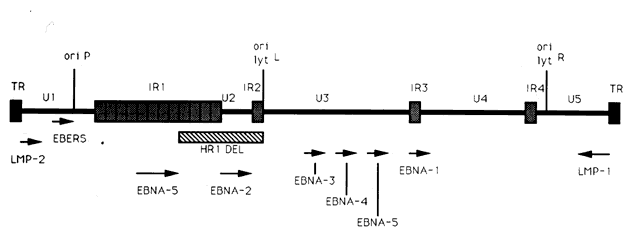
The entire EBV genome has been cloned and sequenced.45–47 Within virus particles, the EBV genome is linear and contains terminal repeats on either end of the genome (putatively thought to be important in circularization of the virus and/or integration of the EBV genome into cellular DNA), four internal repeat (IR1-IR4) regions, and five unique (U1-U5) sequence domains (Fig. 1). Approximately 100 open reading frames have been identified within the EBV genome that may potentially be transcribed and translated into proteins that are essential for the maintenance of the different phases of the EBV life cycle.44 Molecular biology techniques have allowed for the manipulation of many of these EBV genetic sequences. As a result, these in vitro studies have provided insight into the potential function of many of the EBV genes and how infection may be controlled.
|
Although it is known that adsorption of EBV to a permissive cell occurs by interaction of the EBV gp350/220 envelope glycoprotein with the EBV/complement receptor, CD 21, on the surface of the cell,48 it is not clear how the envelope or nucleocapsid is dissolved or how the EBV DNA penetrates the cell nucleus. It is known that once the EBV genome enters the nucleus, it circularizes and that the cellular phenotype is a key factor in determining the pattern of EBV gene transcription and the consequences of virus infection on the cell.49
EXPRESSION OF EBV GENES DURING THE LATENT STAGES OF INFECTION
Two types of latent EBV infection have been identified: (1) active latent infection with transcription of EBV oncoproteins that transform (or immortalize) B cells into lymphoblasts,12 and (2) passive latent infection that is typically utilized by the virus after infecting small resting B cells and during which transcription of only one gene (EBNA-1) has been identified.
A limited number of EBV genes have been reported to be expressed during the active latent (or growth transformation) phase of EBV infection (see Fig. 1).44 These include (1) six EBV nuclear antigens (EBNA-1, -2, -3, -4, -5, -6); (2) the latent membrane proteins (LMP-1 and LMP-2); and (3) two small, nonpolyadenylated RNAs (designated EBER-1 and EBER-2).11 mRNAs encoding each of the EBNA proteins are generated by individual splicing of long rightward transcripts initiated at one of two promoters, one located in the Bam HI C region within the U1 region of the genome, and the other in the IR1 region of the genome. The LMP transcripts are expressed from separate promoters which run in opposite directions, but share the same bidirectional control region.
The EBNA-2 protein appears to be important in the transformation of B lymphocytes into immortalized cells because mutant EBV strains lacking this gene (such as the HR-1 strain with the deletion noted in Fig. 1) are incapable of immortalizing B lymphocytes.44 One of the mechanisms by which EBNA-2 may modulate growth transformation is through its ability to upregulate latent membrane protein-1 (LMP-1) transcription.50
LMP-1 is one of the most abundantly transcribed genes in EBV infected lymphoblastoid cell lines (LCLs).51 LMP-1 has recently been reported to be essential for B-lymphocyte growth transformation. EBV strains with deletion mutations in the LMP-1 gene were found to be incapable of immortalizing B lymphocytes.52 LMP-1 has been shown to be responsible for many of the phenotypic changes observed in EBV-infected B-lymphocytes including upregulation of expression of B-cell activation markers (CD21 and CD23), the cell membrane adhesion molecule ICAM-1, and the bcl-2 protooncogene.53 Increased expression of adhesion molecules facilitates cell clumping which may enhance B-cell growth and proliferation via paracrine growth factors. T cells also adhere to B cells expressing these adhesion molecules, and such T-cell adherence may represent the initial step in the host immune response to EBV infection that ultimately leads to elimination of EBV infected lymphoblasts in vivo.54
EBNA-1 is the only protein transcribed in the passive latent state. EBNA-1 has been reported to play an important role in maintaining EBV latency.44 EBNA-1 has been shown to bind to specific DNA sequences located at three sites within the EBV genome. One of these sites is located within the oriP domain of the EBV genome (see Fig. 1). Binding of EBNA-1 to the oriP sequences maintains EBV DNA as a closed circular episomal molecule, allows replication of EBV episomal DNA, facilitates transcription of latent cycle genes, and prevents transcription of lytic cycle genes.12 Expression of EBV latent associated genes apparently does not necessarily ensure maintenance of the latent state, because a small percentage (approximately 1%) of latently infected B cells in vitro spontaneously enter the lytic phase of the life cycle and produce infectious virus.1
EBER-1 and EBER-2 are the most abundantly transcribed latent genes with approximately 10)7 copies of each per cell.55 EBER-1 and EBER-2 are small (158 and 178 base pairs, respectively), nonpolyadenylated RNAs that are not translated into proteins. These small RNA molecules are thought to be important in RNA splicing which is an integral function in maintaining latency because EBNA mRNAs are extensively spliced.56 Their abundant copy number per cell makes EBERs excellent targets for in situ hybridization techniques that demonstrate cellular sites of EBV latent infection.
EXPRESSION OF EBV GENES DURING THE LYTIC STAGES OF INFECTION
In general, most normal B cells that are permissive for latent EBV infection cannot support EBV replication. Epithelial cells have been found to support lytic EBV infection in humans11; however, at the present time an EBV-permissive epithelial cell line for in vitro studies has not been identified.16 Because of the lack of an EBV-permissive cell line, the expression of EBV genomic sequences during the lytic phase of the virus infection has been investigated predominantly in latently infected B lymphocytes induced into the lytic phase using phorbol esters, the most reliable and reproducible inducers of viral replication.57 The most widely used cell line for these experiments, Raji, is derived from a Burkitt's lymphoma and contains a deletion which does not allow for expression of genes associated with the late phases of lytic infection although most immediate-early and early genes may be expressed.58 Superinfecting Raji cells with the P3HR-1 EBV strain results in expression of most lytic EBV genes in superinfected cells.59,60
The majority of genes expressed in the immediate-early phase of EBV infection are the latency-associated genes described above.44 It has been reported that there are several abundant EBV mRNAs expressed approximately 4 hours after induction into the lytic phase that may be key trans-activators of early lytic gene transcription.61 One immediate-early gene in particular, BZLF1 (often termed ZEBRA), is a “switch” gene that has been shown to play an important role in the switch from the EBV latent phase to the lytic phase.62
There are at least 30 EBV early genes that are defined by their constant synthesis (even when inhibitors of DNA synthesis are present) once a cell is induced into the lytic cycle by superinfection.44 The genomic locations of many of the genes detected during the early phase of EBV replication have been mapped. Although the function of many of these genes is currently unknown, the functions of some of the early EBV proteins have been identified by comparing the genomic location and sequence of these early genes to their counterparts in other herpesviruses. Most of the early proteins identified thus far are involved in DNA replication and include DNA polymerase, thymidine kinase, DNA binding proteins, alkaline reductase, and ribonucleotide reductase.44
EBV genes expressed during the late phase of the lytic cycle predominantly encode EBV structuralproteins and proteins that allow for the transport of the virus out of the cell. Several late genes have been mapped within the EBV genome although not all of the RNA transcripts for these genes have been detected in vitro. Most of the late EBV proteins that have been identified are heavily glycosylated and are associated with the outer envelope of the virus particle. One example is gp350/220, an abundantly expressed glycoprotein that is found in the cell membrane as well as the outer envelope of the virus particle.63 The gp350/220 protein is the predominant envelope protein recognized by neutralizing antibodies.64