CA), and AmBisome (NeXstar Pharmaceuticals, Boulder, CO) are now available and we can expect new formulations of amphotericin B in other lipid vehicles. These promising new preparations eliminate the need for the deoxycholate solubilizer, the prime cause of eye pain and poor compliance when administered topically. Consequently, reduced toxicity allows the use of more drug both systemically and locally in the eye.3,11 A number of studies are under way with a variety of treatment modalities.12–14
|
Amphotericin B (Fungizone, E.R. Squibb and Sons, Princeton, NJ) is available for intravenous use in 20-mL vials containing 50 mg of amphotericin B powder, 41 mg of sodium deoxycholate, and a sodium phosphate buffer. Before intravenous injection, the powder is reconstituted to a concentration of 5 mg/mL in 10 mL of distilled water.
Although intravenous amphotericin B has been used since its discovery for the treatment of ocular infections, its pharmacokinetic profile for ocular tissues is not particularly good. Its most useful application appears to be for the treatment of endogenous and exogenous endophthalmitis caused by Candida species. Levels achievable within the vitreous and posterior eye structures appear to be sufficient for these highly susceptible organisms, although not for other filamentous fungi. Concentrations of biologically active amphotericin B in the cornea and the anterior eye after intravenous administration have consistently been low.15–17
Topical Administration
Although there is no preparation of amphotericin B specifically designed for ophthalmic use, this agent has been used topically for the treatment of fungal keratitis since the late 1950s. Toxicity is a troublesome complication. Because of this problem, Wood and Williford18 evaluated amphotericin B (Fungizone) that had been diluted with distilled water to a concentration of 0.15% in a series of patients with mycotic keratitis. They demonstrated both efficacy and markedly reduced toxicity. In a further series, reduction of the concentration of amphotericin B to 0.05% virtually abolished toxic reactions. Animal studies likewise showed that concentrations as low as 0.03% were remarkably efficacious against susceptible Candida species.19,20 The use of topical concentrations greater than 0.15% has been reported, but this is not recommended because of toxicity; there is little evidence of superior efficacy in the higher concentrations. The topical preparation, when stored at 36°C, retains potency for 1 week. Care should be taken to handle the solution aseptically, because it does not contain a bacteriostatic agent.21
There have been numerous clinical and experimental studies of the efficacy of topical amphotericin B in concentrations ranging from 0.05% to 1%. The pharmacokinetic profile is not particularly good (Table 1). In particular, the corneal epithelium appears to be a powerful barrier to corneal penetration. This is an important issue because the epithelium may heal over fungal lesions in the cornea.22,23 In animal studies, topical amphotericin B 0.15% was virtually unable to penetrate the corneal stroma when the epithelium was intact. Removal of the epithelium was associated with greatly increased penetration, and the degree of penetration was found to be proportional to the size of the epithelial defect.24 Efficacy was also greatly enhanced in these models.
TABLE 1. Ocular Drug Concentrations of Polyenes*
| Levels | ||||||||
| Source | Animal | Drug | Dosage | Route | Assay | Cornea | Aqueous | Vitreous |
| Green et al15 | NZW rabbits | Amphotericin B | 1 mg/kg | Intravenous—normal eye | Bioassay | Not done | 0 μg/mL | Not detected |
| Green et al15 | NZW rabbits | Amphotericin B | 1 mg/kg | Intravenous—inflamed cornea | Bioassay | Not done | Trace | Trace |
| Green et al15 | NZW rabbits | Amphotericin B | 1 mg/kg | Intravenous—inflamed eye | Bioassay | Not done | 0.18 μg/mL | Trace |
| Green et al15 | NZW rabbits | Amphotericin B | 150 μg/0.3 mL | Subconjunctival—normal eye | Bioassay | Not done | Trace | Not detected |
| Green et al15 | NZW rabbits | Amphotericin B | 150 μg/0.3 mL | Subconjunctival—inflamed cornea | Bioassay | Not done | Trace | Trace |
| Green et al15 | NZW rabbits | Amphotericin B | 150 μg/0.3 mL | Subconjunctival—inflamed eye | Bioassay | Not done | Trace | Trace |
| Fisher et al16 | Human | Amphotericin B | 0.6 mg/kg | Intravenous | Bioassay | Not done | 0.24 μg/mL | 0.23 μg/mL |
| O'Day et al17 | Human | Amphotericin B | 15–50 mg/day | Intravenous | Bioassay | Not done | 0.17 μg/mL | 0.17 μg/mL |
| O'Day et al23 | Dutch-belted rabbits | Amphotericin B | 0.15% 1 drop q 5 min × 13 | Topical—intact epithelium | Radioassay | 3.4 μg/g wet wt | 0.38 μg/mL | Not done |
| O'Day et al23 | Dutch-belted rabbits | Amphotericin B | 0.15% 1 drop q 5 min × 13 | Topical—debrided epithelium | Radioassay | 33.5 μ/g wet wt | 2.7 μg/mL | Not done |
| Schwartz et al28 | NZW rabbits | Amphotericin B | Shield soaked in 0.15% | Collagen shield—debrided epithelium | HPLC | 17.5 μg/g wet wt | 0.3–0.5 μg/mL | Not done |
| Schwartz et al28 | NZW rabbits | Amphotericin B | 0.15% 1 drop q 1 min × 5 | Topical—debrided epithelium | HPLC | 8.0 μg/g wet wt | 0.3–0.5 μg/mL | Not done |
| O'Day et al30 | Dutch-belted rabbits | Amphotericin B | 1500 μg/0.3 mL | Subconjunctival—intact epithelium | Bioassay | 80.8 μg/g wet wt | 1 μg/mL | Not done |
| O'Day et al30 | Dutch-belted rabbits | Amphotericin B | 1500 μg/0.3 mL | Subconjunctival—debrided epithelium | Bioassay | 90.1 μg/g wet wt | 0.4 μg/mL | Not done |
| Ellison85 | NZW rabbits | Natamycin | 5 or 10 mg/kg | Intravenous | Photometric | Not done | 2–3 μg/mL | Not done |
| O'Day et al25 | Dutch-belted rabbits | Natamycin | 5% 1 drop q 5 min × 13 | Topical | Radioassay | 2576 μg/g wet wt | 548 μg/mL | Not done |
| Pleyer et al12 | NZW rabbits | Liposomal amphotericin B | 0.48% × 1 drop | Topical—intact epithelium | HPLC | 1.2 μg/g | Not done | Not done |
| Pleyer et al12 | NZW rabbits | Amphotericin B | 0.48% × 1 drop | Topical—intact epithelium | HPLC | 19.0 μg/g | Not done | Not done |
| Pleyer et al12 | NZW rabbits | Liposomal amphotericin B | 0.48% × 1 drop | Topical—debrided epithelium | HPLC | 2.5 μg/g | Not done | Not done |
| Pleyer et al12 | NZW rabbits | Amphotericin B | 0.48% × 1 drop | Topical—debrided epithelium | HPLC | 52.5 μg/g | Not done | Not done |
NZW, New Zealand white, HPLC, high-performance liquid chromatography.
* Concentrations of polyene antifungal agents in ocular tissues that have been reported in the literature.
These data represent peak levels measured and are not comprehensive. Each cited work should be read in its entirety for complete understanding of the exact meaning of these excerpted values.
When the epithelium is absent, amphotericin B also appears to pass through the cornea to the anterior chamber. Penetration of inflamed corneas is also good. Amphotericin B (0.15%) administered topically was present in the cornea and aqueous of rabbit eyes inflamed by the intrastromal injection of clove oil, even in the presence of an intact epithelium.
The high concentrations of drug are mitigated by a very low bioactivity due to tissue binding. In Dutch-belted rabbits, this bioactivity has been measured at 7% of total drug in the cornea and 5% of total drug in aqueous humor.23 In spite of this, a therapeutic concentration of drug is achievable in the cornea with intense topical therapy.
The optimal frequency for the topical administration of amphotericin B is unknown. However, animal studies suggest that an initial loading approach of one drop every 5 minutes for 1 hour leads to a rapid accumulation of drug within the cornea, with drug still detectable 24 hours after the last drop.25 In the clinical setting, a practical approach is to give one drop every half hour initially with a gradual reduction to six to eight drops per day. After the infection is under control, this can be further reduced to three to four drops daily.
In some studies, collagen shields impregnated with amphotericin B were applied directly to infected corneas as a means of enhancing drug levels.26–28 The pharmacologic advantage of this approach over less expensive topical therapy is uncertain. In one study, the use of topically applied liposome-encapsulated amphotericin B was not associated with significantly increased corneal drug concentration.12
Subconjunctival Administration
Subconjunctival injections of amphotericin B have been recommended for the treatment of fungal keratitis and scleritis. Experience with human cases29 and experimental studies in animals30 demonstrate the severe toxicity of this approach. Ulceration and necrosis of the conjunctiva may occur, with inflammation of the underlying sclera and ciliary body. This appears to be due to both the deoxycholate salt that is used as an emulsifying agent and the amphotericin B itself.30 The injections are also associated with severe pain. Studies in animal models suggest that the amount of drug that enters the cornea, anterior chamber, or vitreous cavity is negligible (see Table 1).31 This form of therapy is no longer recommended.
Intracameral Administration
With knowledge of the limited efficacy of systemic and topical amphotericin B in severe intraocular infections, clinicians have advocated the use of intravitreal injections of amphotericin B. There is good evidence that the intravitreal injection of 5 μg of amphotericin B can be efficacious in the treatment of Candida sp. infections,32,33 alone or in combination with systemic amphotericin B or other drugs. The toxicity of intravitreal amphotericin B in a single injection appears to be low and within the acceptable range.33–35 Repeated injections do carry an increased risk of toxicity, although a number of cases have been reported in which repeated injections in these concentrations led to no aftereffects in the eye.36–39 For cases in which there is evidence of anterior endophthalmitis due to progression of infection from the cornea into the anterior chamber, amphotericin B can be irrigated into the anterior chamber.40 Our experience using this drug in concentrations of 5 μg/mL is largely limited to cases at surgery when the chamber is irrigated before penetrating keratoplasty is performed. Injection of liposomal amphotericin B has the theoretic potential to increase the half-life while reducing toxic side effects.11
Susceptibility of Organisms to Amphotericin B
It is generally accepted that amphotericin B is most effective against yeasts, particularly Candida and Cryptococcus species. It appears much less useful for the treatment of filamentous fungus infections. This experience with systemic mycoses is confirmed mostly in the treatment of ocular mycoses. Although clinical and experimental evidence suggests that Candida albicans keratitis is very susceptible to topically applied amphotericin B in concentrations of 0.03% to 0.15%, and that Candida endophthalmitis can be effectively treated by intraocular and/or systemic amphotericin B, there is little evidence that endophthalmitis due to filamentous organisms can be treated by this route. Amphotericin B does exert some antifungal activity against Aspergillus, but experience in saving eyes with Aspergillus endophthalmitis has been disappointing.41,42
NATAMYCIN (PIMARICIN)
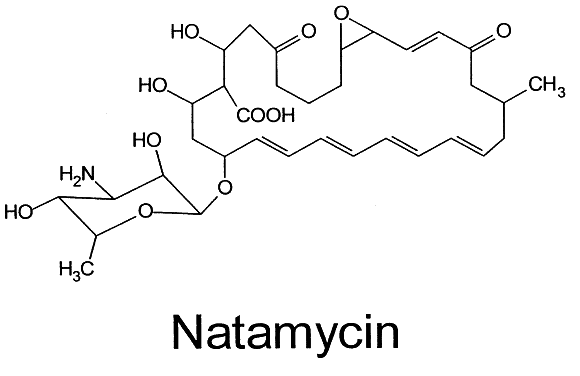
The other important polyene for use in ocular disease is natamycin (Fig. 3). This tetraene polyene is the only antifungal agent commercially available in the United States for use as an ophthalmic preparation. It is marketed by Alcon Laboratories as a 5% suspension (Natacyn). It can be stored at room temperature or refrigerated, but it should not be frozen or exposed to light or high temperatures.
|
Natamycin was discovered in 1958. It is arguably the most important and valuable antifungal agent for ophthalmic use discovered to date. Because of its physical characteristics, natamycin is extremely difficult to study. Much of what is known about the drug has been learned, therefore, from clinical experience. However, some laboratory work does provide information about its pharmacokinetics and pharmacology (see Table 1). Natacyn is available in volumes of 15 mL, stored in glass bottles. As is the case with most polyenes, natamycin is insoluble in water, and the commercial preparation is a microfine suspension that must be shaken well before each use. After topical administration, the drug often adheres to the area of corneal ulceration and may be seen in the fornices.
Pharmacokinetics and Pharmacology
Natamycin is easily absorbed into the cornea; however, it exhibits a high degree of binding in tissue, which severely limits its bioactivity.25 In addition, the corneal epithelium is a substantial barrier to corneal penetration. These characteristics account for the poor pharmacokinetic profile of this drug. However, extensive clinical experience with several fungi,43,44 in addition to laboratory studies,22,45–47 supports its value as an effective antifungal agent. In experimental pharmacokinetic studies using radiolabeled natamycin, high concentrations of the agent were detected in the cornea by radioassay after intensive topical administration. High levels were also detected in the aqueous.25 Because of tissue binding, only 2% of this drug is bioactive. The relatively high total corneal drug levels achievable by intensive administration assure that adequate therapeutic levels in the cornea are available. In experimental studies, it has been demonstrated that removal of the corneal epithelium dramatically increases penetration and efficacy.22,46,47
Topical Administration
As with amphotericin B, an initial loading approach is recommended. This consists of the administration of one drop every half hour with a gradual reduction to six to eight times and then three to four times a day. No other routes of administration are practical with this drug.
Toxicity
Topical natamycin 5% suspension is well tolerated in the cornea. Corneal toxicity does occur, however, with prolonged use, usually in the form of a punctate keratitis. Diffuse conjunctival inflammation is also seen, and a consistent low-grade inflammation has been reported.21,48 In an animal model, natamycin did not retard the healing of corneal epithelial defects.49 Natamycin has been shown to be most effective against the filamentous fungi, especially Fusarium and Aspergillus infections,43,48 which are the most common causes of fungal keratitis worldwide. Numerous clinical series in the United States and elsewhere have established the primacy of natamycin in the treatment of fungal infections caused by filamentous fungi; however, it is less effective against yeast infections.21 It should be noted, however, that treatment failures presumably due to organism resistance occur in both Fusarium and Aspergillus keratitis as well as with other organisms. Large series of cases describing therapy for filamentous fungi other than Fusarium or Aspergillus are lacking.