NICOTINIC DRUGS
Direct-Acting Nicotinic Agonists
This class of drugs does not play a role in ophthalmology.
Indirect-Acting Nicotinic Agonists
Cholinesterase inhibitors increase cholinergic activity in both muscarinic and nicotinic structures. With two exceptions, these drugs are used by the ophthalmologist for their muscarinic effects. One exception is their use in the diagnosis of myasthenia gravis.
Intravenous edrophonium (Tensilon) and, to a lesser extent, intramuscular neostigmine221 have been the cholinesterase inhibitors used most frequently.
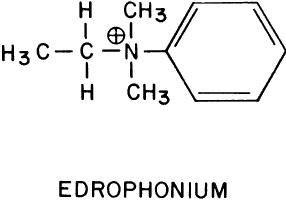
Edrophonium (Fig. 1) is a short-acting cholinesterase inhibitor. It carries a positive charge, enabling it to bind with the negatively charged “anionic” site of acetylcholinesterase. The bulky cyclic structure of edrophonium physically blocks the approach of acetylcholine to the “esteradic” site. In addition, electrostatic hydrogen bonding of the hydroxyl group of edrophonium causes inactivation of the esteradic site. Acetylcholine hydrolysis is inhibited because it must compete with edrophonium for both the “anionic” and “esteradic” sites. Edrophonium's binding to both enzyme sites is weak and short lived.
|
The side effects of intravenous edrophonium are the result of its action on muscarinic structures (bowel, bladder, and heart). They include abdominal cramping, urinary incontinence, bradycardia, and cardiac arrest.222,223 However, the diagnostic value of edrophonium in myasthenia gravis resides in its nicotinic action: the striated musculature contracts more vigorously. Therefore, the side effects of edrophonium can be prevented without interfering with the drug's diagnostic efficacy by simultaneously administering a muscarinic blocking agent, such as atropine. This could be done as follows: Two syringes are used. One contains saline; the other contains atropine, 0.4 mg, and edrophonium, 10 mg. Neither the patient nor the physician evaluating the response knows which of the two solutions is being injected. The nurse gives one syringe to the physician for injection. Three tenths of the volume is injected rapidly, and the response is evaluated during the next minute. If this is well tolerated, the remainder is injected and the response is evaluated. The contents of the second syringe are then injected in the same manner. Only after the patient and physician have committed themselves to an evaluation of the relative efficacy of the two syringes does the nurse identify which contained the edrophonium.
Several different parameters have been suggested for evaluating the edrophonium response. The objective improvements in ocular and lid motility have been of primary interest to the ophthalmologist. Glaser and co-workers224,225 used tonography to document the increased extraocular muscle tonus. They reported a mean 2-mmHg increase in intraocular pressure within 1 minute of injecting 10 mg edrophonium in 15 myasthenic patients. All 15 had unilateral, but not always bilateral, responses. The maximum elevation was 5 mmHg. The response could occur in less than 10 seconds, but the mean response interval was approximately 20 seconds226 to 35 seconds.227 Although there were no false-positive results in the 10 control patients in Glaser and co-workers' study, false-positive results can occur.227,228 Wray and Pavan-Langston228 found that the intraocular pressure increase was greater in myasthenic patients without ophthalmoplegia than in those with ophthalmoplegia.
Kornbleuth and colleagues229 used electromyographic recordings of the extraocular muscles when testing with edrophonium. In their small series, only patients with myasthenia gravis showed an increase in electrical activity. Campbell and associates227 used electronystagmography to study the effect of edrophonium on the fatigue induced by optokinetic nystagmus. A beneficial response occurred in 50% of 17 myasthenia gravis patients given edrophonium injections. There were no false-positive reactions among 18 normal subjects. However, these authors found the test to be of only limited value because of the marked variations in amplitude and rate of optokinetic nystagmus among subjects.
Edrophonium increases the peak saccadic velocity in myasthenic patients but decreases it in normal controls.230 The use of topical cholinesterase inhibitor drops in the treatment of ocular myasthenia gravis has been suggested,231 but their value is not well documented.
Cholinesterase inhibitors are also used for their nicotinic effects in the treatment of pediculosis of the lashes. The cholinesterase inhibitors are pesticides and kill by producing respiratory arrest. Cholinesterase inhibitors used primarily for their intraocular effects are relatively ineffective pesticides. Physostigmine 0.1% in peanut oil and 0.25% ointment kill the adult form of the crab louse (Phithirus pubis), but the nits are more resistant and must be removed with forceps.232,233 Malathion is more effective and relatively safe because it is rapidly inactivated by human serum carboxylesterases. Within 5 minutes of exposure, 100% of lice are killed, and after 10 minutes of exposure 96% of eggs will not hatch. Two problems encountered when using malathion around the eyes are that commercial preparations contain a high (e.g., 78%) alcohol content and the drug gives off a foul-smelling sulfhydryl degradation product.
Demodex folliculorum is another pest infesting the lids. It is quite resistant to ophthalmic preparations. It can survive 3 or more days in 0.125% echothiophate, 0.25% demecarium, or 0.1% diisopropyl fluorophosphate.234
Nicotinic Antagonists
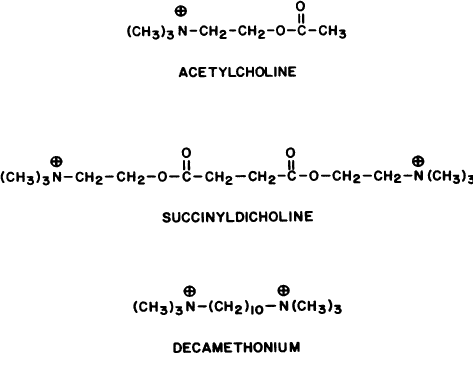
Two classes of nicotinic blocking agents exist: nondepolarizing antagonists (e.g., curare) and depolarizing antagonists (e.g., succinyldicholine).
NONDEPOLARIZING ANTAGONISTS. Neuromuscular Blockade. Duke-Elder and Duke-Elder235 showed that acetylcholine and nicotine caused contraction of dog extraocular muscles and that curare, but not atropine, could block these responses. Muscarine was subsequently shown to have a very weak contractile effect that could be blocked by atropine.236 Until the work of Katz and Eakins,237 it was believed that the ocular muscles were more sensitive to the blocking effects of nondepolarizing agents, such as curare and gallamine, than were the peripheral skeletal muscles.238 Katz and Eakins found that the reverse seemed true (e.g., a systemic dose of curare that paralyzed the cat anterior tibial muscle produced only a 50% block of the twitch response of the superior rectus). Their work was supported by Sanghvi and Smith.236 Systemic administration of nondepolarizing relaxants, such as alcuronium,239 atracurium,240,241 fazadinium,242 metocurine, and pancuronium,243 is associated with a lowering of intraocular pressure. Some of these have a rapid onset and short duration of action, which makes them useful as induction agents for intubation. These drugs also differ in their cardiovascular effects. For example, alcuronium and curare lower arterial blood pressure, whereas pancuronium increases blood pressure and heart rate.244
An effective neuromuscular block with atracurium or vecuronium lasts approximately 30 minutes, making these agents intermediate in their duration of action. Atracurium, unlike the shorter-acting depolarizing antagonist succinyldicholine, undergoes rapid ester hydrolysis at physiologic pH independent of serum butyrylcholinesterase activity. Large doses of atracurium have been suggested as a substitute for succinyldicholine induction because the former does not produce extraocular muscle contraction.245,246 Atracurium 0.5 mg/kg did not produce a significant rise in intraocular pressure in subjects under steady-state nitrous oxide general anesthesia, but succinyldicholine 1 mg/kg did, from a mean baseline of 5.6 mmHg to 13.2 mmHg. Vecuronium seems to act in a manner similar to that of atracurium.247,248
Mivacurium, at a dose of 0.15 mg/kg, has a slower onset of action (approximately 2.5 minutes) than succinyldicholine. At this dose, mivacurium's striated muscle block lasts approximately 16 minutes. Mivacurium, unlike atracurium and vecuronium, is inactivated primarily by butyrylcholinesterase.
Traction on the extraocular muscles during general anesthesia produces slowing of the heart. As stated earlier, pancuronium produces a tachycardia, which is protective. Strabismic children under general anesthesia who received pancuronium had heart rates that dropped from 120 ± 17 beats per minute to 94 ± 25 beats per minute.249 Patients receiving atracurium had pulse rates that fell from 86 ± 20 beats per minute to 34 ± 22 beats per minute, and 40% developed an additional arrhythmia besides the bradycardia. From a cardiac viewpoint, pancuronium has advantages in strabismus surgery.
Ganglionic Blockade. Transmission in sympathetic ganglia (e.g., superior cervical ganglion) and parasympathetic ganglia (e.g., ciliary ganglion) is primarily nicotinic. Several investigators have looked at ocular effects that might be related to partial blockade at this level. Gallamine250 and pancuronium251 administration have been associated with a lowering of intraocular pressure. This may have been due, in part, to a reduction in systemic blood pressure. Intubation for general anesthesia produced tracheal irritation and a reflex increase in blood pressure. When throats were not anesthetized with topical anesthesia, the intraocular pressures 5 minutes after gallamine injection remained at baseline; when throats were anesthetized, the intraocular pressures were below baseline. The reductions in intraocular pressures produced by pancuronium were significant but were not dose related with the use of 0.01 to 0.08 mg/kg. Trimetaphan, a short-acting ganglionic blocking agent, administered intravenously produced reductions in intraocular pressure from preoperative values of 13 to 17 mmHg to intraoperative values of 3 to 4 mmHg as the systolic blood pressure fell to 60 mmHg.252
Hexamethonium and pentamethonium, 2 mg/kg, given intramuscularly to conscious subjects caused a mean maximum fall in intraocular pressure of 9 mmHg.253 The decline was present within 30 minutes of injection and persisted for 3 hours. This effect was attributed to a blockade of sympathetic ganglia and a resultant lowering of systemic blood pressure. Barnett253 treated hypertensive patients for 9 to 11 weeks with these drugs. He reported an improvement in the hypertensive retinopathy. However, these drugs produced an orthostatic hypotension that was disabling, which limited their use.
Retrobulbar tetraethylammonium injection in human subjects has produced mydriasis.254 The pupil remained dilated longer than 6 hours and did not constrict after topical physostigmine. Ciliary ganglion blockade also produced cycloplegia.255,256 Decreased tearing has been attributed to blockade of the sphenopalatine ganglion after systemic administration of tetraethylammonium.255
Based on the assumption that sympathetic superior cervical and stellate ganglia fibers innervate and constrict the retinal arterioles, ganglionic blocking agents have been used in the treatment of central retinal artery occlusions, in the hope that the vessels would dilate and the embolus pass to a more distal arterial branch. However, there is little evidence that they are beneficial, and it can be argued that they may further reduce the perfusion blood pressure to the eye.
Botulinum Toxin. Botulinum toxins are produced by the anaerobic bacteria Clostridia botulinum. Each of eight different strains produces an immunologically distinct form of the toxin: A, B, C1, C2, D, E, F, and G. Four, A, B, D, and E, have been shown to block cholinergic function. They bind to unmyelinated cholinergic nerve terminals and prevent the release of quantal acetylcholine.257,258 Death occurs by paralysis of the respiratory muscles; doses as low as 1 ng per kilogram body weight can be lethal in humans.259
Botulinum toxins are zinc-dependent proteases that attack proteins linking the acetylcholine-containing vesicles to the synaptic axon membrane. Calcium is no longer able to trigger a functional release of the transmitter.28 The structure of botulinum A toxin has been fully sequenced.260 Type A botulinum toxin attacks a soluble 25-kd cytoplasmic attachment protein, SNAP-25,29,261,262 abolishing quantal (i.e., functional) acetylcholine exocytosis; some nonquantal acetylcholine release does persist. The toxin does not block the neuron's action potential, the synthesis or storage of acetylcholine, the vesicle number, or calcium entry.263 The initial binding site of botulinum A toxin is a sialic acid-containing site on the external neuron surface; this attachment can be antagonized by lectins.264 Once inside the neuron, botulinum toxin can no longer be neutralized by antitoxins.
Type A botulinum toxin is more effective at nicotinic neuromuscular junctions than at nicotinic ganglion synapses or parasympathetic neuromuscular junctions. It has been used to treat essential blepharospasm, blepharospasm associated with facial dyskinesias, hemifacial spasm, nonparalytic and thyroid ophthalmopathy-induced strabismus, paralytic strabismus, nystagmus, corneal exposure, and entropion.265–278
The toxin is injected into the muscle to be rendered paretic or subcutaneously in the adjacent area. The effect is dose dependent. Multiple small doses (e.g., 0.001 to 0.003 μg; 2.5 to 7.5 U) or fewer but larger doses may be given. The onset of effect is within 48 hours, and the maximum effect may take 5 to 6 days to develop. The results are usually transient and average 2 to 3 months in duration.
Histologic examination of orbicularis muscle fibers after prolonged treatment of blepharospasm failed to show morphologic changes. However, the motor neurons had many unmyelinated axon sprouts, most of which ended blindly; those ending on muscle fibers were able to elicit new and multiple motor end plates at previously nonsynaptic sites.279,280
In general, botulinum A treatment is of little value in strabismus in which the eye muscle movements are restricted, and it is of less value than traditional surgery in comitant deviations.281 Retrobulbar injections, used to treat nystagmus and oscillopsia, may be of temporary value depending, in part, on the amount and duration of ptosis produced.282–285 Complications occur from local spread of toxin to adjacent muscle fibers and from an inability to accurately predict the magnitude of the effect. Ptosis is one of the most common complications caused by local spread of toxin.286 Ptosis, partial or complete, can at times be of therapeutic value (e.g., before recovery from Bell's palsy).287–289 Deliberate ptosis can be achieved by injecting botulinum A toxin along the superior orbital roof, approximately 25 mm behind the superior orbital rim.
Clinical signs of systemic toxicity have not been reported, but there is evidence of subtle systemic effects (e.g., abnormal electromyographic recordings in arm muscle fibers of patients receiving 0.005 μg of toxin).290 Neurologists treat dystonias with relatively large doses of botulinum A toxin; these may produce only subtle distant electromyographic changes but are likely to elicit neutralizing antibodies that limit the toxin's usefulness.291–293 A distinction should be made between non-neutralizing antibodies, which may have no clinical significance, and neutralizing antibodies.294
DEPOLARIZING ANTAGONISTS. Extraocular Muscle Effects. The depolarizing antagonists, such as succinyldicholine, decamethonium, and hexacarbacholine (Fig. 2), paralyze striated voluntary muscles throughout the human body but have a different effect on the extraocular muscles. The extraocular muscles respond with a sustained contraction. This was first noted by Hofmann and Holzer,295 who reported that the contraction of the extraocular muscles caused an increase in human intraocular pressure. This observation was supported by Lincoff and co-workers.296,297 The maximum intraocular pressure reported is 55 mmHg.298
|
The shortening effect and increased muscle tension created by succinyldicholine have been measured in humans.299 The drug always produced contraction in the horizontal and vertical recti muscles. The results in the oblique muscles were variable.
The explanation is that there are two types of extraocular muscle fibers. In the center of each muscle, typical striated muscle fibers, with a single myoneural junction (en plaque fibers), are most numerous. These are paralyzed by succinyldicholine. However, the surface layers contain fibers with multiple myoneural junctions. These are the “en grappe” fibers. Their innervation has the appearance of a bunch of grapes. It is these fibers that respond to succinyldicholine with a sustained contraction.300–302 The strength of contraction depends on the dose of succinyldicholine and the number of multiply innervated fibers. In sheep superior oblique muscle, succinyldicholine produces only 7% of the force generated by maximum electrical stimulation of the entire muscle.303 In cats, up to one third of the whole muscle force has been generated by succinyldicholine.304 Succinyldicholine causes a partial reduction in the resting membrane potential and a localized contraction in the area around each myoneural junction.305 Because these junctions are found along the entire length of the en grappe fiber, most of each fiber is depolarized and shortened.306 However, unlike en plaque fibers, which are capable of generating action potentials, the en grappe fibers are believed to respond to both neuronal impulses and succinyldicholine with slow, graded contractions (i.e., they are unable to conduct action potentials).307 This may explain why Kornbleuth and colleagues229 found a dissociation between ocular muscle electrical activity and the elevation of intraocular pressure. They performed simultaneous tonography and electromyography on subjects given succinyldicholine. They expected to find that the increase in intraocular pressure was proportional to the degree of extraocular muscle electrical activity. Electromyography was performed on antagonistic vertical or horizontal recti of the same eye in subjects receiving 5, 10, 15, or 20 mg succinyldicholine. These doses were too small to produce apnea. A 2- to 4-mmHg increase in intraocular pressure occurred in patients under general anesthesia, and a 5- to 14-mmHg increase occurred in conscious volunteers. The duration, but not the magnitude, of the ocular pressure elevation appeared to be dose related. The rise in intraocular pressure elevation occurred just as the electrical activity of the extraocular muscle was reduced. The inability of the en grappe fibers to conduct action potentials may explain this dissociation.
Curare, but not atropine, blocked the sustained contraction produced by succinyldicholine in animal studies.308 Katz and Eakins237 found evidence that the en plaque fiber response to depolarizing antagonists was quantitatively different for eye muscles and skeletal muscles (e.g., the dose of succinyldicholine needed to depress the twitch response of the superior rectus muscle was greater than that needed to block skeletal muscles). This difference was less when decamethonium was used.
Each of the six human extraocular muscles contains multiply innervated and singly innervated fibers in about the same proportions: in the periphery, 92% ± 10% are multiply innervated, and in the center, 33% ± 12% are multiply innervated.309 The contractile effect of succinyldicholine on the en grappe fibers is generally considered undesirable. However, Hannington-Kiff310 suggested that the anesthesiologist use the resultant change in interlimbal distance to monitor the action of succinyldicholine. The interlimbal distance in 35 patients under general anesthesia before succinyldicholine injection was 52.9 mm ± 4.3 mm. Ninety seconds after injection, this distance had decreased to 49 mm ± 5.5 mm. One minute after the return of spontaneous respirations, the interlimbal distance was near baseline values, 51.4 mm ± 3.3 mm. The duration of succinyldicholine stimulation of en grappe fibers and the duration of succinyldicholine paralysis of en plaque fibers appeared similar. Taylor and associates311 also found that the intraocular pressure returned to baseline with the onset of spontaneous respirations. Pandey and co-workers312 studied the time course of the ocular pressure elevation in 34 patients given succinyldicholine in a dose of 1.5 to 2 mg/kg. An increase in intraocular pressure was present in less than 1 minute, peaked in 2 to 4 minutes, and returned to baseline in 6 minutes.
Succinyldicholine stimulation of the extraocular muscles results in a variable amount of enophthalmos; as much as 3.25 mm has been reported.313 The conjunctival vessels dilate,314 and the forced-duction test may suggest a mechanical restriction for up to 20 minutes.315
Mindel and colleagues316 theorized that the basic deviation of the eyes was produced by the balance of forces exerted by the multiply innervated muscle fibers. They used intravenous succinyldicholine to stimulate these fibers and to remove the influence of the singly innervated fibers, which were paralyzed. Under general anesthesia, esotropic patients became esotropic when 2 mg/kg was injected. Exotropic patients became exotropic.317 Despite this qualitative success, the measured amount of drug-induced ocular deviation did not agree closely with that in the conscious patient. These authors believed that this incongruity was due to A and V pattern effects, that is, in conscious patients the eyes could be maintained in the primary position by fixation, whereas in anesthetized patients the eyes could move vertically as well as horizontally. Lingua and associates318 have reported similar findings. In addition, they found a correlation between the ocular position induced by succinyldicholine immediately after strabismus surgery and the 1-week postoperative result.
Intraocular Muscle Effects. Abramson319 used ultrasound to measure anterior chamber depth and lens thickness in human subjects given 1 mg/kg succinyldicholine intravenously. Succinyldicholine caused a rapid increase in anterior chamber depth and a decrease in lens thickness. This effect was present within 20 seconds, was maximal in 45 to 210 seconds, and disappeared in 3 to 5 minutes. Abramson attributed this effect to a relaxation of accommodation. However, there was no change in the diameter of the pupil. It is unlikely that succinyldicholine would decrease the activity of only one of the two parasympathetic structures innervated by the ciliary ganglion.
Side Effects. Succinyldicholine may produce a bradycardia. This is usually associated with multiple injections. However, asystole has been reported after a single dose.320 Succinyldicholine and, to a lesser degree, its first hydrolysis product, succinylmonocholine, are responsible for this cardiac effect.321
Succinyldicholine is inactivated by esterase hydrolysis. The apnea produced by respiratory muscle paralysis may be prolonged in patients with genetically322 or acquired reduced serum cholinesterase activities.323,324 Chronic systemic absorption from topical cholinesterase inhibitor eyedrops can result in such an effect.325 In one report, a reduction of serum cholinesterase activity 62% below normal from echothiophate eyedrops permitted endotracheal intubation using a total of only 9.5 mg succinyldicholine.326 Decamethonium, because it is excreted unmetabolized, is not dependent on the level of serum cholinesterase activity for termination of its effect.
There are at least four alleles, at a single locus, that control the type of serum cholinesterase made by the liver: normal, dibucaine resistant, fluoride resistant, and silent genes. Approximately 1 in 4000 in the population will develop prolonged apnea after succinyldicholine injection. This is the incidence of a person having both genes abnormal. Approximately 1 in 400 in the population will be a normal dibucaine-resistant heterozygote and will develop a shorter but still significantly prolonged apnea. Other factors reducing serum cholinesterase activity are hepatic disease,327 malnutrition, anemia, severe dehydration, late pregnancy,328 and systemic absorption of cholinesterase inhibitors. In one study, 5% of a general surgical population had decreased serum cholinesterase activity.329
The sustained contraction of the extraocular muscles after succinyldicholine injection poses a threat if there is a preexisting perforating laceration or if an incision is to be made into the eye (e.g., cataract extraction). Expulsion of the intraocular contents can result from the steady squeezing of the eye muscles. Lincoff and colleagues297 mentioned three personal communications linking the use of succinyldicholine to this complication. There are several ways it can be avoided: After succinyldicholine injection, the intraocular pressure is monitored with a tonometer until it returns to normal. Use of depolarizing nicotinic antagonists is avoided. Alternatively, a pharmacologic antagonist can be used to prevent the effect. With regard to this last suggestion, cholinesterase inhibitors are not of value. Although they reverse the action of nondepolarizing muscle relaxants (e.g., curare), they prolong and enhance the action of depolarizing drugs.330 Theoretically, pretreatment with small doses of curare or succinyldicholine itself might prevent succinyldicholine-induced ocular muscle contraction. However, the results are conflicting.331,332 Macri and Grimes333 found that prior curare injection prevented ocular hypertension in cats given succinyldicholine. Because both drugs competed for the same receptor, multiple doses of succinyldicholine overcame the curare effect. Miller and associates334 found that 1 mg/kg succinyldicholine produced a mean intraocular pressure increase of 8.5 mmHg in 10 control subjects. Parenteral gallamine, 20 mg, or curare, 3 mg, administered 3 minutes before succinyldicholine, prevented this effect in both glaucomatous and normal subjects. However, when Meyers and co-workers335 studied the value of curare, 0.09 mg/kg, or gallamine, 0.3 mg/kg, given 3 minutes before 1 to 1.5 mg/kg succinyldicholine, they found that ocular pressure elevations still occurred (e.g., in 10 patients with a mean intraocular pressure of 13 ± 1 mmHg, curare did not prevent an elevation to 24 ± 1.3 mmHg). Pancuronium, 0.14 mg/kg, also was ineffective.336 Katz and colleagues337 found that hexafluorenium, which is both a nondepolarizing (curarelike) blocking agent and a plasma cholinesterase inhibitor, in doses of 0.4 mg/kg, prevented the rise in intraocular pressure from succinyldicholine, 0.3 mg/kg. Hexafluorenium was effective only if it was injected before succinyldicholine, not after it.
Perhaps the preceding inconsistencies result from the multiplicity of factors that can affect the intraocular pressure (e.g., depth of anesthesia, serum O2 and CO2 levels, venous pressure, and tracheal stimulation). Barbiturates, given to induce anesthesia, can reduce muscle endplate currents.338 When thiopental was injected 2 minutes before succinyldicholine, 1 mg/kg, there was no significant increase in ocular pressure.339 Macri and Grimes333 disinserted all the extraocular muscles in cats. This reduced, but did not prevent, an increase in intraocular pressure after succinyldicholine. Taylor and associates311 reported that in 25 of 29 patients, the blood pressures 1 minute after succinyldicholine injection were elevated. These may have contributed to the short-term elevations in ocular pressure.
The blood pressure elevations could be because succinyldicholine raises blood catecholamine levels. Succinyldicholine, but not curare, increased plasma norepinephrine levels immediately after injection.340 Succinyldicholine, 1 mg/kg, produced a peak norepinephrine elevation 3 minutes after injection, raising levels from a mean of 301 pg/mL at baseline to 647 pg/mL. The increase had disappeared by 10 minutes. The elevation in epinephrine levels was less marked and became insignificant by 2 minutes.
Wynands and Crowell341 found that 50% of patients given succinyldicholine had a rise in intraocular pressure. In 3 of 18 subjects, the elevation was more than 10 mmHg. However, endotracheal intubation alone produced a rise of more than 10 mmHg in 10 of 23 subjects. Lewallen and Hicks342 and Craythorne and co-workers298 reported conflicting results in subjects without intubation. The former found that injection of 40 mg succinyldicholine did not raise human intraocular pressure; the latter found that continuous infusion of 0.1% to 0.2% succinyldicholine did.
A most intriguing study was that of 15 patients having uniocular enucleations.343 These subjects were anesthetized with 3 to 4 mg/kg thiopental intravenously and maintained with halothane or isoflurane and nitrous oxide/oxygen. No premedications were used. All six extraocular muscles were severed from the eye to be enucleated. Intraocular pressures were then obtained bilaterally. Succinyldicholine 1.5 mg/kg was administered and the intraocular pressures were measured every 30 seconds for 5 minutes. The systolic and diastolic blood pressures were not significantly different before and 90 seconds after succinyldicholine administration, but the intraocular pressures were significantly and maximally elevated bilaterally at this time. Surprisingly, there was no significant difference in the mean ± SE intraocular pressure rise between those eyes with severed muscles and those eyes with muscle insertions intact. The presuccinyldicholine injection intraocular pressures of the normal and muscle-severed eyes were, respectively, 15.1 ± 1 mmHg and 16.1 ± 1 mmHg. At 90 seconds after succinyldicholine injection, the respective intraocular pressures were 25.2 ± 1.6 mmHg and 24.7 ± 1.8 mmHg. Perhaps an increase in venous blood pressure produced these bilateral results, but the apparent lack of an extraocular muscle effect is inexplicable.
MUSCARINIC DRUGS
Direct-Acting Muscarinic Agonists
GENERAL CONSIDERATIONS. The ocular effects of this class of drugs include reduction in intraocular pressure; stimulation of the iris sphincter, producing miosis; and stimulation of the ciliary muscle, producing accommodation. Examples of direct-acting muscarinic drugs are muscarine, pilocarpine, aceclidine (3-acetoxyquinuclidine), arecoline, and acetyl β-methylcholine (methacholine, Mecholyl) (Figs. 3 through 5). In high concentration, pilocarpine also stimulates choline acetyltransferase synthesis of acetylcholine,344 but this is probably of little clinical significance.345
|
|

|
Isopilocarpine is a naturally occurring stereoisomer of pilocarpine present in very small amounts in most preparations of the drug.346 Isopilocarpine has one tenth the binding affinity of pilocarpine for bovine ciliary muscle and has very little pharmacologic activity.
Carbachol (carbamylcholine) is both a direct-acting muscarinic agonist and a direct-acting nicotinic agonist. In addition, it has indirect agonist activities (i.e., it is a cholinesterase inhibitor). This last action results from the NH2 substitution on the acetyl group of carbachol. It markedly reduces the molecule's susceptibility to hydrolysis by acetylcholinesterase but does not prevent carbachol from competing with acetylcholine for the anionic and esteradic sites of acetylcholinesterase. This interference with acetylcholine hydrolysis results in a further increase in cholinergic activity. Aceclidine, too, is slightly cholinesterase resistant and has weak anticholinesterase activity.
Pilocarpine's structure is quite different from that of acetylcholine. Either of the nitrogen atoms in the imidazole ring can become positively charged and bind to the anionic receptor site. Pilocarpine does not have the classic muscarinic agonist structure of acetylcholine (N-C-C-O-C-C). However, pilocarpine's interatomic spacing (N-C-C-C-C) may mimic it.347 Evidence exists that the intrinsic muscarinic activity of pilocarpine is less than that of acetylcholine and carbachol.348,349 However, additional factors play a role in determining clinical efficacy. These are primarily pharmacokinetic. For example, acetylcholine and acetyl β-methylcholine are hydrolyzed by tissue esterases and may not reach the site at which their action is desired. Inhibiting tissue cholinesterases increases the efficacy of both drugs. Arecoline, aceclidine, and pilocarpine are tertiary amines and can penetrate the corneal epithelium more readily than the positively charged quaternary amines such as acetylcholine, carbachol, and acetyl β-methylcholine. This correlates with the lipid solubilities of these drugs. As measured with toluene: pH 6.5 buffer, the partition coefficient of arecoline is 0.1; pilocarpine, 0.033; aceclidine, 0.002; and carbachol, <0.0001.350 The pHs of eyedrops and tears alter the proportions of nonionized drug (e.g., the ability of arecoline and oxotremorine to reverse mydriasis increases as a function of the pHs of their solutions).351 Drugs that are ionized at all pHs, such as carbachol, are concentration dependent, not pH dependent. The poor corneal penetration of carbachol resulted in Clarke352 finding marked variability in its ability to lower intraocular pressure. O'Brien and Swan353 performed a corneal massage through the lids of human subjects. This allowed a 0.09% carbachol solution to be as effective a miotic as a 1.5% to 4.5% carbachol solution. Topical anesthetics and surfactants also aided penetration (e.g., the addition of 0.03% benzalkonium to 1.5% carbachol produced significant accommodation for 48 hours).
Accurate estimates of corneal penetration by pilocarpine have been impaired by the use of tritiated molecules. Asseff and co-workers354 gave anesthetized monkeys eyedrops of 1% to 8% tritiated pilocarpine. They reported approximately 3% corneal penetration at 5 minutes. Chrai and Robinson,355 studying rabbits, found similar levels after applying unpurified tritiated pilocarpine but only 0.13% corneal penetration at 5 minutes after applying purified material. Maximum penetration occurred at 20 minutes and was 0.18%. At 2 hours, the aqueous humor concentration was 0.03% of the administered dose. These authors attributed the previous higher values to tritium exchange between pilocarpine and its solvent during storage. Penetration of the tritiated solvent resulted in falsely high estimates of corneal penetration. Krohn and Breitfeller356 applied two drops of 2% pilocarpine HCl (approximately 2000 μg) to corneas of patients under general anesthesia; the pilocarpine concentration in the aqueous humor was not more than 5 μg/mL.
Pilocarpine can be administered from continuous-release membranes (Ocusert) made of polymerized ethylenevinylacetate. Ideally, the low-dosage form (Ocusert P20) releases 20 μg/h and the high-dosage form (Ocusert P40) releases 40 μg/h. However, Armaly and Rao357 found that the in vitro release rates from membranes supposedly dispensing 20 μg/h, 50 μg/h, and 80 μg/h were two and one half to three times their projected rates during the first 7 hours of use. Between 7 and 24 hours, the release rates remained slightly elevated, but within the next 24 hours they fell to slightly below their projected levels and continued to decline. After 3 to 4 days, the release rates were 78%, 88%, and 81% of those expected. These results may explain why the glaucoma of two of Armaly and Rao's patients was controlled for the first 2 days of membrane use but not thereafter.
Pilocarpine has also been applied as a polymer emulsion358,359 and as a gel. Both provide prolonged drug delivery to the eye and, compared with drops, longer duration of action. The gel is available commercially as 4% pilocarpine HCl in a viscous aqueous acrylic gel vehicle; this concentration of pilocarpine approaches the binding limit of the polymer used to make the gel. A single daily application of the gel has a hypotensive effect for 24 hours. For the first 18 hours, this effect is indistinguishable from that elicited by 4% pilocarpine eyedrops given four times a day in terms of both pressure and pupillary diameter.360 For hours 18 to 24, the drops are more effective.361
Pathology can alter pharmacokinetics and drug response. Patients with such diverse pathology as atopy and trisomy 21 are said to be more sensitive to muscarinic agonists and antagonists.362,363
Patients with intraocular inflammation inactivate pilocarpine. Schonberg and Ellis364 found that the primary aqueous humor of humans and rabbits did not inactivate pilocarpine, but serum and secondary aqueous did. Ten percent of 500-μg pilocarpine was inactivated by 200 μL of secondary aqueous humor in 1 hour. Inactivation could be prevented by heating serum to 60°C for 10 hours, by dialysis, or by the addition of some (e.g., penicillamine and ethylenediaminetetraacetic acid), but not all, chelating agents. Inactivation could not be inhibited by anticholinesterase drugs. Rabbit serum was more active against pilocarpine than human serum. Only at high pilocarpine concentrations were human ocular tissues (cornea, iris-ciliary body, lens, retina, and choroid) more active than the corresponding rabbit ocular tissues.365 Lee and colleagues366 found that pigmented rabbit corneas inactivated pilocarpine much more effectively than did albino rabbit corneas.
OCULAR PHARMACOLOGY Iris Sphincter Muscle. ANIMAL STUDIES. Pilocarpine has less intrinsic muscarinic activity than the endogenous transmitter acetylcholine. As a result, Swan and Gehrsitz367 found that rabbits given topical 0.25% physostigmine or 0.1% DFP had less miosis if 4% pilocarpine was previously administered. If pilocarpine were injected into the cornea after maximum DFP miosis, a transient dilatation occurred. These results were consistent with pilocarpine-acetylcholine competition for the same receptors.
Miosis in mammals is associated with M3 receptor activation and increased phospholipase C activity.368,369 Feedback inhibitory prejunctional muscarinic receptors on sympathetic neurons of the iris dilator muscle seem to be of a different subtype.199 In rabbits, these prejunctional receptors respond to methacholine, oxotremorine, muscarine, and carbachol by inhibiting norepinephrine release; atropine can block this effect.370
Denervation supersensitivity of the rat iris sphincter is not associated with a change in muscle fiber resting potential but is associated with increased sensitivity to calcium.371,372 Thus, supersensitivity could be due to an increased release of intracellularly bound calcium and/or an increased influx of extracellular calcium.
Bito and Banks373 administered DFP drops chronically to monkeys and then discontinued the drug. They observed several interesting pupillary phenomena, one of which was a diminished response to direct-acting muscarinic agonists during the recovery period. For at least 3 days after discontinuing DFP, the pupils responded to light but did not respond to carbachol or pilocarpine. Similar results were obtained in the cat.374 These results may have been produced from an acquired subsensitivity of the sphincter muscarinic receptors (i.e., a subsensitivity produced by the prior high acetylcholine concentrations). Another possibility is that the greater intrinsic agonist activity of acetylcholine, compared with that of pilocarpine and carbachol, became manifest. Other data from this laboratory,375 confirmed by Claesson and Barany,376 suggested that an alteration of muscarinic receptor sensitivity had occurred. Exposure of feline eyes in vivo to continuous light resulted in subsensitivity to pilocarpine, whereas light deprivation resulted in increased sensitivity. After 7 days in ambient light of 1200 lumens/m2, the irides became insensitive to pilocarpine. Surprisingly, subsensitivity to carbachol or acetyl β-methylcholine could not be demonstrated in vitro. Hemicholinium (which reduces acetylcholine synthesis) injected intravitreally in vivo resulted in increased sensitivity to administered muscarinic agonists. Barany377 reported that chronic administration of pilocarpine drops did not produce subsensitivity in experimental animals challenged with intravenous pilocarpine. However, after prolonged treatment in the form of continuous-release membranes, pilocarpine did produce subsensitivity.
Perhaps the explanation for the aforementioned phenomena is miosis from peptide transmitters. There is evidence that the neurotransmitter substance P, identified in trigeminal nerve fibers, produces miosis.378 Mandahl379 has suggested that, in the rabbit, the miosis elicited by cholinesterase inhibitors and pilocarpine is primarily the result of release of substance P from trigeminal nerve endings in the iris. According to this theory, continuous application of cholinesterase inhibitors and pilocarpine would deplete the stores of substance P, resulting in a reduced response, whereas the acetylcholine release by light-stimulated parasympathetic fibers would remain intact.
HUMAN STUDIES. Lowenstein and Loewenfeld380 and Newsome and Loewenfeld381 studied the iris using pupillography. Drops of 1% to 2% pilocarpine were given to volunteers without ocular disease. The onset of miosis, maximum miosis, and duration of miosis varied greatly from person to person. Maximum miosis occurred in 25 to 45 minutes, was maintained for 1 to 3 hours, and required 24 to 36 hours for full recovery. The pupillary reflex to light persisted unless miosis was maximal. However, during the recovery phase, the light response was markedly diminished. When submaximally effective concentrations of pilocarpine (e.g., 0.1%) were followed by submaximal concentrations of a cholinesterase inhibitor (e.g., 0.1% physostigmine), the results were additive.
Ogle and associates382 and Morgan and co-workers383 also used pupillography to study miosis in normal subjects. They reported latencies after 1% pilocarpine of 9.8 ± 0.9 minutes. The latencies for 0.75% carbachol and 0.25% physostigmine were 13.7 ± 2.2 minutes and 13.9 ± 1 minutes, respectively. The rates of miosis and amounts of maximum miosis were less for carbachol than for pilocarpine and physostigmine, yet all three drugs required about the same time to produce their maximum effect. The times required for recovery to 50% of the predrop pupil diameters were: carbachol, 164 ± 46 minutes; pilocarpine, 271 ± 109 minutes; and physostigmine, 456 ± 28 minutes. The latency of mydriasis from 0.5% tropicamide (Mydriacyl) was similar to the latency of miosis from pilocarpine. The changes in pupillary diameter due to light stimuli during the latency periods of both pilocarpine and tropicamide were similar; the amounts and maximum rates of pupil constriction after onset of action also were similar.
After the short-term application of various direct-acting muscarinic agonists, the response produced by the light reflex was reduced.384 This may have represented competition between residual exogenous agonists, with weaker intrinsic activity, and the endogenous agonist acetylcholine, with more potent intrinsic activity. The light reflex required approximately 24 hours to recover after 2 drops of 2% pilocarpine, 7 hours to recover after 10 drops of 0.2% arecoline hydrobromide, and 24 hours to recover after 10 drops of 0.2% aceclidine hydrochloride.
Ciliary Muscle. ANIMAL STUDIES. Tornqvist385–387 studied accommodation in monkey eyes. Intramuscular pilocarpine over a dose range of 0.2 to 2 mg/kg appeared to have equal affinity for iris and ciliary muscle. The lowest dose caused 2 diopters (D) or less of accommodation, and the highest dose always caused more than 12 D of accommodation. One monkey was given higher doses of pilocarpine, and the myopia paradoxically decreased: 2 mg/kg produced 20.3 D of accommodation, 5 mg/kg produced 11.7 D of accommodation, and 50 mg/kg produced 8 D of accommodation. Similarly, small doses of systemic carbachol, 0.005 to 0.1 mg/kg, produced increasing accommodation, but larger doses, 0.1 to 0.5 mg/kg, produced decreased accommodation. This phenomenon could not be reproduced by using high concentrations of topical muscarinic agonists. Retrobulbar anesthesia did not prevent this paradoxical effect, indicating that central nervous system and ganglionic effects were not the cause. Intravenous atropine prevented, or reversed, the paradoxical effect. A topical dose of pilocarpine that produced half the maximum accommodative effort was 100 times more than that needed to produce half the maximum miosis. Because systemically administered pilocarpine did not exhibit this difference, it was concluded that pharmacokinetic factors resulted in more drug reaching the iris than reached the ciliary muscle after topical application.
Barany388 studied subsensitivity of the monkey's ciliary muscle after a single drop of topical carbachol given unilaterally. For approximately 1 week, the accommodative response in the treated eye was less than that in the untreated eye after systemic administration of pilocarpine, carbachol, or bethanechol. The reasons for this effect were not clear, but the author did not believe the answer was a reduction in the number of muscarinic receptors or a reduction in their binding characteristics.
In monkeys, the loss of accommodation that occurs with aging is not associated with a reduction in the number of ciliary muscle muscarinic receptors.389
The agonist aceclidine is less effective in producing accommodation than in increasing miosis and outflow facility. Studies in monkey eyes found that M3 receptors predominate in ciliary muscle, iris sphincter muscle and trabecular meshwork (i.e., the differences in pharmacologic responses could not be attributed to different muscarinic receptor subtypes).390,391
After ciliary ganglionectomy, monkey ciliary muscle supersensitivity to pilocarpine was associated with a 12% to 84% reduction in muscarinic receptors.392 The magnitude of this seemingly paradoxical finding is difficult to explain simply by attributing it to the loss of prejunctional muscarinic receptors on nerve endings.
HUMAN STUDIES. Ciliary body thickness was measured in volunteers by the use of ultrasound.393 Two hours after applying a drop of 2% pilocarpine, the increase in mean thickness was 0.06 mm. Anterior chamber depth was measured after single and multiple drops of 2% or 4% pilocarpine.394 All subjects showed a decrease in anterior chamber depth after a single drop of pilocarpine. This shallowing remained significant for the 6 hours that measurements were made. When compared with the untreated eye, a single drop of 2% pilocarpine produced a mean maximum shallowing of 0.09 mm that occurred 3 hours after instillation; 4% pilocarpine produced a mean maximum shallowing of 0.07 mm that occurred 4.5 hours after instillation. Pilocarpine (2%), every 6 hours, produced a mean maximum anterior chamber narrowing of 0.12 mm. This was not significantly different from the single-drop effect. One week after chronic therapy was discontinued, the mean depth of the treated eye was not significantly different from that of the control eye. Abramson and colleagues395 confirmed that the anterior chamber shallowing effect from 2% pilocarpine, as well as the increase in lens thickness, was similar after single-drop and chronic (10-day) therapy.
Hallden and Henricsson396 produced with-the-rule astigmatism in 10 young men by asymmetrically contracting the ciliary body. This was achieved by applying a cotton pledget containing 10% pilocarpine to the temporal bulbar conjunctiva for 3 minutes, creating 0.75- to 2 D myopia with an axis between 0° and 20°.
Intraocular Pressure. The site at which muscarinic drugs act to lower intraocular pressure is a subject of controversy. Their effect is believed to be local, although an occasional report raises the possibility of a systemic effect (e.g., Willetts397 reported that normotensive subjects given unilateral pilocarpine drops had a bilateral decrease in ocular pressure). However, subjects with a mean plasma level of 2.9 ng/mL, delivered by skin patches, did not have a significant reduction in intraocular pressure.398
The different mechanisms of action that have been proposed include a decrease in aqueous humor secretion; an increase in removal of aqueous humor by active transport; an increase in removal of aqueous humor by passive trabecular outflow (from increased iris sphincter tone, increased ciliary muscle tone, or decreased episcleral venous pressure); and an increase in removal of aqueous humor by way of nontrabecular routes.
AQUEOUS HUMOR SECRETION. The data from different laboratories are inconsistent. Becker399 found that pilocarpine drops increased rabbit aqueous humor formation in vivo. Green and colleagues400 reported that acetylcholine and pilocarpine produced an increase in rabbit aqueous humor secretion in vitro. Macri and Cevario401 administered pilocarpine intra-arterially to enucleated cat eyes and also found an increase in aqueous humor secretion; however, outflow facility was increased, producing an overall reduction in intraocular pressure. Berggren402,403 found the opposite results. He used in vitro thinning of rabbit ciliary processes as a measure of secretion. Pilocarpine 10-9 mol/L, physostigmine 10-7 mol/L, acetylcholine 10-5 mol/L, and carbachol 10-5 mol/L inhibited secretion. The therapeutically inactive stereoisomer of pilocarpine, isopilocarpine, did not block secretion. Atropine 10-7 mol/L alone had an inhibitory effect402 and also prevented the more potent pilocarpine effect.403 However, when intravenous pilocarpine, 2 mg/kg, was injected in vivo 5 minutes before the in vitro studies, there was no evidence of altered secretion.
Edwards and colleagues,404 using constant-pressure perfusion, measured the effect of unilateral topical 4% pilocarpine on monkeys under general anesthesia. Pilocarpine caused an increase in aqueous humor production. However, Walinder and Bill405 found in vervet monkeys that infusion of pilocarpine 10-4 mol/L into the anterior chamber produced a reduction in aqueous humor formation of approximately 1 μL/min; atropine infusion 3 × 10-5 mol/L abolished the pilocarpine effect.
Barsam406 found that the hypotensive effect of pilocarpine lasted longer than the duration of increased outflow facility in human subjects. He believed that pilocarpine was producing a reduction in aqueous humor secretion. However, fluorophotometry of normotensive eyes has provided evidence that pilocarpine has only a minor effect on aqueous humor formation, which is to increase secretion by approximately 14%.407 This may or may not be true for the glaucomatous eye as well.
Muscarinic agonists might alter aqueous humor secretion by altering ciliary blood flow. Stjernschantz and Bill408 studied cholinergic control of uveal blood flow in anesthetized animals. Stimulation of the intracranial third nerve altered the blood flow to the iris and ciliary body but not to the retina and choroid. In the cat, the ciliary body blood flow was increased; in the rabbit, it was decreased; and in the monkey there was no statistically significant effect. In all three species, the iris blood flow was decreased by third-nerve stimulation. Intravenous atropine blocked this iris effect in all three species and prevented the increased ciliary body blood flow in cats.
AQUEOUS HUMOR ACTIVE TRANSPORT. Large vacuoles have been observed in the endothelium of the inner wall of Schlemm's canal. It has been suggested that they represent the result of active transport of aqueous humor. Holmberg and Barany409 found that monkey eyes treated with pilocarpine had a reduced number of vacuoles. Lutjen-Drecoll410 placed 20 μg pilocarpine hydrochloride into the anterior chambers of monkeys and then studied the eyes with electron microscopy. Large vacuoles were rarely encountered. Grierson and associates411 studied the enucleated eyes of eight patients with melanomas. Pilocarpine drops were given before their removal. These were compared with 11 control eyes but not in a masked manner. There were more than twice the number of large vacuoles in the pilocarpine-treated eyes. However, the authors' results from baboon eyes led them to conclude that the increased vacuolization was not due to a direct effect of pilocarpine on the endothelium of Schlemm's canal. Instead, they attributed the vacuoles to a secondary effect from increased trabecular flow.412
INCREASE IN PASSIVE AQUEOUS HUMOR OUTFLOW. Most investigations have dealt with this mechanism of muscarinic action. Edwards and co-workers404 found that topical 4% pilocarpine caused more than a doubling in monkey eye outflow facility, from 0.41 ± 0.04 μL/min/mmHg to 1.09 ± 0.14 μL/min/mmHg. Atropine alone had no effect. Barany413 found that pilocarpine, administered topically or injected into the anterior chamber, increased vervet monkey facility of outflow. Two components appeared to be present, because atropine, injected intravenously or intraocularly, reversed part of the increase in 3 to 5 minutes and the remainder in approximately 30 minutes. Lutjen-Drecoll410 studied electron micrographs of monkey eyes and found that the trabecular pore area in contact with the inner wall of Schlemm's canal correlated, after pilocarpine administration, with the outflow facility.
In humans, the hypotensive effect of pilocarpine is usually, but not always, associated with an increase in outflow facility. Outflow facility, accommodation, and pilocarpine-induced accommodation decrease with age. However, the pilocarpine-induced increase in outflow facility and reduction in intraocular pressure were found not to decrease with age.414 Pilocarpine, using gonioscopy to evaluate the drug's effect, seems to produce a bowing inward of the trabecular meshwork toward the anterior chamber and away from the canal of Schlemm.415
Gaasterland and colleagues416 found that normotensive subjects given a single drop of 4% pilocarpine had, within 2 hours, a reduction in mean intraocular pressure from 14.6 to 11.2 mmHg and an increase in outflow facility from 0.24 to 0.40 μL/min/mmHg. Willetts417 studied the effect of 2% pilocarpine, three times a day for 4 to 5 days, in normal eyes. Although intraocular pressures declined, he found no significant alterations in outflow facility. Krill and Newell418 found that normal and glaucomatous eyes responded to four-times-a-day pilocarpine therapy with a reduction in pressure that was proportional to the initial pressure (i.e., the higher the initial intraocular pressure, the greater the absolute amount of pressure reduction). However, the percent declines were similar: 8% to 38% in normal eyes and 12% to 40% in glaucomatous eyes.
Although pilocarpine increased the initial outflow facility, there was no correlation between the magnitude of the increase and the fall in pressure. In two normal and five glaucomatous eyes there was little or no outflow facility increase. Grant,419 Becker and Friedenwald,420 and Harris and Galin421 reported similar findings. Barsam406 and Flindall and Drance422 reported a temporal dissociation between the effects of muscarinic agonists on outflow facility and intraocular pressure. Barsam406 found that the effect on outflow facility reached a maximum at 2 hours and then dissipated so rapidly that it appeared that pilocarpine was acting through a decrease in aqueous humor formation. Flindall and Drance422 reported a dissociation in the opposite direction. Five hours after a single unilateral drop of 0.75% or 1.5% carbachol, the intraocular pressure reduction was no longer significant. However, the outflow facility remained increased, by 24.5% and 24.1%, respectively, compared with that of the control eye. Kronfeld423 found that a single drop of pilocarpine produced, in general, more lowering of intraocular pressure than could be explained by the increase in outflow facility. This was true whether the drop was given to normotensive subjects or to glaucoma patients. In the latter, therapy had been discontinued 1 week before the test drop. Lieberman and Leopold424 reported that aceclidine lowered intraocular pressure without increasing outflow facility. Investigations of the mechanism by which increased outflow facility occurs have dealt with the following actions:
IRIS SPHINCTER CONTRACTION. The hypotensive action of muscarinic agonists could result from miosis if the iris base pulled on the scleral spur and widened the trabecular meshwork pores. However, there is little evidence to support such a mechanism. In glaucomatous beagles, the miosis from muscarinic agonists is maximal at 45 to 120 minutes postinstillation, after which recovery begins; however, the hypotensive action is greatest 2 to 5 hours after instillation. Thorburn425 gave normal human eyes a 4% pilocarpine drop. The pupillary diameter was minimum at 30 minutes, but the intraocular pressure was not significantly decreased. O'Brien and Swan353 reported that three eyes with surgical sphincterotomies continued to respond to 1.5% carbachol with a reduction in intraocular pressure below 20 mmHg. Mapstone426 found that 2% pilocarpine followed by 10% phenylephrine lowered intraocular pressure in normal eyes from a baseline of 14.9 mmHg to 13.7 mmHg and increased outflow facility from 0.25 μL/min/mmHg to 0.33 μL/min/mmHg; measurements were made 90 minutes postinstillation. When a second drop of phenylephrine was given 2.5 hours after the first, there was an insignificant decrease in intraocular pressure and the outflow facility increased from 0.33 μL/min/mmHg to 0.38 μL/min/mmHg. Bito and Merritt427 dissociated the miotic effect of pilocarpine from the hypotensive effect in monkeys. Chronic treatment with echothiophate resulted in pilocarpine producing full miosis and an increase in intraocular pressure instead of the pretreatment reduction.
CILIARY MUSCLE CONTRACTION. Flocks and Zweng428 investigated the possibility that ciliary muscle contraction pulled on the scleral spur and widened the trabecular pores. Pilocarpine drops were given to one eye of monkeys and atropine to the other. The monkeys were killed with intraperitoneal pilocarpine. Eyes were examined histologically and by gonioscopy. These investigators believed the histologic appearance was that of trabecular lamellae being stretched and separated in the eyes receiving pilocarpine. Gonioscopy was stated to be too insensitive to allow evaluation. When Jampel and Mindel429 unilaterally stimulated the nucleus of accommodation in the midbrain of macaque monkeys, up to 10 D of accommodation was achieved. Maintenance of this marked accommodation for 100 seconds did not alter intraocular pressure relative to the control eye; however, Armaly and Burian430 found that accommodation in normal human subjects caused a more rapid decline in the intraocular pressure during tonography. Thorburn425 reported that the maximum increase in outflow facility in normal human eyes occurred 30 to 60 minutes after a 4% pilocarpine eyedrop and coincided with the refractive change that occurred from ciliary muscle contraction. However, the temporal course of the ocular hypotensive effect did not coincide with either the outflow facility or the degree of accommodation. Abramson and colleagues395,431 found that individual pilocarpine drops in glaucoma patients on chronic therapy produced maximum lens thickening and shallowing of the anterior chamber in 1 hour. By 2 hours, these effects were almost gone, but the hypotensive action was maintained far longer. Grierson and associates411 studied human eyes given pilocarpine before enucleation. The scleral spurs in treated eyes appeared to be pulled more posteriorly, widening the scleral sulcus by a mean value of approximately 15° but narrowing the lumen of Schlemm's canal.
Kaufman and Barany432 disinserted and recessed surgically the ciliary muscles of monkey eyes. Not only did this not result in elevated intraocular pressures, but the intraocular pressures remained lower than in control eyes during the more than 1-year period after disinsertion. Perhaps cyclodialysis clefts had been created. However, because outflow facility did not increase when intravenous and intraocular pilocarpine was injected 12, 33, and 56 weeks postoperatively, these authors believed that their model supported the ciliary muscle-scleral spur traction theory.
It has also been proposed that the pull of the ciliary muscle might act to lower intraocular pressure by pulling open a collapsed canal of Schlemm.433,434
In final consideration of the ciliary muscle-scleral spur traction theory, it is interesting to note that one of the beneficial aspects of pilocarpine sustained-release therapy (Ocuserts) is that a dissociation is created between the degree of hypotension and the degree of induced myopia. Maximum hypotension occurs with minimal stimulation of the ciliary muscle. François and colleagues435 reported that the miosis, myopia, and shallowing of the anterior chamber were less with the Ocusert P20 than with 2% pilocarpine drops. The mean accommodative myopia was: age 20 to 40 years, 5.8 D with drops versus 0.8 D with Ocusert; age 40 to 60 years, 1.3 D with drops versus 0.3 D with Ocusert; and age greater than 60 years, 0.2 D with drops versus 0.0 D with Ocusert.
EPISCLERAL VENOUS PRESSURE. The aqueous humor collection channels eventually empty into the episcleral venous circulation. If the episcleral venous pressure were increased or decreased by muscarinic agonists, the rate of aqueous humor outflow would be decreased or increased, respectively. Wilke436 found that topical 2% pilocarpine produced a transient rise in intraocular pressure in normal human eyes. The elevation was 2 to 5.5 mmHg and lasted 20 to 40 minutes. It was followed by a reduction in intraocular pressure beginning 40 to 60 minutes postinstillation. The early period of pressure elevation was associated with dilatation of conjunctival and episcleral vessels and an increase in episcleral pressure of 3.5 to 5 mmHg lasting 9 to 17 minutes. Gaasterland and associates416 gave normotensive human subjects a single drop of 4% pilocarpine and reported that 1 hour later, during the hypotensive phase, there was virtually no change in episcleral venous pressure (i.e., it was 9.3 ± 0.3 mmHg before pilocarpine and 9.5 ± 0.4 mmHg 1 hour after pilocarpine administration).
NONTRABECULAR OUTFLOW PATHWAYS. The pseudofacility seen on tonographic tracings may represent, at least in part, aqueous humor outflow by routes other than the trabecular meshwork. Bill and Walinder437 instilled 10-4 mol/L pilocarpine unilaterally in monkey eyes. Uveoscleral flow was decreased, compared with that of the control eye, whereas drainage by way of the trabecular system was increased and intraocular pressure was elevated by a mean value of 2.6 ± 0.7 mmHg. These authors theorized that contraction of the ciliary muscle caused the diminished uveoscleral flow. Gaasterland and co-workers416 found that topical 4% pilocarpine, given to normotensive humans, increased pseudofacility during the hypotensive response.
Lacrimation. The lacrimal gland receives a parasympathetic innervation that may control secretion. In familial dysautonomia there is decreased lacrimation, and it has been suggested that this innervation may be deficient. However, topical solutions of 2.5% and 20% acetyl β-methylcholine failed to increase lacrimation in 12 and 6 dysautonomic subjects, respectively, although miosis occurred. Failure was defined as less than a 5-mm increase in Schirmer paper strip wetting. The phenotypically normal parents of these patients also failed to respond with increased lacrimation.438 Smith and colleagues439 injected acetyl β-methylcholine intravenously. The effect on lacrimation was determined by observation only. Increased lacrimation occurred in normal controls at infusion rates of 2.5 to 11.7 μg/kg/min. In patients with dysautonomias, increased lacrimation occurred at infusion rates that were lower, 0.4 to 2.8 μg/kg/min. This confirmed the earlier observation by Kroop,440 who produced lacrimation in patients with dysautonomias with subcutaneous acetyl β-methylcholine. These results were consistent with the finding that acetyl β-methylcholine was ineffective topically because it could not reach the appropriate lacrimal receptors.
DeHaas441 reported three patients with unilateral sensory and/or motor paresis of the lacrimal reflex. Pilocarpine, 7 to 10 mg parenterally, produced lacrimation on the damaged side. In only one subject was the effect on the contralateral normal side stated; it also was increased.
THERAPEUTICS. Open-Angle Glaucoma. A number of studies found that lowering the intraocular pressure preserved the vision of patients with chronic open angle glaucoma.442–448
Drance and Nash449 gave 1%, 2%, 4%, and 8% pilocarpine hydrochloride in 0.5% hydroxypropyl methylcellulose, pH 4.5, to 12 ocular hypertensive patients without field loss. Intraocular pressures were recorded every 15 minutes for the first hour and hourly thereafter for 7 more hours. Each concentration of pilocarpine was given as a single drop a week apart, and the order of concentrations was randomized except for the 8% solution, which was always given last. During the first hour, there was usually a pressure rise. A statistically significant intraocular hypotensive effect occurred between hours 1 and 2 and lasted for nearly 8 hours. The reduction from 1% pilocarpine was about half maximal and was of shorter duration. Although higher concentrations gave dose-related increased reductions in ocular pressure, the 8% solution was no more effective than the 4% soultion. However, a maximum increase in outflow occurred in some patients at 8%; in some patients, the maximum outflow effect occurred at 1% or 2%. Harris and Galin421 also found that the 8% pilocarpine solution was no more effective than the 4% solution in lowering ocular pressure.
Worthen450 studied the effect of pilocarpine, 2% or 4%, four times a day, on the diurnal pressure curve of open-angle glaucoma patients. Pilocarpine not only lowered the intraocular pressure by a mean value of 9 mmHg but it also flattened the diurnal curve by decreasing the maximum fluctuation from 18.5 mmHg before treatment to 8.5 mmHg during treatment. The maximum pressure-lowering effect of pilocarpine occurred between 9 AM and 6 PM. Pratt-Johnson and colleagues451 found that 4% pilocarpine four times a day was less effective in controlling the diurnal variation than 0.06% echothiophate twice a day. The mean variation while on pilocarpine was 12 mmHg and that while on echothiophate was 7 mmHg.
Rothkoff and associates452 investigated whether the hypotensive response to a single drop of 2% pilocarpine would predict the efficacy of chronic treatment. There were three parts to their study: (1) one drop of 2% pilocarpine hydrochloride was instilled at 8 AM and the pressure was measured hourly for 4 hours; (2) all subjects were subsequently admitted and administered pilocarpine every 4 hours beginning at 6 AM and ending at 10 PM, with intraocular pressures measured hourly from 7 AM to 11 PM; and (3) all subjects were discharged on 2% pilocarpine and followed at monthly intervals for 3 months. Rothkoff and co-workers found that they could not predict whose intraocular pressure would be controlled consistently below 24 mmHg but that they could, from the single-drop response, predict whose would not be controlled. These authors also found that hypotensive efficacy did not correlate with initial intraocular pressure. Subjects responding to the single drop with a pressure below 24 mmHg had baseline pressures of 30.7 ± 5.2 mmHg; these were not significantly different from those of the failures, which were 33.4 ± 4.6 mmHg. Data from other studies leave doubt that a single application of pilocarpine will predict the efficacy of chronic therapy. Van Hoose and Leaders453 found that pilocarpine accumulated in the cornea (i.e., the cornea acted as a reservoir). This might explain why Harris and Galin421 did not find significant dose-related differences in the hypotensive action of single drops of 1% to 10% pilocarpine. These were given to 25 open-angle glaucoma patients off therapy for 3 weeks. However, on chronic therapy, 4% and 8% concentrations were more efficacious than a 1% concentration. The mean percent decline in ocular pressure at 90 minutes from a single drop was 1% pilocarpine, 18.25%; 4% pilocarpine, 18.78%; and 10% pilocarpine, 20.85%. One week of chronic therapy produced mean declines of 1% pilocarpine, 17.48%; 4% pilocarpine, 26.76%; and 8% pilocarpine, 29.07%. These authors noted that there seemed to be two populations of responders to the single drops. One group responded with a marked fall in intraocular pressure (i.e., 34%) to a single drop of 1% pilocarpine; this group responded similarly to the 10% solution (i.e., with a 35% decrease in pressure). The other population responded relatively poorly to 1% pilocarpine (8% decline) and to the higher concentrations as well (16% decline).
In subjects with maximum intraocular pressures of 24 to 37 mmHg, Flindall and Drance422 found that a single drop of carbachol had a maximum hypotensive effect at 4, 4, and 2 hours for 0.75%, 1.5%, and 3% solutions, respectively. The two lower concentrations produced similar maximum responses. Recovery was complete at 6 hours for 0.75% carbachol and at 8 hours for 1.5% carbachol, whereas a near maximal response remained 8 hours after the 3% solution. The absolute reduction in intraocular pressure was greater, in millimeters of mercury, for eyes with higher baseline values. However, when expressed as percent reduction, there was no difference.
There appeared to be no consistent advantage to substituting carbachol for pilocarpine. When 26 patients on 2%, 4%, or 6% pilocarpine four times a day were changed to 3% carbachol three times a day, 18 had either no additional benefit or an elevation in intraocular pressure. The remaining 8 patients had an additional mean 4.3-mmHg reduction in intraocular pressure.454
Barsam406 found that open-angle glaucoma subjects taken off 2% pilocarpine therapy for 48 hours and given a single drop of 2% pilocarpine or 0.03% or 0.06% echothiophate had maximum pressure reduction 2 to 4 hours after the pilocarpine and 4 to 6 hours after the echothiophate. The maximum hypotensive effect was similar for all three solutions, but the duration of effect was greater for the echothiophate solutions.
Romano455 used a double-masked cross-over study to compare 3 weeks of three-times-a-day instillation of 2% aceclidine and 2% pilocarpine in open-angle glaucoma patients. Both were effective, and the differences between them were insignificant with regard to hypotensive effect, outflow facility effect, and miosis.
The hypotensive action of a sustained-release form of pilocarpine (Ocusert) was simulated using eyedrops every 3 minutes.456 The minimum effective dose was between 10 and 30 μg/h. Armaly and Rao357 found that membranes dispensing 50 μg/h were more effective in open-angle glaucoma therapy than were 20-μg/h membranes. This was confirmed by Drance and associates.457 Release rates higher than 40 μg/h were no more effective.357,457
Intraocular pressure control 4 hours after insertion of a 20-μg/h membrane was similar to that 4 hours after instillation of a 2% pilocarpine eyedrop.357 The duration of hypotensive efficacy of this membrane was at least 1 week; that of a membrane releasing 40 μg/h was at least 10 days.
The concept of “tolerance” to muscarinic agonists and the beneficial effects of “rest therapy” are occasionally encountered. For example, Clarke352 found that subjects no longer responsive to pilocarpine therapy would respond after a pilocarpine-free period. Inactive solutions and patient noncompliance were not considered in explaining this phenomenon. Pharmacologically, it is difficult to understand why tolerance would develop so infrequently and why reinstitution of therapy would not result in a reappearance of tolerance. Sustained-release therapy (Ocusert) provides continuous exposure to pilocarpine and would be expected to lead to an increased incidence of tolerance, but this has not been reported (e.g., Worthen and co-workers458 found that membrane therapy for periods up to 8 months was as effective as pilocarpine drops).
Muscarinic agonists and β-sympathetic receptor blocking drugs have an additive effect. A solution containing 4% pilocarpine and 0.5% timolol administered twice a day produced a greater mean ± SD reduction in intraocular pressure (9.2 ± 5.1 mmHg) in glaucoma patients than did 4% pilocarpine four times a day (5.6 ± 3.6 mmHg) or 0.5% timolol twice a day (7.5 ± 5 mmHg).459–461 Similar results have been reported for combination solutions of pilocarpine-carteolol.
Transient Ocular Hypertension. Yttrium-aluminum-garnet laser capsulotomies and argon laser trabeculoplasties are often followed by a transient elevation in intraocular pressure. Pilocarpine (4%) drops administered immediately after laser therapy reduce the frequency and degree of these complications.462,463 In pilocarpine-treated trabeculoplasties, the mean intraocular pressure 2 hours after the procedure was 22 mmHg, whereas in the untreated group the mean was 28 mmHg.
Twenty-five glaucoma patients never treated pharmacologically had laser trabeculoplasties preceded 1 hour before by two drops of 2% pilocarpine; their mean ± SD maximum increase in intraocular pressure in the first 2 hours post-treatment was 2.4 ± 4.4 mmHg.464
The use of viscoelastic substances during cataract surgery is often associated with a transient postoperative elevation in intraocular pressure. Intracameral carbachol (0.01%), given in volumes of 0.1, 0.25, or 0.5 mL at the conclusion of surgery, was more effective than saline in controlling the intraocular pressure during the following 48 hours.465 Most effective was the 0.5-mL volume. This was confirmed in another study in which 8 of 30 eyes receiving saline had intraocular pressures exceeding 25 mmHg at 6 hours and 4 eyes exceeded 25 mmHg at 18 hours.466 None of the 30 patients receiving 0.5 mL 0.01% intracameral carbachol exceeded 25 mmHg at 6 hours, and only 1 of these 30 eyes exceeded 25 mmHg at 18 hours. One inch of 4% pilocarpine gel, placed in the cul-de-sac at the conclusion of surgery, was also effective.467 Intracameral acetylcholine (1%) was effective at 3 and 6 hours after surgery but not at 9 hours and 24 hours.468
Closed-Angle Glaucoma. Pilocarpine drops have been used to break attacks of acute closed-angle glaucoma. The rationale behind their frequent success is that the lax iris base is pulled taut and away from the trabecular meshwork. A similar rationale has been used to explain their value in preventing future attacks. Of Winter's469 patients with unilateral closed-angle glaucoma, 68% had involvement of the second eye. Although this occurred from hours up to 18 years after involvement of the first eye, most had contralateral involvement within 5 years. In 20 patients who maintained a continuous pharmacologic miosis, only 40% had a contralateral attack. In 8 patients maintained on intermittent therapy, 88% had a contralateral attack. In 19 patients without any therapy, 89% had a contralateral attack.
However, a narrow line exists between prophylactic miosis and provocative pupillary block. Thus, Lowe470 reported that two patients developed a contralateral attack shortly after stopping prophylactic pilocarpine but a third patient developed acute glaucoma just after starting pilocarpine. He believed 2% pilocarpine would be less likely to produce pupillary block than higher concentrations and suggested that, because most spontaneous attacks seemed to occur during the night, prophylactic drugs should be taken after the evening meal and again at bedtime. He cautioned that the miosis produced by physostigmine was more intense than that of pilocarpine and more likely to produce pupillary block. This is supported by Bain's471 results, in which 21% of 137 patients treated prophylactically with physostigmine or pilocarpine developed acute closed-angle glaucoma. Only 17% of 103 patients using pilocarpine had an attack, whereas 32% of 34 patients using physostigmine had an attack. Fifty-four percent of an untreated control group of 39 patients developed a second attack.
Mapstone472 used pilocarpine as part of a provocative test to predict which eyes with narrow angles were likely to develop acute narrow-angle glaucoma. He observed that the increase in ocular pressure after provocative testing tended not to occur when the pupil was widely dilated but, rather, when the pupil was mid-dilated. He believed that mid-dilation was the optimal position for maximizing the two crucial factors: trabecular block by the iris base and pupillary block by the iris sphincter. He found that the combination of a muscarinic agonist, pilocarpine, with an adrenergic agonist, phenylephrine, gave a greater incidence of positive provocative testing than did muscarinic atagonists. In 70% of 49 subjects with prior acute closed-angle glaucoma, pressure elevations of 8 mmHg or more were obtained within 2 hours of administering phenylephrine plus pilocarpine. Only 33% of subjects responded to 0.5% tropicamide. The response was reversed within 90 minutes of injecting acetazolamide, 500 mg, intravenously and applying one drop of 2% pilocarpine topically. However, this provocative test with a combination of pilocarpine and phenylephrine lacks sensitivity. The test was repeated at yearly intervals in the contralateral eyes of 104 patients who previously presented with unilateral acute narrow-angle glaucoma.473 Mean follow-up was 10 ± 5.7 years. Of 56 eyes with consistently negative tests, 14 developed acute glaucoma and 8 developed chronic or subacute glaucoma (i.e., 40% of eyes with consistently negative tests developed some form of closed-angle glaucoma).
Accommodative Esotropia. The near synkinesis consists of accommodation, convergence, and miosis. Decreased accommodative effort results in decreased convergence. This can be achieved by muscarinic agonists: pharmacologic stimulation of the ciliary muscle results in lens accommodation and reduces the need for central nervous system input. This rationale also applies to a nonpharmacologic technique of therapy, convex lenses. By reducing the neurogenic component of accommodation, both forms of therapy reduce accommodative convergence.
Pilocarpine is effective in reducing accommodative esotropia.474 Whitwell and Preston475 used it for a limited period beginning 2 to 3 days after strabismus surgery. Pilocarpine (1%), instilled in each eye three times a day, was gradually tapered over an 8-week period and then discontinued. Its use allowed fusion to develop, with or without the aid of orthoptics, if a residual accommodative component to the esotropia remained. Used in this manner, therapy was of permanent value in only those patients capable of stereopsis.
The longer-acting indirect muscarinic agonists have largely replaced the direct agonists in the therapy of accommodative esotropia. The former's use will be discussed in the section on cholinesterase inhibitors.
Intraoperative Miosis. Ray,476 Harley and Mishler,477 and Rizzuti478 injected acetylcholine into the anterior chamber after cataract extraction to protect the vitreous face and prevent iris incarceration into the surgical wound. Harley and Mishler477 dilated the eyes of patients preoperatively with 5% homatropine and 10% phenylephrine. Approximately 0.5 mL of 0.1%, 0.2%, 0.5%, or 1% acetylcholine was used. All concentrations were effective but short lived. The use of α-chymotrypsin did not affect the miosis. The stability of acetylcholine solutions was dependent on the pH and temperature. At 25°C and pH 5, the half-life of acetylcholine solutions was 14 years; at pH 7, 50 days; at pH 8, 5 days; and at pH 10, 1 hour.479 The pH of the commercially available 1% acetylcholine solution (Miochol) was approximately 8.5.
Douglas480 compared the efficacy of 1% acetylcholine with that of 0.01% carbachol (Miostat). In eyes dilated preoperatively with 5% homatropine and 10% phenylephrine, the miotic response to either solution injected into the anterior chamber was similar at 2 and 5 minutes. Several hours after surgery, the reduction in pupillary diameter, 4.4 ± 0.3 mm, was significantly greater in the eyes receiving carbachol than in the eyes receiving acetylcholine (2.9 ± 0.5 mm). Twenty-four hours after surgery there was no significant difference.
Drugs that inhibit prostaglandin synthesis provide increased mydriasis during intraocular surgery. Preoperative treatment with 0.03% flurbiprofen eyedrops did not prevent the miotic response 1, 2.5, and 5 minutes after intracameral acetylcholine.481
Reversal of Pharmacologic Mydriasis. α-Adrenergic agonists, such as 10% phenylephrine and 1% hydroxyamphetamine, produced mydriasis that was reversed by 1% pilocarpine.482 The mydriatic drops were given 65 to 70 minutes before pilocarpine. The pilocarpine-induced mean recovery time after phenylephrine was 22 minutes and that after hydroxyamphetamine was 14 minutes; however, 1% pilocarpine produced myopia as well as overcorrected mydriasis (i.e., miosis occurred).
The material deposited in exfoliation syndrome may impede iris movement. In 20 patients with exfoliation syndrome, phenylephrine produced less dilation than it did in 20 age-matched controls, and there was less constriction from a 10-μL drop of 4% pilocarpine given to reverse this mydriasis.483
Variable results have been reported when pilocarpine was used to reverse the mydriasis of muscarinic antagonists. One study found that one drop of 1% pilocarpine administered 30 minutes after tropicamide dilation not only returned the pupil to baseline diameter 3.5 hours later but continued to produce constriction, resulting in an overcorrection.482 However, another study of 23 volunteers found that one drop of 2% pilocarpine given unilaterally 30 minutes after bilateral tropicamide dilation was no more effective in reversing the mydriasis and cycloplegia 30, 120, and 180 minutes later than was the drop of saline placed in the other eye.484 Pilocarpine (1%), administered 72 minutes after homatropine dilation, was of little if any beneficial effect.482 Mean pupil recovery time was 28 hours. Reversal of mydriasis from a combination of 0.1% dilute tropicamide and 1% hydroxyamphetamine was “easily produced” by 2% pilocarpine. 485 Recovery from the mydriasis produced by three drops of 0.5% cyclopentolate was reduced from less than 20 hours to less than 6 hours by 1% pilocarpine.486
Occasionally, a patient will present with a dilated fixed pupil. If the patient is a nurse, he or she will be especially upset because of the known associations with aneurysms and tentorial herniation. In this situation, however, a likely cause is inadvertent self-administration of an atropinic agent: the nurse's fingers were contaminated when atropine drops were administered to a patient. If inadvertent or deliberate self-administration is suspected, a 0.5% or 1% pilocarpine drop administered bilaterally will be diagnostic. The pharmacologically dilated pupil will not respond as rapidly or as markedly, if at all. However, a dilated pupil due to an acquired efferent (third nerve) defect will respond. Rarely, congenital defects, such as absence of the sphincter muscle, will produce a mydriasis unresponsive to pilocarpine.487
Reversal of Pharmacologic Cycloplegia. Muscarinic agonists have been used to reverse cycloplegia. Although cholinesterase inhibitors have been used for this purpose with some success,488 most of the experience is with pilocarpine. In patients who had an initial dose of two drops of 0.5% cyclopentolate, followed 5 minutes later by a third drop, 1% pilocarpine reduced recovery time from 24 hours to 6 hours.489 One drop of 1% pilocarpine allowed subjects 14 to 35 years of age to read 2 to 4 hours after receiving 1% tropicamide.490 Pape and Forbes491 reported a retinal detachment after a single drop of 1% pilocarpine that was given to reverse mydriasis and cycloplegia.
Diagnosis of Supersensitivity. TONIC PUPIL (ADIE'S SYNDROME). In this entity the iris sphincter and ciliary muscle exhibit supersensitivity to direct-acting muscarinic agonists. The underlying pathology is believed to be degeneration of the postciliary ganglion fibers.492 A similar pathology is believed to occur in the tonic pupils associated with dysautonomias, such as Shy-Drager syndrome. The earliest pharmacologic test of sphincter function used physostigmine.493 However, it was not until the work of Scheie494 that pharmacologic testing of tonic pupils became routine. Scheie demonstrated supersensitivity with acetyl β-methylcholine. The supersensitivity appeared to be a permanent phenomenon: Laties and Scheie495 reported that it persisted in a subject for 25 years. It was initially believed that a drop of 20% acetyl β-methylcholine would constrict all pupils but that a drop of 2.5% would constrict only a supersensitive pupil. Subsequently, 41% of normal pupils were found to constrict more than 0.2 mm after 2.5% acetyl β-methylcholine,496 whereas only 63% of patients with a clinical diagnosis of Adie's syndrome had a positive response.497 Ocular hypertensive patients, previously untreated, had significantly greater pupil constriction to 2.5% acetyl β-methylcholine than did age- and sex-matched controls.498 Concentrations of acetyl β-methylcholine as high as 25% did not always constrict normal pupils.499 This variability in responses is, at least in part, attributable to differences in drug corneal penetration.500 False-negatives can result from increased tearing and blinking.
False-positive tests can result from abnormalities in the corneal epithelium that increase drug penetration (e.g., patients with exophthalmos and blink defects may give positive results,501,502 the interpretation of which must await repeated testing after the recovery of the corneal epithelium).503 For similar reasons, prior administration of eyedrops containing local anesthetics or surfactants should be avoided.
Despite these difficulties, the 2.5% acetyl β-methylcholine test remained in clinical use until the drug was no longer readily available for topical use. Another direct-acting muscarinic agonist was looked for, and found. Pilocarpine became heir to the acetyl β-methylcholine test even though the latter subsequently reappeared in a commercial preparation.
The pilocarpine test is best performed under conditions in which ambient lighting and accommodation are controlled, because both can affect pupil diameter. The pupil diameter can be evaluated by observation, photographs, or pupillography. A drop of test solution is instilled bilaterally. Thirty to 60 minutes after instillation, the pupillary diameters are reevaluated under the same conditions as before.
What concentrations of pilocarpine are optimal for demonstrating a tonic pupil? Pilocarpine 0.125% to 0.25% has been advocated even though the normal pupil will almost always constrict. Because the drops are instilled bilaterally, it does not matter if a submaximal constriction of the normal pupil is elicited. The relatively greater miotic response of the tonic pupil identifies it as supersensitive. Advocates of 0.0625% to 0.1% pilocarpine argue that tonic pupils can occur bilaterally and that relative miosis is, therefore, difficult to interpret. Instead, the less concentrated solutions should be used so that any constriction can be interpreted as evidence of supersensitivity. Unfortunately for this argument, not only will an occasional tonic iris not respond to a drop of 0.1% pilocarpine (i.e., there are false-negatives), but normally innervated irides may respond to 0.0625% (i.e., false-positives can occur). For example, 6 of 10 normal irides showed some constriction 30 minutes after a drop of 0.04% pilocarpine.504
Because 0.03125% to 0.05% concentrations of pilocarpine did not consistently constrict supersensitive irides, Cohen and Zakov505 and Young and Buski506 suggested using 0.0625% to 0.1% solutions. The 0.0625% pilocarpine solutions produced a mean 0.5-mm constriction in normal pupils and a mean 3.5-mm constriction in patients with Adie's syndrome. Pilley and Thompson497 and Bourgon and colleagues496 found that 85% of normal irides constricted more than 0.2 mm after 0.125% pilocarpine solutions. However, more constriction occurred in eyes with Adie's syndrome. This permitted the authors to make the correct diagnosis of supersensitivity 80% of the time. The mean constriction was 2.4 mm in normal eyes and 4.2 mm in supersensitive eyes.505 Hedges and Gerner507 advocated using a 0.25% pilocarpine concentration, whereas Young and Buski506 found 0.2% pilocarpine too potent for supersensitivity testing. In summary, the pharmacologic tests of iris sphincter denervation appear somewhat unreliable.
The relative pharmacologic supersensitivity of the ciliary muscle in Adie's syndrome has been examined. Single drops of 0.25% pilocarpine were instilled bilaterally, followed by a second set of drops 30 seconds later.508 An interocular difference of 1 D or more was considered significant. Seventy-three percent of patients with a clinical diagnosis of a unilateral lesion developed more myopia in the abnormal eye. An asymmetric increase in astigmatism occurred in one third of positive tests; this was attributed to sector supersensitivity of the ciliary muscle. However, pharmacologic testing appeared to be of little clinical value because 0.5 D or more of asymmetry in accommodation could be detected in 66% of Adie's syndrome patients simply by measuring the near point. Furthermore, the lens has lost almost all accommodative power by age 50, so neither pharmacologic nor near-point testing is of value in these older patients.
Ciliary Ganglion Denervation. Several investigators have examined whether damage to the oculomotor nerve fibers innervating the ciliary ganglion (i.e., preganglionic denervation) resulted in iris sphincter muscle supersensitivity to muscarinic agonists. Ponsford and colleagues509 found that oculomotor nerve palsies resulted in supersensitivity to 2% acetyl β-methylcholine. Fourteen normally innervated eyes developed a mean 0.25-mm constriction, whereas the contralateral eyes with oculomotor nerve palsies responded with a 0.96-mm constriction. In a second study, only 3 of 5 patients showed supersensitivity.510 One potential confounding factor in interpreting pharmacologic testing is that aberrant regeneration of extraocular muscle nerve fibers to the ciliary ganglion can produce pupil diameter changes when eye movements are made.511 Jacobson512 applied two drops of 0.1% pilocarpine bilaterally. Supersensitivity was defined as being present if, at 30 minutes, either the eye with the oculomotor nerve paresis constricted 0.5 mm more than the contralateral eye or the involved eye became the more miotic of the two. Supersensitivity was present in 5 of 11 eyes with tumor compressions, 4 of 5 eyes with traumatic palsies, 2 of 2 eyes with congenital palsies, and 0 of 5 eyes with idiopathic pupil-involving palsies. Supersensitivity, when present, did not correlate with the degree of anisocoria.
TOXICITY AND SIDE EFFECTS. The clinical use of direct-acting muscarinic agonists has been relatively free of significant side effects and toxicities.
The inadvertent 2-mL subcutaneous injection of an ophthalmic preparation of 4% pilocarpine (80 mg) in a 39-year-old woman was not fatal. It produced diaphoresis, salivation, nausea, diarrhea, and dyspnea; these symptoms gradually cleared over a 4-day period.513 However, deaths have been attributed to systemic pilocarpine toxicity.514
O'Brien and Swan353 stated that systemic and local reactions to carbachol were rare. The eyeache and headache that occurred initially were usually gone by the third day of therapy. Direct-acting muscarinic agonists produce fewer side effects than cholinesterase inhibitors.451 Drance and Nash449 used 1% to 8% pilocarpine solutions and found that browache occurred irrespective of concentration. Romano455 found conjunctival hyperemia, browache, and myopia more common with 2% aceclidine than with 2% pilocarpine. Contact dermatitis and urticaria are allergic reactions produced by topical pilocarpine.515
Miosis and narrowing of the anterior chamber may occur less often with sustained-release membranes (Ocusert), but this difference tends to decrease with age.435 However, Drance and associates516 found the decrease in visual acuity from induced myopia to be greater from Ocusert P20 than from 2% pilocarpine 90 minutes after placing the drop in one eye and the membrane in the other. They were reluctant to blame the difference in acuity purely on an accommodative cause because the spherical equivalents in the two eyes were similar. There was no interocular difference in acuity 90 minutes after placing a 4% pilocarpine drop in one eye and an Ocusert P40 membrane in the other. Worthen and co-workers458 reported that the membranes often were irritating and associated with browache, tearing, and discharge. Meyer517 reported the development of acute glaucoma after the membrane was lost from an eye treated prophylactically with an Ocusert.
Poinoosawmy and colleagues518 found that pilocarpine drops did not change human corneal thickness but did reduce the radii of the horizontal and vertical meridians of the cornea from 7.86 to 7.78 mm and from 7.90 to 7.81 mm, respectively. In addition to the corneal changes, there was a reduction in the radius of curvature of the anterior lens surface. In all 78 eyes in which pilocarpine produced a decrease in acuity, concave lenses were effective in returning the vision to predrop levels. Drance519 found that pilocarpine and echothiophate altered scleral rigidity. In 58 human eyes, 68% had a decrease of less than 30% in scleral rigidity, 17% had a decrease of more than 30%, and 15% had an increase of more than 30%.
Kennedy and associates520 reported that patients receiving pilocarpine developed an atypical band keratopathy. However, it is believed that this was the result of the mercury-containing preservative, not the pilocarpine.521–523 Mercury-containing preservatives are absorbed onto the rubber and plastic components of bottles and undergo degradation.524,525
Miosis from pilocarpine drops significantly reduced the visual field area measured by Goldmann or automated techniques.526,527 This is presumably due to reduced retinal illumination. Goldmann visual field area reductions for smaller isopters (I-2e = 64.5% ± 22.3%) were greater than for larger isopters (I-4e = 23.9% ± 16%) 30 minutes after normotensive subjects received 2% pilocarpine. In open-angle glaucoma subjects, a drop of 2% pilocarpine reduced automated visual field sensitivity by -1.49 dB.
Pilocarpine eyedrops increase the permeability of the blood-aqueous humor barrier.528 Normotensive subjects with darkly pigmented irides were given a single drop of 1% or 3% pilocarpine unilaterally and the vehicle contralaterally. Pilocarpine (1%) produced a significant increase in the mean ± SE anterior chamber protein content 3, 8, and 10 hours later. The increase at 8 hours, 21% ± 10%, was maximum. Pilocarpine (3%) produced significant elevations 1 to 4 and 6 hours postinstillation. The increase at 1 hour, 55% ± 11%, was maximum.
Chronic therapy of glaucoma with topical agents may alter the bulbar conjunctiva and reduce the success of subsequent filtering procedures. Rabbit eyes treated 4 months with pilocarpine, timolol, or saline had increased fibroblast proliferation after filtering procedures; epinephrine treatment did not produce this response. Pilocarpine produced the largest increase.529 One retrospective study could not confirm that long-term topical treatment of patients had a significant negative impact on subsequent filtering surgery.530 Another study was more suggestive of a deleterious effect but could not determine whether topical therapy in general, a specific drug class, or length of disease was the culprit.531 In this study, 106 patients had conjuctival biopsies at the time of filtering surgery and were followed a minimum of 6 months. Specimens from those whose trabeculectomies failed had significantly more fibroblasts, macrophages, and lymphocytes. The surgical success rate in subjects treated less than 2 months, 86%, was not significantly different from that in subjects treated less than 3 years, 92%. However, patients treated with topical drugs for more than 3 years before surgery had a significantly lower surgical success rate, 30%.
The possibility of pilocarpine-induced lens opacities has been examined by several workers. de Roetth179 reported anterior subcapsular vacuoles in 16% of eyes treated with 1% to 6% pilocarpine for a mean of 66 months and in 51% of eyes treated with 0.03% to 0.25% phospholine iodide for a mean of 20 months. In an age-matched group of normal eyes, 15% had vacuoles. In a retrospective study of patients treated for 3 years, Shaffer and Hetherington532 found an 8% incidence of cataracts in an untreated control group, a 6% incidence in a group treated with pilocarpine, and a 38% incidence in a group treated with an anticholinesterase (DFP, demecarium, or echothiophate). Axelsson and Holmberg533 found about a fivefold increase in development and progression of cataracts in eyes treated with echothiophate compared with eyes treated with pilocarpine. These results do not support a cataractogenic role for pilocarpine.
Muscarinic agonists can produce a closed-angle glaucoma if the lens accommodates sufficiently to push the iris base against the trabecular meshwork534–536 or sufficiently to block the miotic pupillary opening. Eyes with shallow chambers are more at risk, but occasionally the latter occurs in eyes with angles that were initially open.537
Specular and electron microscopic investigations of toxicity to rabbit endothelium from 1% acetylcholine and 0.01% carbachol (Miostat) failed to show permanent pathology.538 There were no temporary changes after in vitro corneas were bathed for 15 minutes with acetylcholine. After carbachol, changes in cell junctions were apparent within 1 hour but not thereafter. Intraocular 1% acetylcholine caused acute, reversible anterior subcapsular lens opacities in rabbits.539 Higher concentrations caused iritis in dogs.477 However, Rosen and Lazar540 pointed out that these effects probably were due to hyperosmolarity (e.g., the 1% acetylcholine solution was 390 mOsm). Similarly, Jay and MacDonald541 found that intraocular injection of 1% pilocarpine or acetylcholine was not toxic to bovine endothelial cells unless the solutions were made significantly hyperosmolar or hypo-osmolar relative to aqueous humor (300 mOsm). Systemic absorption of acetylcholine after injection into the anterior chamber has, rarely, been accused of causing hypotension and bradycardia.542 However, the inability of atropine to reverse the bradycardia makes it unlikely that acetylcholine absorption was at fault.
A cause-and-effect relationship between the topical use of muscarinic agonists and a resultant retinal detachment cannot be established because of the rarity of occurrence of the latter despite the frequency of use of the former; however, suspicions of causality remain. Both 1% pilocarpine 543 and membrane (Ocusert) pilocarpine delivery have been associated with retinal detachment.544,545 A macular hole developed in an eye with a nondetached posterior vitreous face when pilocarpine therapy was initiated; the hole resolved on discontinuation of the drug.546
Indirect-Acting Muscarinic Agonists
GENERAL CONSIDERATIONS. The ocular effects of this class of drugs, the cholinesterase inhibitors, are qualitatively similar to those of the direct-acting agonists: miosis, ciliary muscle contraction, and ocular hypotension. The indirect agonists can be subdivided into anticholinesterases that carbamylate the enzyme and anticholinesterases that phosphorylate it. Physostigmine, neostigmine, pyridostigmine, and demecarium are examples of the former, and DFP and echothiophate are examples of the latter (Figs. 6 and 7). The carbamylating drugs are usually shorter acting and more acetylcholinesterase specific than the phosphorylating compounds (e.g., DFP is more active against serum cholinesterase [pseudocholinesterase, butyrylcholinesterase], whereas physostigmine is about equally active against acetylcholinesterase and serum cholinesterase). An exception is demecarium, which, although a carbamylator, is long acting.547 This is because it contains two carbamate groups; it can carbamylate at two sites and cause permanent structural alteration of the enzyme. Demecarium is somewhat more active against acetylcholinesterase than against serum cholinesterase.
|
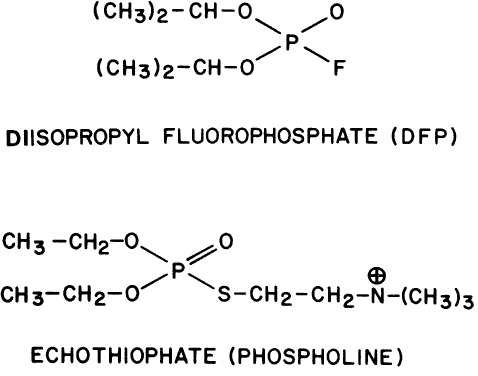
|
There is evidence that multiple molecular forms of acetylcholinesterase exist and not all forms are equally susceptible to organophosphate inactivation.548 Topical application of organophosphates seems to produce complete inhibition of acetylcholinesterase in the anterior structures (e.g., the cornea and iris) but not the retina.549 The retina, probably because of its distance from the cornea, is partially protected. Recovery of acetylcholinesterase activity is characterized by more rapid synthesis of lower-molecular-weight forms of the enzyme, which in turn are assembled into the higher-molecular-weight forms.550
Echothiophate was first marketed in 1959, after Leopold and colleagues551 had shown its clinical usefulness. DFP had been the phosphorylating anticholinesterase used previously, but it was unstable in water. One-tenth percent DFP in water was ineffective within 7 days.552 The clinical efficacy of DFP in peanut oil was superior to that of DFP in petrolatum.553 In peanut oil, DFP remained active after 1 year of storage at room temperature and after 1 hour of autoclaving,554 and this was the usual method of dispensing it. Echothiophate, being a positively charged quaternary amine, penetrated the corneal epithelium less readily than DFP but had the advantage of much greater stability in water. After 5 weeks at room temperature, the solution retained 75% activity: at 10 weeks, there was 62% activity, and at 30 weeks, there was 55% activity.551 Lawlor and Lee555 found that bottles of echothiophate solution refrigerated at 5°C for 8 weeks and opened daily lost only 4% to 5% of their activity. If stored at room temperature and opened daily, a 19% decrease in activity occurred in 8 weeks. Unopened solutions refrigerated 5 months retained 64% of their activity.
The phosphoryl group can be hydrolyzed off the esteradic site of cholinesterase by the oximes (RCH<cmb15>NOH). The oximes are much less effective in removing carbamyl-inactivating groups. By removing the phosphorylating group, the cholinesterase molecule is reactivated. However, if the phosphorylating agent becomes dealkylated while it is attached to the enzyme, that is, through hydrolytic loss of (CH3)2CH for DPF or CH3CH2 for echothiophate, then the drug cannot be removed from the enzyme. This process, called aging, results in a permanently inactivated cholinesterase molecule.556 An example of an oxime used to reactivate cholinesterase is pralidoxime (2-pyridine aldoxime methochloride, protopam, 2-PAM) (Fig. 8).
|
Like other oximes, pralidoxime can competitively bind to muscarinic receptors, but this is probably of no clinical significance.557 The drug is highly stable in solution; after storage at 37°C for 12 years, 75% of pralidoxime remained active.558
Pralidoxime is eliminated rapidly in the urine (80% to 90% of an orally administered dose is excreted in 60 to 90 minutes).559 Ocular penetration of pralidoxime after topical or systemic administration is limited because the molecule carries a positive charge at all times. Human volunteers given repeated oral and intramuscular doses of pralidoxime mesylate experienced diplopia and blurred vision.560 There was a 60% incidence of visual effects when subjects were given 4 g every 6 hours for 24 hours followed immediately by three 500-mg intramuscular injections, each injection being separated by 20 minutes. When rabbits were injected intramuscularly with pralidoxime mesylate 10 mg/kg, no drug was detected in the aqueous humor.561 When the concentration was increased to 40 mg/kg, the drug was detectable in aqueous humor. Diacetylmonoxime (DAM) does not carry a positive charge and can readily penetrate the blood-eye and blood-brain barriers (see Fig. 8). However, it is not commercially available in the United States.
OCULAR PHARMACOLOGY. Iris Sphincter Muscle. The carbamylating and phosphorylating anticholinesterases have no intrinsic cholinergic agonist activity. They act only by preventing acetylcholine hydrolysis. In cats with ciliary ganglionectomies, DFP did not produce miosis, nor did a 10% acetylcholine drop have significant effect.552 However, after DFP application, a 1% acetylcholine eyedrop produced miosis for 9 to 13 days.
Because cholinesterase inhibitors act through the accumulation of acetylcholine at the muscarinic receptor, drugs that compete with acetylcholine can alter the response to these indirect agonists. Pilocarpine has less intrinsic muscarinic activity than acetylcholine. Thus, when maximally effective doses of physostigmine or DFP are given, the subsequent addition of pilocarpine causes a diminution of both miosis and hypotension.367 Kaufman and Barany562,563 found similar results in primate eyes pretreated with cholinesterase inhibitors. However, if the inhibition of acetylcholinesterase is sufficiently submaximal (e.g., as when produced by low concentrations of physostigmine), then additional pupillary constriction can be achieved by giving pilocarpine drops.380 The mydriasis and cycloplegia from 3 drops of 1% atropine, given within a 36-hour period, are partially reversed by 2 to 3 drops of 0.5% echothiophate, given within a 30-minute period.551 Single instillations of 0.5% demecarium overcome mydriasis from either 1% atropine or 4% homatropine; within 1 to 3 hours of demecarium administration, the pupil is smaller than normal. This miosis becomes maximum at 24 hours and requires up to 7 days to recover. However, if the 0.5% demecarium were given first and the atropine or homatropine were instilled 6 hours later, the initial miosis would be abolished within 10 minutes.
Bito and Banks373 observed that monkeys given 0.1% DFP drops twice a day developed maximum miosis on the second day of treatment, but by the sixth day of treatment, the pupils had returned to pretreatment diameters in both dark and light. Increased iris dilator tone was not producing this phenomenon because it occurred despite prior superior cervical ganglionectomy.564 Subsensitivity of the muscarinic receptors may have been the cause, or a toxic effect of DFP on the iris sphincter may have been at fault.565 Soli and associates566 reported a similar finding in guinea pigs. Single drops of cholinesterase inhibitor produced marked miosis, but by the third day of daily treatment, a decreased response was found. They could not explain this effect either by a reduced number of muscarinic receptors, using quinuclidinyl benzilate binding, or by a more rapid synthesis of acetylcholinesterase. Furthermore, the miotic response to light stimulation remained, throwing into question the theory that drug toxicity of iris musculature was at fault. The rat iris dilator muscle has been shown to relax when exposed to low acetylcholine concentrations but to constrict, producing mydriasis, at 1 μmol/L and higher concentrations.567 Perhaps this would explain the decreased miosis. Another theory relates to choline uptake. Both carbamylating and phosphorylating cholinesterase inhibitors reduce rat iris choline uptake.568 Such a reduction could eventually impair acetylcholine synthesis. However, administration of an exogenous agonist should result in miosis. When Bito and Baroody569 topically applied pilocarpine to monkey eyes made subsensitive by daily cholinesterase inhibitor drops, they found a reduced miotic response in echothiophate-treated eyes and no miotic response in DFP-treated eyes.
Dunphy571 provided evidence that acetylcholine is continuously being released by ciliary ganglion neurons even in the absence of light stimulation or the near synkinesis. A volunteer had a single drop of 0.05% DFP instilled. He was kept in the dark, yet miosis occurred. Mindel570 demonstrated spontaneous acetylcholine release by denervated ciliary ganglia. Patients whose oculomotor nerves had been severed intracranially responded to an eyedrop of cholinesterase inhibitor with a marked miosis. Presumably, the amount of spontaneous acetylcholine released was insufficient to stimulate the iris sphincter until the cholinesterase inhibitor allowed the transmitter to accumulate. Loewenfeld and Newsome572 found that 8 minutes after a drop of 0.5% physostigmine, the increased effectiveness of released acetylcholine allowed the iris to react more rapidly and completely to light stimulation.
In normal subjects, miosis begins 5 to 10 minutes after instillation of 0.1% DFP, 10 to 45 minutes after instillation of 0.5% echothiophate, and 45 to 60 minutes after instillation of 0.1% to 0.5% demecarium. Maximum miosis occurs within 20 minutes of DFP application, whereas miosis from demecarium requires 2 to 4 hours. Concentrations of DFP below 0.01% have no effect. Recovery from the miosis of DFP, echothiophate, or demecarium requires 3 to 28 days.547,551,552
If physostigmine were given 15 minutes before DFP, the miotic effect would be that of the former (i.e., recovery requires only 2 to 3 days).553 This demonstrates that both carbamylating and phosphorylating cholinesterase inhibitors attack the same site on acetylcholinesterase and in a noncompetitive manner. The first drug to reach the esteradic site will inactivate it until spontaneous hydrolysis removes the carbamyl or phosphoryl group.
Ciliary Muscle. Strips of ciliary muscle from cats, chickens, frogs, rabbits, and monkeys exposed to acetylcholine had their contractions increased by factors of 10 to 10,000 if physostigmine 1 μg/mL was in the bathing solution.573 Atropine was effective in relaxing muscle strips that had previously received physostigmine.
Monkeys treated for 5 to 6.5 months with topical echothiophate had approximately a 65% reduction in their ciliary muscle muscarinic binding sites and a decreased response to pilocarpine. The binding affinity of the remaining muscarinic receptors was unchanged.574,575 The iris response to pilocarpine was unaffected. After echothiophate treatment was discontinued, the number of ciliary muscle muscarinic binding sites and the response to pilocarpine returned in 4 to 8 weeks. Eyes permitted to recover for 5 to 6.5 months exhibited a rebound effect (i.e., their ciliary bodies had more than double the number of muscarinic bindings sites compared with untreated control monkeys). When 0.05% physostigmine was applied topically to human subjects, an accommodation of the lens could be elicited that was above physiologic levels.576 In three subjects, aged 27 to 30 years, this increase was more than 2 D. Physostigmine (0.05%), by facilitating accommodation, also produced a decrease in the accommodative convergence/accommodation (AC/A) ratio (e.g., in one subject the AC/A ratio was 1.2 to 1.3 before physostigmine and 0.8 to 0.9 after physostigmine). Wilkie and co-workers394 measured human anterior chamber depth in response to echothiophate. A single drop produced little shallowing, but there was a progressive decrease in depth during the 10-week treatment period.
Intraocular Pressure. DFP causes an initial rise in rabbit intraocular pressure.577,578 The pressure elevation begins with the onset of miosis and at its maximum, approximately 30 minutes after instillation, is 15 to 30 mmHg above baseline. The pressure remains above baseline 4 to 6 hours later. Intravenously administered fluorescein accumulates in the anterior chamber, indicating that the blood-aqueous barrier has been impaired.579 Histologic examination shows epithelial blisters on the ciliary processes, engorgement of the iris and ciliary vessels, and increased protein in the anterior and posterior chambers.577 Prior phenylephrine, injected subconjunctivally, prevents the elevations in anterior chamber protein and intraocular pressure, presumably by constricting the uveal vessels.578
Monkey eyes treated for 5 or more months with topical echothiophate drops had elevated intraocular pressures associated with histologic evidence of damage to the aqueous humor outflow system.580 Extracellular material was found in the trabecular meshwork, Schlemm's canal was altered in shape, and there was discontinuity between ciliary muscle bundles and trabecular beams.
McDonald581 reported that 10% of human eyes treated with DFP also had an initial rise in intraocular pressure of 3 to 7 mmHg. Krishna and Leopold547 and Drance and Carr582 found similar rises after demecarium and echothiophate administration. Some of the theories offered to explain these increases were that congestion of the ciliary body vessels caused a narrowed filtration angle, that increased protein in the aqueous humor drew water into the anterior chamber by osmosis, and that ciliary muscle contraction caused a forward shift of the lens-iris diaphragm.583
The eventual hypotensive effect of cholinesterase inhibitors is variable in normotensive eyes. Leopold and colleagues551 found it to be rarely more than 2 to 3 mm. Drance and Carr582 reported a 52% decrease in pressure. In most normotensive subjects the intraocular pressure after a single instillation begins to decline in approximately 30 minutes without transient pressure rise. Although miosis begins about the same time as the hypotensive action, the two do not always occur together.582 With 0.1% to 0.5% demecarium administration, the maximum hypotensive effect occurs 24 hours after instillation and may remain below baseline for as long as 9 days. A significant increase in tonographic outflow facility is usually present 2 hours after instillation and is almost always present 24 hours after instillation.547,584 However, the hypotensive action is not always associated with an increase in outflow facility.585 Drance585 found that instillation of single drops of 0.1% or 0.25% demecarium in normotensive eyes did not produce hypotensive effects until after latency periods of 12 hours. The tonographic increases in outflow facility lasted up to 5 days. However, in four eyes there were no increases in outflow facility despite large decreases in intraocular pressure.
Retina. Infusion of physostigmine into the rabbit retinal circulation abolished the directional specificity of both on-center and on-off retinal ganglion cells.586
Extraocular Muscles. Topical application of DFP to rabbit eyes resulted in increased sensitivity of the superior recti muscles to solutions of acetylcholine and carbachol (i.e., the muscles produced larger contractions than in control eyes).587 Recovery was gradual. More than 7 days without DFP treatment was needed for sensitivity to return to normal. DFP is highly lipid soluble. It was believed that DFP penetrated the conjunctiva and inhibited acetylcholinesterase in the neuromuscular junctions.
THERAPEUTICS. Glaucoma. Physostigmine in concentrations of 0.25% to 1% increases outflow facility and lowers intraocular pressure in subjects with initial readings of 22 mmHg or greater.423
Demecarium is a potent hypotensive agent.588,589 Drance585 used demecarium to treat 40 eyes with chronic simple glaucoma. In 38, there was a decrease in intraocular pressure. The mean time for maximal decrease in pressure was 34 hours, and the mean percent fall in pressure for all 40 eyes was 48%. In 34 eyes tested by tonography, the mean increase in outflow facility was 121%.
Phosphorylating agents are also effective. DFP, used by Leopold and McDonald553 over a period of 4 or more months, lowered the ocular pressures of 71% of eyes with chronic narrow-angle glaucoma, 74% of eyes with aphakic glaucoma, and 54% of eyes with open-angle glaucoma. It was of value in controlling 61% of eyes with uveitic (inflammatory) glaucoma and in terminating acute glaucoma attacks. Only 14% of acute attacks were terminated by the use of pilocarpine, acetyl β-methylcholine, physostigmine, or neostigmine, whereas 57% ceased after the application of DFP. However, Leopold and McDonald553 also noted six glaucoma patients in whom DFP produced an unexpected increase in intraocular pressure. Four had preexisting narrow angles. The maximum effective concentration of DFP in peanut oil was 0.2%. These authors found that pilocarpine and/or physostigmine was as effective as 0.1% DFP in lowering ocular pressures below 30 mmHg in 17 glaucoma subjects. However, in 29 other subjects, DFP was successful when the other two were not.
Echothiophate has been the most widely used phosphorylating cholinesterase inhibitor. In 59 chronic simple glaucoma patients given a single drop of 0.1% to 0.25% echothiophate, the mean decrease in ocular pressure was 48% 3 hours postinstillation, and the mean increase in outflow facility, measured in 28 eyes 24 hours after instillation, was 127%.582 Echothiophate, unlike pilocarpine and carbachol, produced a sustained increase in outflow facility.406 In dilutions as low as 0.01%, given twice a day, it was as effective as pilocarpine in 42% of patients.592 Harris593 studied 15 glaucoma patients and found that maximum improvement in intraocular pressure and outflow facility occurred with 0.06% echothiophate; increasing the concentration to 0.25% did not significantly improve either (Table 1). The maximum effect on intraocular pressure and outflow facility has been reported to occur from 4 to 27 hours after application.406,594
TABLE 1. Results of Studies of Echothiophate in 15 Glaucoma Patients
| Echothiophate Versus Control | ||||||||
| 0.03% | Control | 0.06% | Control | 0.12% | Control | 0.25% | Control | |
| Intraocular pressure (mmHg) | 26.4 ± 2.6 | 31.8 ± 2.2 | 24.3 ± 2.2 | 32.0 ± 2.4 | 22.4 ± 2.1 | 31.6 ± 2.5 | 21.1 ± 2.2 | 31.0 ± 2.4 |
| Outflow facility | 0.10 ± 0.01 | 0.06 ± 0.02 | 0.12 ± 0.02 | 0.05 ± 0.02 | 0.13 ± 0.02 | 0.06 ± 0.02 | 0.14 ± 0.01 | 0.07 ± 0.02 |
There is evidence to support Barsam's406 belief that echothiophate and pilocarpine are similar in hypotensive potency and differ only in duration of action. In patients on chronic therapy, the minimum intraocular pressure on the diurnal curve is a measure of drug potency, and the magnitude of the fluctuation of the diurnal curve is a measure of drug duration of action. Drance594 had compared the diurnal hypotensive efficacy of 4% pilocarpine four times a day with that of 0.06% echothiophate two times a day using subjects with ocular hypertension and chronic simple glaucoma. Seventy percent of pilocarpine-treated eyes had diurnal pressure peaks that rose above 24 mmHg as opposed to only 20% of echothiophate-treated eyes. Similar results were reported by Barsam and Vogel,595 comparing 0.06% echothiophate once a day with 2% pilocarpine four times a day. Pratt-Johnson and colleagues451 studied the pressure response to 0.06% echothiophate twice a day and to 4% pilocarpine four times a day in 20 eyes with intraocular pressures greater than 24 mmHg. The mean peak pressure while receiving pilocarpine, 23 ± 5.3 mmHg, was greater than that while receiving echothiophate, 19 ± 5.5 mmHg. However, the mean minimum pressure achieved with pilocarpine, 13 ± 2.7 mmHg, was similar to that achieved with echothiophate, 12 ± 2.8 mmHg. Either the two drugs are equally potent or the 0.06% echothiophate solution is too dilute to evoke a maximum hypotensive effect.
When demecarium or echothiophate was added to the various therapies used by poorly controlled glaucoma patients, the results indicated that the two drugs were about equally effective.596 After 6 months of therapy, 0.25% and 1% demecarium maintained 27% and 36% of eyes, respectively, at a pressure less than 20 mmHg, and 13% and 20%, respectively, had an outflow facility of more than 0.15 μL/min/mmHg. Using these same criteria, 0.25% echothiophate controlled the pressures in 25% of eyes and improved the outflow values in 20% of eyes. Of 42 eyes with pressures uncontrolled on echothiophate therapy and 32 eyes with pressures uncontrolled on demecarium therapy, demecarium was effective in 13% of the former and echothiophate was effective in 24% of the latter.
Echothiophate has been used in combination with epinephrine, acetazolamide, and pilocarpine.555,597 Klayman and Taffet598 found that 0.06% echothiophate, twice a day, controlled for at least 1 year the intraocular pressure of 43% of eyes with pressures greater than 20 mmHg and visual field loss. Another 33% required the addition of acetazolamide or epinephrine to bring the ocular pressures below 21 mmHg. In 7% of eyes the pressures were controlled when all three drugs were used. The pressures in 17% of eyes remained uncontrolled despite therapy with all three drugs.
Accommodative Esotropia. Atropine was occasionally used in the past to treat accommodative esotropia. With the onset of cycloplegia, the esotropia would increase as the patient attempted to focus. Then, as this inability was accepted, accommodative efforts would cease and the eyes would straighten. To varying degrees, however, near work would once again elicit volitional accommodative effort and the eyes would deviate inward. In contrast to atropine, cholinesterase inhibitors correct accommodative esotropia by facilitating peripheral accommodation (i.e., they decrease the need for volitional accommodative effort). Although they also induce miosis and thereby increase the depth of focus, this is not their primary mechanism of action.599 If the AC/A ratio is abnormally high, cholinesterase inhibitors will lower it. Cholinesterase inhibitors would then be expected not only to facilitate accommodation but also to produce an accommodative myopia in nonpresbyopic patients. However, there is some evidence that cholinesterase inhibitors are less likely to produce a spasm of accommodation than are the direct-acting agonists, such as pilocarpine.600,601 Rehany and associates602 found that a single drop of 0.125% echothiophate failed to induce accommodation during the 24 hours after instillation, whereas a single drop of 4% pilocarpine produced a demonstrable accommodation that lasted several hours, being more than 4 D at its peak 30 minutes after instillation. Miller600 found that patients treated with DFP, demecarium, and echothiophate had less than 0.5 D of induced accommodation after 1 week of therapy.
Wheeler603 used cholinesterase inhibitors to diagnose accommodative esotropia, before prescribing spectacles, and to treat the condition. Both carbamylating agents (e.g., 0.25% physostigmine) and phosphorylating agents (e.g., 0.1% DFP) were effective.474 However, the latter were longer acting. Miller found 0.025% DFP,600 0.25% demecarium, and 0.1% echothiophate about equal in controlling accommodative esotropia.
Parks604 used a tapering dose of anticholinesterase to wean children off therapy over a 6-week to 18-month period. As fusional capabilities gradually increased, less medication was required. Lasting improvement occurred in 27% of patients under 5 years of age and in 88% of children 7 to 12 years of age. The overall success rate was 68%. Parks concluded that cholinesterase inhibitors were more likely to give permanent improvement if they were discontinued after age 7 rather than before. Children who develop accommodative esotropia within the first year of life form a special subgroup. Approximately 50% of these patients develop a nonaccommodative esotropia requiring surgery, even though they initially respond to echothiophate or DFP treatment.605
Synechiae. Cholinesterase inhibitors have been reported to occasionally break posterior synechiae that could not be lysed by dilatation. The rationale for their success was that the iris sphincter muscle provided a more forceful contraction than the iris radial muscle. Mamo and Leopold606 initially used anticholinesterases on two subjects with long-standing synechiae; they were unimpressed by the results. Byron and Posner607 reported 10 patients with posterior synechiae to the lens capsule and nine patients with posterior synechiae to secondary membranes. These subjects were initially made miotic with two drops of echothiophate. The echothiophate effect was then reversed by injecting 0.1 to 0.2 mL of 5% pralidoxime subconjunctivally. Maximum mydriasis was then achieved with two drops each of 1% atropine and 10% phenylephrine. Dense synechiae did not respond. There was partial to total lysis of smaller synechiae in 8 of the 10 patients with adhesions to the lens capsule. In 5 of 9 patients with adhesions to secondary membranes, there were increases of at least 2 mm in the pupillary diameters.
Cyclodialysis Clefts. Gonioscopic examination has verified that surgically created cyclodialysis clefts remained open postoperatively when these eyes were treated with cholinesterase inhibitors. Gorin608 found that a drop of 0.1% DFP daily for several weeks was effective in 15 patients.
Refractive Errors. Cholinesterase inhibitors have been used instead of spectacles to correct myopia, presbyopia, and hyperopia.609 Their effectiveness in the first two conditions was attributed to the marked miosis, producing a pinhole effect. Echothiophate (0.06%), twice a day, was the initial dose, with the frequency and concentration increased as needed. In presbyopes, the criterion for success was maintenance of best distance acuity with improvement of near reading acuity to Jaeger 3; in hyperopes, the criterion was 20/25 (6/7.5) distance vision. Fifty-five percent of presbyopes no longer required spectacles and 26% required only a distance prescription. In hyperopes and myopes in the + 2.00 D to -2.00 D range there was a 42% success rate. In patients with errors greater than this, there was a 12% success rate.
Adie's Syndrome. Variable success has been reported with dilute concentrations of physostigmine to reduce the mydriasis and improve the accommodation in eyes with Adie's syndrome.610 Eyedrops of 0.0125% to 0.125% physostigmine were given to five patients. Two patients tolerated long-term treatment, but three patients discontinued their use because of symptoms of miosis, blur, or discomfort.
OCULAR TOXICITY AND SIDE EFFECTS. Since the mid 1960s, there has been considerable emphasis on the toxicities and side effects of the cholinesterase inhibitors.
Cataractogenesis. Harrison611 is credited with first reporting an organophosphate-induced lens opacity. The patient was a 13-year-old girl given 0.025% DFP in peanut oil nightly for 3 months. Anterior subcapsular lens opacities were noted. The medication was stopped and in 9 days the opacifications had partially cleared. In 3 weeks, the lenses appeared normal. Pietsch and co-workers612 found no lens changes in children treated up to 48 months with echothiophate. Although lens opacities rarely occur in children, this is not true of adults. Axelsson and Holmberg533 found that in glaucoma patients treated for a mean of 22 months with 0.06% or 0.25% echothiophate, new opacities developed at a rate more than six times greater than that in glaucoma patients treated for the same period with 2% or 4% pilocarpine. Opacities developed in 40% of the echothiophate-treated patients but in only 6% of the pilocarpine-treated patients. Progressive pathologic changes also occurred in subjects with preexisting opacities. About five times as many patients on echothiophate progressed compared with those on pilocarpine. A positive correlation existed for strength as well as for length of treatment. DFP and demecarium were also cataractogenic.532,613 In monkeys, carbachol, which has some cholinesterase inhibitory action, has produced cataracts.614 In a prospective study of 16 patients observed for 7 to 21 months, there were no lens changes in those 6 subjects receiving 0.06% or 0.125% echothiophate.615 However, 5 of 10 subjects receiving 0.25% echothiophate developed anterior subcapsular vacuoles, nuclear sclerosis, and/or posterior subcapsular cataracts. Some authors have stated that anterior subcapsular changes occurred earliest, beginning with vacuoles and progressing to a mossy-appearing opacification.532 Axelsson616 and Morton and colleagues617 noted that when echothiophate therapy was discontinued, the lens changes progressed in some eyes whereas in others they regressed. Occasionally, the lens changes regressed despite continued treatment.
In monkeys, concomitant treatment with atropine seemed to delay the onset of lens changes and in some instances seemed to partially reverse lens changes produced by echothiophate.618 Levene619 noted that in 10 of 12 patients developing echothiophate-related cataracts, pilocarpine had been used as prior therapy for 2 months or less. He suggested that 3 months or more of pilocarpine pretreatment might protect against lens changes. However, many subjects with cholinesterase inhibitor-induced cataracts had received prior long-term pilocarpine therapy. In Shaffer and Hetherington's532 study, all their subjects received prior pilocarpine therapy, but DFP, demecarium, and echothiophate use was associated with 30%, 40%, and 35% cataract incidences, respectively. Axelsson616 found that there was a 50% incidence of decreased vision in eyes treated initially and solely with echothiophate; in eyes previously treated with pilocarpine or pilocarpine-physostigmine, the incidence was about the same (43%).
The biochemical abnormality responsible for the development of cataracts has not been defined. The phosphorylating agents are highly reactive and may attack multiple enzymes and cell structures. Harkonen and Tarkkanen620 gave albino rabbits 0.25% echothiophate eyedrops twice daily for 15 weeks. No lens opacities occurred, but ATP content decreased by 35% and lactate content by 17%.
Iris Nodules. Abraham621 reported that two thirds of 66 young subjects treated with DFP for strabismus developed iris nodules. Only one subject in the study was over age 14 years. Nodules appeared after a mean of 10 weeks of treatment. A nasal pupil location was favored. In all cases, the nodules disappeared after discontinuation of treatment, leaving shrunken tags. In 83%, the nodules were gone within 15 weeks; by 42 weeks, all had shrunken. The tags usually, but not always, disappeared gradually. Concentrations as low as 0.03% echothiophate have been reported to produce nodules.592 Therapy with pilocarpine and physostigmine also led to nodule formation, but much less often. Microscopic examination revealed hyperplasia of the iris pigment epithelium, forming solid elevations more often than cysts.622 Phenylephrine (10%) given nightly appeared to prevent the development of these nodules.603
Retinal Detachment. Because the human retina stretches during marked ciliary muscle contraction (e.g., 9 D of accommodation can cause 4% to 5% retinal stretching),623 it is not surprising that retinal tears have been associated with cholinesterase inhibitor use. However, the incidence is too low to allow a statistical analysis of causality. Instead, the literature on this subject consists of case reports.596,624 Physostigmine, the first drug used to treat glaucoma, was reported to be clinically effective by Leber in 1876. In 1877, he reported a retinal detachment associated with therapy. Pape and Forbes491 and Alpar625 reported detachments in association with direct-acting muscarinic agonists, such as pilocarpine, as well as with indirect agonists. In the former paper, 41% of detachments occurred within 2 months of the initiation of therapy, but the range extended to 17 years.
Acute Closed-Angle Glaucoma. Leopold and McDonald553 found DFP to be of value in treating narrow-angle glaucoma. However, the cholinesterase inhibitors subsequently have been shown to provoke attacks in predisposed eyes.551,583
Miscellaneous. Headache, browache, eyeache, induced myopia, conjunctival congestion, allergy, punctal stenosis, fibrinous iritis, and posterior synechiae have been associated with the use of cholinesterase inhibitors.547,553,596,626 In over 70% of subjects with side effects, the signs and symptoms appeared within the first 48 hours of treatment. The aching usually did not persist beyond the first week of treatment and was often relieved by salicylates. Side effects caused 14% of demecarium-treated patients and 20% of echothiophate-treated patients to discontinue therapy. Approximately 25% of the former and 50% of the latter were successfully changed to the other drug. Using lower drug concentrations appeared to reduce the frequency of side effects, but all the same problems still occurred.551,592,593
SYSTEMIC TOXICITY AND SIDE EFFECTS. Leopold and Comroe552 reported that 0.1% DFP in peanut oil was absorbed sufficiently after topical eyedrop use to lower serum pseudocholinesterase activity but not red blood cell acetylcholinesterase activity. Pilocarpine therapy, however, was not associated with a reduction in serum or red blood cell cholinesterase activity.627 In glaucoma patients treated with echothiophate twice a day, the 0.03% and 0.06% concentrations reduced serum cholinesterase activity by 35% to 40% and red blood cell acetylcholinesterase activity by 25% to 30% within 2 weeks628; this level of reduction was maintained while therapy was continued for the ensuing year. Twice-a-day application of the more concentrated echothiophate solution, 0.25%, resulted in a 75% to 80% reduction in serum cholinesterase and an 80% to 85% reduction in red blood cell acetylcholinesterase activity. These, too, occurred within 2 weeks. Upon cessation of therapy, cholinesterase activity began to increase within a week. Recovery was complete by 4 months.629 Humphreys and Holmes630 and Wahl and Tyner631 believed that systemic side effects, such as diarrhea632 and nausea, were inversely correlated with blood cholinesterase activity; however, de Roetth and co-workers628 did not (e.g., one of their subjects who was symptom free had only 5% of pretreatment blood cholinesterase activity).
The pupils are usually miotic in patients with systemic anticholinesterase toxicity unassociated with ocular use (e.g., insecticide poisoning). However, Dixon633 reported two patients with organophosphorus toxicity who had dilated pupils. Nattel and colleagues634 studied the effect of intravenous physostigmine, 2 mg, on the pupils of patients with drug-induced coma. In subjects comatose from drugs without atropinic activity (e.g., barbiturates), physostigmine caused pupils to dilate. In subjects comatose from drugs with atropinic activity (e.g., amitriptyline), physostigmine did not cause pupil dilatation. These results are difficult to interpret and may have been due to central nervous system effects. Physostigmine, being a tertiary amine, can cross the blood-brain barrier.
Phosphorylating inhibitors depress serum cholinesterase activity and prolong the effects and increase the toxicities of drugs normally inactivated by this enzyme. Succinyldicholine and ester local anesthetics such as procaine are commonly cited in this regard. However, it is difficult to find instances in which chronic administration of eyedrops has depressed serum cholinesterase activity to a clinically significant degree. With regard to the prolongation of succinyldicholine effect, the report frequently referred to is that by Pantuck.635 It consists of a single case. A 39-kg female, on daily 0.125% echothiophate drops bilaterally for 9 months, was given 40 mg succinyldicholine intravenously. Diaphragmatic activity and tracheal tug did not begin for 10 minutes, but by 20 minutes postinjection her respirations had returned to normal. Serum cholinesterase activity was found to be 30% of normal levels. This relatively mild prolongation of apnea does not seem to justify the degree of fear that is often voiced. With regard to local anesthetic toxicity, there is even less justification. Again, a firm theoretic and experimental basis exists,636 but the amount of cholinesterase depression from systemic absorption of eyedrops seems inadequate to convert a therapeutic dose into a lethal one.637,638 Other drugs shown to be hydrolyzed by serum cholinesterase are methylprednisolone acetate,639 substance P,640 and heroin.641 The evidence that maternal cholinesterase-inhibitor eyedrops reduce fetal serum cholinesterase activity is limited.642
The human blood complement system, consisting of 20 plasma proteins and 9 regulatory proteins, neutralizes and lyses bacteria and viruses. Activation of this system usually requires two serine esterases, C1R and C1S, and an esteradic site on C2. The phosphorylating cholinesterase inhibitors, such as DFP, are potent inhibitors of these serine esterases.643 There are no reports of increased susceptibility to infections in patients receiving topical cholinesterase inhibitors.
ANTIDOTES. A small dose of intravenous physostigmine protects against a subsequent lethal dose of DFP. It also prevents a prolonged decrease in serum cholinesterase activity.644 However, physostigmine does not protect or reactivate serum esterase if it is given after DFP. Oxime therapy will reactivate serum cholinesterase if it is given soon enough. After 9 months of 0.25% echothiophate eyedrops twice a day, 1 week of pralidoxime, 500 mg orally three times a day, did not elevate serum or red blood cell cholinesterase activity.629 This was because the phosphorylated enzymes had aged. After 3 weeks of oxime treatment there was a significant increase in red cell acetylcholinesterase activity.645 This resulted from erythrocyte turnover and synthesis of new enzyme. In chronic simple glaucoma patients receiving 10.5 g pralidoxime orally each day for 3 weeks, there were no changes in pupil size, intraocular pressure, or outflow facility.645 de Roetth and associates629 reported that the intraocular pressures rose in patients given oral pralidoxime, but these subjects had discontinued their echothiophate eyedrops.
Local ocular oxime therapy has been investigated. Pralidoxime, diacetyl monoxime, TMB-4 (1,1-trimethylene bis-4-formylpyridinium oxime bromide), and MINA (monoisonitrosoacetone) have been instilled as 1%, 3%, and 5% aqueous drops, 5% aqueous drops with 1/3000 benzalkonium, and 5%, 10%, and 20% petrolatum ointments.606 In 31 volunteers, they had no effect on the normal pupil or in pupils made miotic with pilocarpine. With a few exceptions, multiple drops, even with benzalkonium, had no effect when given immediately after DFP- or physostigmine-induced miosis. The 10% and 20% ointments were effective antidotes in about half the subjects. Subconjunctival injection of a 5% solution was 100% effective in reversing miosis and was more effective than 1% atropine plus 10% phenylephrine eyedrops.607 Similar results were reported by Becker and associates597 in glaucomatous eyes. In untreated glaucoma patients, subconjunctival pralidoxime had no effect. However, in echothiophate-treated patients and, to the authors' surprise, in two of six pilocarpine-treated eyes, it produced a decrease in outflow facility.
Muscarinic Antagonists
GENERAL CONSIDERATIONS. The muscarinic blocking agents in clinical use act by competing with the physiologic agonist, acetylcholine, for the receptor. This may be too simplistic a view, however. Marshall646 studied the two stereoisomers of atropine: (-) hyoscyamine was 30 times more active than (+ ) hyoscyamine; (-) hyoscyamine was a competitive antagonist, but (+ ) hyoscyamine appeared to be a noncompetitive antagonist. Gupta and co-workers647 studied the binding of an irreversible antagonist, benzilylcholine mustard, to the muscarinic receptor. They found evidence that binding occurred at two different sites. Ellenbroek and colleagues648 believed that atropinic drugs were competitive antagonists but that agonists and antagonists bound to the receptor at different sites.
The muscarinic antagonists can be subdivided into the naturally occurring agents, such as atropine (hyoscyamine) and scopolamine (hyoscine), and the synthetic agents, such as cyclopentolate, homatropine, and tropicamide (Figs. 9 and 10). All are quite stable.
|
|
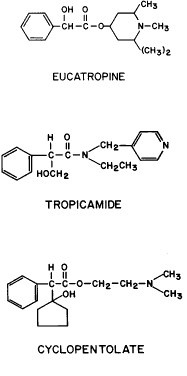
When cyclopentolate spontaneously degrades, it is by hydrolysis of its ester bond with formation of N,N-dimethylaminoethanol and cyclopentanyl benzeneacetic acid. The latter then either breaks down into phenylacetic acid and cyclopentanone or is hydroxylated and becomes hydroxycyclopentyl benzeneacetic acid.649 Atropine spontaneous hydrolysis is pH and temperature dependent. At 20° C, the half-life of atropine in a pH 7 solution is 2.7 years; in a pH 6 solution, 27 years; in a pH 5 solution, 266 years; and in a pH 4.5 solution, 811 years. At 30° C, these values are reduced to 0.61 years at pH 7, 6.1 years at pH 6, 61 years at pH 5, and 194 years at pH 4.5.650 The half-life of tropicamide is even longer (e.g., at 20° C and pH 7.45, it is 2346 years).651 Additional factors, besides pH and temperature, are important for commercial preparations. Unbuffered solutions of atropine and scopolamine are more stable than buffered solutions, and low-density polyethylene containers are preferable to glass.652
Tropicamide, with pKa = 5.37, is the only clinically used muscarinic antagonist that is primarily (approximately 98%) in the nonionized form at physiologic pH. Homatropine, with pKa = 9.9, is only 0.32% nonionized. Atropine, pKa = 9.8, and cyclopentolate, pKa = 8.4, are also primarily in an ionized state.653,654 As a result, tropicamide has much greater access to the muscarinic receptors because it can readily penetrate the corneal epithelium. The relative in vitro muscarinic blocking activities of these drugs is atropine > cyclopentolate > homatropine > tropicamide. However, the greater penetration of tropicamide makes it more effective than homatropine and changes the in vivo order of potency to atropine > cyclopentolate > tropicamide > homatropine.654 Tropicamide consists of two stereoisomers.655 Both appear to be competitive blockers of acetylcholine.656 However, the levorotatory isomer is far more active pharmacologically. In pigmented and nonpigmented irides, it is 75 times and 50 times, respectively, more active than the dextrorotatory isomer.
Muscarinic receptors in different tissues, and even in different mammalian species, tend to bind the non-subtype-specific antagonist atropine in a similar manner. The concentration of atropine needed to half-saturate human iris receptors in vitro is 0.4 to 0.7 nmol/L.657 This value agrees with those reported for rabbit iris,654 guinea pig ileum,658 and guinea pig heart.659 However, the variability in duration of action of muscarinic antagonists at different sites may be due to differences in receptor subtype (e.g., the tachycardia effect of atropine lasts minutes to hours, but the pupil dilation effect lasts for days). Two other factors that may affect duration of action are the pigment binding of drugs and, in some species, the presence of enzymes that inactivate muscarinic antagonists. Chen and Poth660 noted that eucatropine was a more effective mydriatic in white subjects than in blacks; the effect on Orientals was intermediate. Patil and associates661 found that the binding to uveal pigment was not stereoselective. Salazar and co-workers662,663 found that pigmented rabbit iris accumulated much more atropine than nonpigmented iris. On repeated washing, the atropine bound to nonpigmented tissues was removed more readily. Pigmented human iris tissue behaved in a manner similar to that of pigmented rabbit iris tissue. Pigment binding explains several clinical phenomena. There is a delay in onset and a decrease in magnitude of dilation in pigmented subjects. These result from an initial competition for atropine between pigment and receptors. There is a prolonged effect from atropine in pigmented subjects. It results from the melanin-bound drug acting as a reservoir. Emiru664 reported that normally pigmented black Africans responded differently to 4% homatropine plus 4% phenylephrine eye drops than did albino Africans. The latter responded in a manner similar to that of white subjects (i.e., onset of dilation and peak dilation occurred more rapidly).
Many rabbits, but not all, contain enzymes absent in humans that are capable of hydrolyzing and inactivating muscarinic antagonists. Atropinase (atropinesterase) is one of these and may protect rabbits from the toxic effects of belladonna ingested during leaf eating. Atropinase is also found in some strains of Pseudomonas.665 Its existence in mammalian species other than rabbits (e.g., rats and guinea pigs) is not well documented.666
Cauthen and colleagues667 found that rabbit atropinase hydrolyzed atropine and scopolamine but not cocaine. Although all three were esters and had similar structures, a different enzyme inactivated cocaine (Fig. 11). Cocainesterase did not hydrolyze atropine or scopolamine. The effects of both atropinase and pigment binding were studied by Salazar and co-workers.663 In rabbits with atropinase, the molecules bound to pigment were atropine and not its hydrolysis products, tropic acid and tropine. They found that the half-times for recovery of rabbits from atropine mydriasis were: albino rabbits with atropinase, 3.8 hours; albino rabbits without atropinase, 29.7 hours; pigmented rabbits with atropinase, 12.4 hours; and pigmented rabbits without atropinase, 96 hours or more.
|
Hemicholinium is a cholinergic blocking agent that exerts its effect at a site other than the receptor. It prevents uptake of choline by nerve endings. This results in decreased acetylcholine synthesis. It is highly toxic because it prevents acetylcholine synthesis by both nicotinic and muscarinic neurons. Its antinicotinic action results in paralysis of the respiratory muscles and death. Rabbits and cats can be killed from the systemic absorption of three drops of 1% hemicholinium.668 At concentrations below 0.5%, single drops have no effect. At higher concentrations, a single drop instilled unilaterally will produce pupillary dilation bilaterally and respiratory embarrassment within 40 minutes. If 0.001% to 0.0035% benzalkonium is added to 0.25% to 0.50% hemicholinium solutions, sufficient drug is absorbed through the cornea to produce dilation unilaterally without respiratory complications. The pupil does not respond to light or to stimulation of the ciliary nerve. These effects occur within 60 minutes and are usually gone within 6 hours. Hemicholinium is approximately 0.01 as potent a mydriatic agent as atropine (e.g., in mice, 0.1% hemicholinium gives as much dilation as 0.001% atropine sulfate). Topical 0.5% hemicholinium lowers the intraocular pressure in most, but not all, cats. There is an associated decrease in outflow facility. Therefore, the presumed mechanism of the hypotensive effect is a large decrease in aqueous humor production. Subconjunctival and intracameral injections of hemicholinium have produced corneal anesthesia in experimental animals.669,670 This may indicate a role for the corneal epithelial cholinergic system in touch-pain sensation.
OCULAR PHARMACOLOGY. Changes in pupillary diameter are more easily monitored than are changes in lens accommodation. As a result, animal studies of muscarinic antagonists have concentrated on the former. Gordon and Ehrenberg486 demonstrated that dilation from muscarinic antagonists is due to a parasympatholytic effect. The eyes of anesthetized albino rabbits, dilated with cyclopentolate or atropine, no longer responded to third-nerve stimulation but continued to dilate further on stimulation of cervical sympathetic neurons. Lund Karlsen,192 using quinuclidinyl benzilate, which is a muscarinic antagonist with 10 times higher affinity for the receptor than atropine,671,672 demonstrated that muscarinic receptors are found only in the sphincter region of the iris. Denervation did not increase their number. In one study, kittens and cats received daily topical atropine drops for 13 weeks.673 One year later there was a relative miosis and an eightfold increase in the number of muscarinic iris receptors in the kitten eyes, whereas adult cat eyes were unchanged from contralateral untreated eyes. Smith653 dilated the eyes of a volunteer with cyclopentolate before and after guanethidine drops. The pupil diameter after guanethidine paralysis of the dilator muscle was 7.05 mm, only slightly less than that before guanethidine (7.75 mm). This was consistent with the sphincter muscle playing the dominant role in determining the pupil size. Korczyn and Laor,674 after observing the pupillary responses of a patient with Waardenburg's syndrome given different pharmacologic agents (atropine, homatropine, guanethidine, and cocaine), suggested that atropine might have a partial direct α-agonistic action in addition to being antimuscarinic.
Both muscarinic agonists and antagonists, at concentrations that are submaximally effective, alter the pupillary light reflex in a similar manner: the speed and extent of the light response are diminished.383 This suggests that both types of drugs limit the number of muscarinic receptors on the sphincter muscle available for constriction to endogenously released acetylcholine. After a muscarinic antagonist is given, the pupils dilate with a speed proportional to the difference between the maximum pupil diameter and the actual pupil diameter.675
Accommodation has been considered a cause of myopia. Monkeys forced to do only near work develop myopia.676 Preventing accommodation with the use of atropine arrests the development of myopia in experimental animals.677 However, atropine may be working at other sites, either within the eye or as a result of systemic absorption. For example, pituitary secretion of growth hormone is inhibited by low blood levels of atropine. Subcutaneous atropine injections prevent chick eyes from developing myopia even though their ciliary muscles have nicotinic receptors and would not be expected to be affected by the drug.678 Furthermore, myopia can occur in animals whose ability to accommodate has been destroyed by lesions in the Edinger-Westphal nuclei679 and in animals who are normally unable to accommodate.680 Daily administration of the relatively selective M1 antagonist pirenzepine prevents chick myopia,681,682 and alternating daily between pirenzepine eye drops and pirenzepine subcutaneous injections prevents myopia in a mammal, the tree shrew.683 Because mammalian ciliary muscle contains primarily M3 receptors, this effect of pirenzepine implies a site of action other than on the muscle producing accommodation. However, the relatively large doses of pirenzepine applied could result in a loss of M1 receptor selectivity or in a toxic effect.
Monkeys treated unilaterally twice a day with 1% atropine eyedrops from age 10 days to 8 months developed an anisometropic amblyopia that remained present 4 months after discontinuation of the drug.684
The corneal epithelium contains acetylcholine. Its physiologic role is unknown. Umrath and Mussbichler685 and von Oer686 found that atropine drops caused a relative corneal anesthesia in rabbits. Atropine is structurally similar to the anesthetic cocaine. However, van Alphen,155 studying both rabbits and humans, could not confirm that atropine produced anesthesia.
THERAPEUTICS. Mydriasis. Mydriasis aids the visualization of those intraocular structures behind the iris. The amount of mydriasis produced by a drug can be altered by the light reflex and by intraocular inflammation. α-Adrenergic agonists, such as phenylephrine, produce mydriasis by stimulating the iris dilator muscle. However, much of this dilation is lost when the retina is stimulated by a bright light (e.g., that of an ophthalmoscope or slit lamp) because of sphincter muscle constriction. When muscarinic antagonists produce mydriasis, light-induced relaxation of the dilator muscle does not result in a significant decrease in pupillary diameter. Most clinical studies of mydriatic agents do not measure pupil size in response to bright lighting. This is an especial failing when the mydriasis from an α-adrenergic agonist is being compared with that from a muscarinic antagonist.
Mydriasis also plays a major role in preventing posterior synechiae to the anterior lens capsule. The anterior lens surface is convex (Fig. 12). As mydriasis increases, the area of posterior iris surface in contact with the lens capsule decreases. Cycloplegia is of additional value because it reduces both the convexity and the thickness of the lens. If posterior synechiae do develop while the iris is dilated, there is less chance of iris bombé (as the pupil becomes more dilated, more synechiae must form if aqueous humor flow is to be blocked). This relationship is expressed by πd, where d is the diameter of the pupil. There is some controversy as to whether a mobile iris is or is not superior to a dilated fixed iris when intraocular inflammation is present.687 The rationale of those who believe mobility is superior is that the base of the dilated iris is thicker and may be in contact with the trabeculum. Prolonged mydriasis may, therefore, lead to anterior synechiae. A mobile iris would effectively prevent both anterior and posterior synechiae.
|
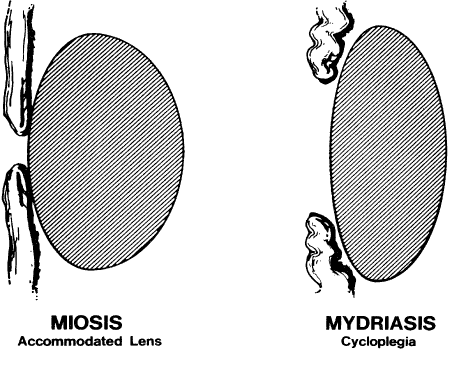
The results from investigations of mydriatic efficacy will be presented individually. Comparisons between studies are virtually impossible because different brands, concentrations, numbers of instillations, and drop intervals are used. In addition, patient populations are not uniform in age, race, and iris pigmentation. If one generalization does emerge, it is that all the commercially available muscarinic blocking agents produce adequate mydriasis if given either frequently enough or in high enough concentration. Eucatropine holds a unique position. Its muscarinic blocking activity is sufficient to produce mydriasis but too weak to produce significant cycloplegia. It would be an ideal agent when mydriasis alone is desired were it not for its prolonged latency, especially in darker irides. In these, it is not uncommon for maximum mydriasis to require several drops and well over 1 hour.
The efficacy of 1% atropine drops three times a day for 3 days was demonstrated by Marron688 in subjects 15 to 40 years of age. He noted that 5 minutes after the first drop there was a mean decrease in pupil diameter of 0.6 mm. By 40 minutes, maximum mydriasis had been achieved. In 16 of 107 subjects, maximum mydriasis was no longer present by the end of the second day or the beginning of the third day. However, compliance was not controlled, and these subjects may not have taken all their drops. In general, 4 to 6 days after the last drop, the pupils began to react to light, but 12 days were required for full recovery. In 21 subjects, with a mean age of 23 years, a single drop of 0.5% scopolamine also produced an initial miosis. By 20 minutes, maximum mydriasis had occurred. This lasted at least 90 minutes. Full recovery occurred by the eighth day. Early miosis also occurred when pupils were dilated with 5% homatropine, followed in 5 minutes by 1% hydroxyamphetamine, followed in 5 minutes by a second drop of homatropine. Maximum mydriasis occurred by 30 minutes and remained for 1 hour. Pupillary diameters were normal in 48 hours.
Wolf and Hodge689 compared single drops of 1% atropine, 1% methylatropine, and 1% homatropine in subjects 16 to 37 years of age. The respective times until maximum mydriasis were 40, 50, and 40 minutes. However, the amounts of maximum mydriasis differed, the respective ratios being 1.4:1.3:1. The onset of recovery for all three drugs was 6 hours.
Milder and Riffenburgh690 compared 0.5% cyclopentolate (two drops, each given 10 minutes apart to one eye) with 5% homatropine (two drops, each given 10 minutes apart to the other eye). The eye receiving homatropine also received a single drop of 1% hydroxyamphetamine. Cyclopentolate produced dilation sooner, but the mean maximum mydriasis achieved with the homatropine-hydroxyamphetamine combination was greater: 6.7 mm for the former versus 7.4 mm for the latter. Recovery also occurred sooner with cyclopentolate, often beginning by 3 hours and usually not complete until the next day. The mydriatic effect was less in black subjects in both magnitude and duration.
Gordon and Ehrenberg486 gave two drops of 0.5% cyclopentolate followed by a third drop 5 minutes later. Maximum mydriasis occurred at approximately 30 minutes, and the mean final pupillary diameter (7 mm) was greater than that achieved by 2% homatropine (6.1 mm) given in a similar manner. Recovery occurred within 20 hours of cyclopentolate administration but required up to 48 hours with homatropine administration.
Two drops of 1% cyclopentolate were administered to 48 children whose mean age was 7 years.691 Instilling the drops one minute apart produced as much mydriasis as instilling them 5 minutes apart.
Five volunteers were given (at different times) one drop of each of four different muscarinic antagonists in pH 6.5 physiologic saline: 5% eucatropine, 0.5% tropicamide, 0.5% homatropine, and 0.5% cyclopentolate.692 At 20 minutes postdrop, the mean mydriatic effects, compared with baseline, were: eucatropine (1.19 mm) < homatropine (1.99 mm) < tropicamide (3.72 mm) < cyclopentolate (3.96 mm). The maximum mydriasis was in the same order: 2.89 mm, 3.80 mm, 4.02 mm, and 4.42 mm. Percent recovery at 8 hours was: tropicamide (95%) > eucatropine (72%) > homatropine (30%) > cyclopentolate (12%). At 24 hours, recoveries from eucatropine and tropicamide were essentially complete, but not from homatropine (79%) or cyclopentolate (44%).
Merrill and colleagues693 found that 0.5% or 1% tropicamide almost always produced maximum mydriasis within 30 minutes, whereas 1% cyclopentolate, 5% homatropine, or 10% phenylephrine almost always required 60 to 90 minutes. The magnitude of dilation from tropicamide at 30 minutes was greater than that from the other drugs at 60 minutes. Tropicamide 1% was more effective than 0.5% tropicamide. Mydriasis disappeared 6 hours after 1% tropicamide in a group of 17 white subjects, aged 8 to 20 years; however, mydriasis from 1% cyclopentolate was still present in 82% of these subjects.
Gettes485 found single drops of 4% cocaine and 0.05% tropicamide about equal in mydriatic potency. So, too, were 1% hydroxyamphetamine and 0.1% tropicamide when compared 30 minutes postinstillation. Indirect ophthalmoscopy could be performed after a single drop of 0.5% tropicamide or after a combination of 0.2% tropicamide and 2% phenylephrine.
Gambill and associates694 compared the mydriatic effects of 0.5% tropicamide and 2% homatropine in 15 subjects using pupillography (Table 2). The maximum amount of mydriasis from tropicamide was approximately 5% greater than that from homatropine in eyes with light irides, and approximately 14% greater in eyes with dark irides.
TABLE 2. Comparisons of Mydriatic Effects of Tropicamide 0.5% and Homatropine 2%*
| Light Irides | Dark Irides | |||
| Tropicamide | Homatropine | Tropicamide | Homatropine | |
| Latency | 7.1 | 14.3 | 6.4 | 14.2 |
| Maximum effect | 36.8 | 68.0 | 42.4 | 78.4 |
| Half recovery | 151.8 | 420.0 | 186.6 | 425.4 |
| 90% recovery | 451.8 | 1192.8 | 489.0 | 1190.4 |
*Average time in minutes.
(Gambill HD, Ogle KN, Kearns TP: Mydriatic effect of four drugs determined with pupillograph. Arch Ophthalmol 77:740, 1967)
A single eyedrop containing tropicamide and phenylephrine in various combinations (i.e., 0.5% to 1% tropicamide and 2.5% to 10% phenylephrine) produces adequate but not maximum pupil dilation.695–697
Carpel and Kalina698 dilated the eyes of 25 premature infants with birth weights of 800 to 2900 g. Three applications of 1% cyclopentolate, one drop in each eye every 5 minutes, were given, and the pupils were measured 30 minutes after the last drop. Then 10% phenylephrine was given to the right eye only and the pupils were remeasured 30 minutes after the last drop. The mean pupillary diameter after cyclopentolate alone was less than 70% of the corneal diameter at both readings. In the eyes receiving cyclopentolate and phenylephrine, the mean pupillary diameter increased to approximately 90% of corneal diameter.
Alzheimer's dementia is associated with degeneration of central nervous system cholinergic neurons. It is unknown if this is a primary or secondary finding. Because of this association, iris supersensitivity to tropicamide has been suggested as a diagnostic test. Pupil supersensitivity to a drop of 0.01% tropicamide was reported to differentiate non-Alzheimer patients from those who had the disease or were suspect.699 However, multiple studies have failed to confirm this finding.700–702
Cycloplegia. Ciliary muscle paralysis prevents the eye from increasing the convex power of the lens. This occurs because the curvature of the intraocular lens cannot be altered. It is not that a change in the corneal curvature is prevented. This was shown by Daily and Coe,703 who performed keratometry readings before and after cycloplegia. The two were identical. Poinoosawmy and co-workers518 reported that stimulation of accommodation with pilocarpine did alter corneal curvature.
With the use of ultrasound, ciliary body thickness was found to decrease by 0.06 mm 2 hours after volunteers received a drop of 1% cyclopentolate.393 This was believed to be due to relaxation of the ciliary muscle.
Cycloplegia is of clinical importance because it allows a baseline evaluation of the refractive status of the eye. In subjects capable of accommodation, a dynamic focusing system is present. As the young child looks from physician, to retinoscope light, to mother, to distant fixation target, the diopteric power of the intraocular lens changes. Accurate refraction becomes difficult or impossible. This is not acceptable in children with accommodative esotropia or anisometropic amblyopia. In orthotropic children with normally developing vision, it might be argued that cycloplegia is unnecessary because small errors in the measurements are unimportant. However, this defense assumes that the refractionist knows his or her errors are insignificant without having measured them. On the other hand, in younger subjects, cycloplegia will occasionally induce errors in the amounts and axes of astigmatism. These may result from the taut zonules tilting the lens in a nonphysiologic manner. The art of refraction lies in blending both cycloplegic and noncycloplegic findings.
The assessment of the relative cycloplegic activity of different drugs suffers from the same limitations mentioned for mydriatic activity. Different concentrations and commercial preparations are used in populations that differ in race, age, and iris pigmentation. Furthermore, the techniques for determining residual accommodation differ from paper to paper. Some measure the near point of accommodation with print or a Prince rule; others use lens techniques that incorporate near-point determinations; and others use retinoscopic findings. If blur-point techniques are used to measure residual accommodation, then depth of focus is being evaluated as well.
Atropine (1%) drops, three times a day for 3 days, in 107 subjects ranging in age from 15 to 40 years produced maximum cycloplegia 23 to 48 hours after the first drop. Accommodation returned rapidly enough that most subjects could read newsprint 3 days after the last drop. Giving drops five times a day did not increase cycloplegia.688 A single drop of 0.5% scopolamine in 21 subjects with a mean age of 23 years produced maximum cycloplegia in 40 minutes; this persisted for at least 90 minutes. By the third day, most could read. Normal accommodation returned within 10 days. Homatropine (5%), two applications given 5 minutes apart to 25 subjects with a mean age of 28 years, resulted in maximum cycloplegia 50 minutes after the first drop. This was maintained for 30 minutes. By 6 hours, subjects could read, and full return of accommodation occurred within 48 hours.
Atropine (1%), as an ointment, was given twice a day for 4 days to 648 1-year-olds.489 The cycloplegia induced was compared with that from a single drop of 1% cyclopentolate given to each of 355 1-year-olds 30 minutes before retinoscopy. A third group of 415 1-year-olds received two instillations of 1% cyclopentolate, 10 minutes apart, with retinoscopy performed 30 minutes after the last drop. There were no significant differences in the results obtained from one or two drops of cyclopentolate. However, there was significantly more (p < 0.01) cycloplegia, averaging approximately 0.4 D per subject, in the group receiving atropine. Of esotropic children under age 5.5 years, 22% had 1 D or more of hyperopia uncovered by atropine rather than cyclopentolate.704
Single drops of 1% atropine, methylatropine, or homatropine were given to subjects 16 to 37 years of age. The times until maximum cycloplegia were 5 hours, 5 hours, and 25 minutes, respectively.689 However, the amounts of maximum cycloplegia were not the same for all three drugs. Residual accommodation was least after atropine and most after homatropine. The duration of maximum cycloplegia was 1 day for atropine, 6 hours for methylatropine, and 1 hour for homatropine. The times until complete cycloplegia recovery from atropine (9.2 ± 3.6 days) and from methylatropine (6.8 ± 3.8 days) were not significantly different.
Twelve black and 19 white subjects, whose ages ranged from 8 to 48 years, were given 0.5% cyclopentolate to one eye and 5% homatropine to the other. Two instillations of each drug, 10 minutes apart, produced no significant differences in residual accommodation. Cyclopentolate had a more rapid onset but shorter duration: 40% of cyclopentolate-treated eyes achieved maximum cycloplegia in less than 15 minutes; 22% of cyclopentolate-treated eyes required 45 minutes or more. By 24 hours, the cyclopentolate effect, but not that of homatropine, was gone. In 30% of eyes receiving cyclopentolate, maximum cycloplegia lasted less than or equal to 30 minutes. It was concluded that the optimum time for refraction was 45 minutes or less after 0.5% cyclopentolate was given in this manner.690 Blacks tended to have less cycloplegia from both drugs. In subjects initially given two drops of 0.5% cyclopentolate to the eye, followed by a third drop 5 minutes later, similar results were obtained.486 Maximum cycloplegia occurred 30 to 60 minutes after the drops. The residual accommodation 60 minutes after the cyclopentolate drops was less than that from 4% homatropine, 1.1 D versus 2.0 D. In both studies, cyclopentolate recovery occurred within 24 hours, whereas that from homatropine lagged.
Single drops of 1% cyclopentolate, 1% tropicamide, and 0.5% tropicamide, were compared with use of a Prince rule.693 Tropicamide (0.5%) produced much less cycloplegia. In subjects under 10 years of age, the mean residual accommodation 30 minutes after a 0.5%, or 1% tropicamide drop was, respectively, 4.22 or 0.35 D. The maximum cycloplegic effect from 1% tropicamide was greater than that from 1% cyclopentolate or 5% homatropine but of much shorter duration (Table 3). Six hours after instillation of 1% tropicamide, 90% of accommodation had returned, whereas 6 hours after instillation of cyclopentolate, 42% of accommodation had returned.
TABLE 3. Residual Accommodation (Prince Rule) in Subjects 4 to 9 Years
of Age After a Single Drop
| Diopters of Residual Accommodation Minutes After Drop | |||||||
| Cycloplegic | Race | No. Subjects | 15 | 30 | 60 | 90 | 120 |
| 1% Tropicamide | White | 22 | 4.2 | 0.4 | 3.7 | 6.4 | 5.6 |
| Black | 14 | 4.4 | 2.6 | 5.3 | 8 | 8.2 | |
| 1% Cyclopentolate | White | 8 | 4.8 | 4 | 4.1 | 4.7 | 4.8 |
| Black | 8 | 4.8 | 4.2 | 3.3 | 3.1 | 5 | |
| 5% Homatropine | White | 5 | 7.6 | 6.3 | 5.9 | 5.5 | 5.4 |
| Black | 5 | 8 | 6.4 | 5.9 | 5.6 | 5.5 | |
However, a study by Gettes490 found that the cycloplegia produced by a single instillation of 1% tropicamide was inadequate. In 53% of the eyes of subjects whose ages ranged from 14 to 35 years, more than 2.5 D of residual accommodation remained 20 to 25 minutes after drug application. In the 20- to 35-minute period postinstillation, 38% of eyes had more than 2.5 D residual accommodation. If a second drop of 1% tropicamide was given 5 to 25 minutes after the first, 100% of these subjects had less than 2.5 D of residual accommodation 20 to 25 minutes after the second drop. If refractions were performed up to 35 minutes after the second drop, 10% of eyes had more than 2.5 D of residual accommodation. The effect of 1% tropicamide, 1% cyclopentolate, or 4% homatropine-1% hydroxyamphetamine given to one eye was compared with the cycloplegic effect of one of the other two solutions placed in the contralateral eye. Two instillations of each medication were given, 5 minutes apart.705 Refractions were performed 20 to 40 minutes after the second drop. Residual accommodation was less than 2.5 D in 79% of the eyes of white subjects and 69% of the eyes of black subjects given tropicamide, 83% of the eyes of white subjects given cyclopentolate, and 51% of the eyes of white subjects and 36% of the eyes of black subjects given homatropine. By 40 to 60 minutes after the second drop, residual accommodation was more than 2.5 D in over half of the eyes receiving tropicamide. However, 91% of eyes receiving cyclopentolate and 59% of eyes receiving homatropine still had less than 2.5 D of residual accommodation.
Milder706 performed similar studies in a masked manner. Two instillations of one drop of 1% tropicamide, 1% cyclopentolate, or 5% homatropine were given to each eye of the subjects. Tropicamide drops were given 5 minutes apart and the others were given 10 minutes apart. Residual accommodations were measured at 30 and 60 minutes. The values 30 minutes after the tropicamide were compared with those 60 minutes after cyclopentolate or homatropine. In 80% of subjects, homatropine was superior to tropicamide, and in 92% of subjects, cyclopentolate was superior to tropicamide. The magnitude of residual accommodation was inversely related to age, and it was concluded that tropicamide should not be used for cycloplegia in patients under 40 years of age.
Miranda707 preceded 1% cyclopentolate drops to each eye with bilateral proparacaine anesthetic drops. In one eye, a second drop, 1% tropicamide, was given 25 seconds after the cyclopentolate. Refractions were performed 30 to 40 minutes later. Residual accommodation was measured by holding a reading card at 16 inches and gradually overminusing a + 2.50 lens. In a test group of 185 subjects ranging in age from 7 to 36 years, the cyclopentolate-tropicamide combination was superior in heavily pigmented eyes. In blue, green, light brown, and medium brown irides, the residual accommodation of the two eyes ranged from 0.25 to 1.00 D. However, the residual accommodation in eyes with dark brown irides receiving cyclopentolate alone was 1.25 to 5.50 D, whereas that in eyes receiving cyclopentolate-tropicamide was 0.50 to 1.25 D. Age was not as important a variable as was iris pigmentation. Equivalent cycloplegia was achieved in a group of children with a mean age of 7 years by instilling two drops of cyclopentolate 1 minute apart or 5 minutes apart.691
Five volunteers were given, at different times, one drop of the following made up in pH 6.5 physiologic saline: 5% eucatropine, 0.5% homatropine, 0.5% tropicamide, and 0.5% cyclopentolate.692 The mean maximum reduction in accommodation was eucatropine (2.85 D) < homatropine (6.17 D) < tropicamide (7.14 D) < cyclopentolate (8.64 D). The percent recoveries at 8 and 24 hours, respectively, were: eucatropine, 89% and 100%; tropicamide, 99% and 98%; homatropine, 39% and 89%; and cyclopentolate, 46% and 66%.
Analgesia. The source of pain during intraocular inflammation is unknown. The cornea is innervated by sensory fibers of the trigeminal nerve that pass near or through the uvea. In primates, trigeminal innervation is also present in the anterior stroma of the ciliary body and, to a lesser degree, in the iris and trabecular meshwork.708,709 Cycloplegic agents relieve the discomfort associated with intraocular inflammation. The degree of relief is variable but often dramatic.
Malignant Glaucoma. Slack zonules may be a cause of malignant glaucoma. The lens comes forward and flattens the anterior chamber. This mechanism is resistant to peripheral iridectomy and muscarinic agonists. The latter may initiate or aggravate the condition. Chandler and Grant534 reported seven patients in whom malignant glaucoma developed after a peripheral iridectomy for narrow-angle glaucoma. Muscarinic antagonists, such as atropine, homatropine, and scopolamine, were of value in breaking these attacks. Presumably they acted by paralyzing the ciliary muscle, pulling the zonules taut, and drawing the lens posteriorly. Frezzotti and Gentili710 found that long-term muscarinic antagonists also prevented recurrences of malignant glaucoma. However, Chandler and colleagues711 reported a more complex situation. Not all patients with malignant glaucoma responded to muscarinic antagonists. Those who did were not always controlled during long-term therapy. Although malignant glaucoma usually returned when muscarinic blocking agents were discontinued, one subject could be maintained on acetazolamide alone. Overall, their short-term success rate in 17 subjects was 60% and their long-term success rate was just under 50%.
Provocative Testing. Muscarinic antagonists can elevate intraocular pressure. Sometimes this is an undesired side effect. At other times, it is of diagnostic value.
CLOSED-ANGLE GLAUCOMA. Muscarinic antagonists have been used to predict which eyes with narrow angles are likely to develop acute closed-angle glaucoma. The rationale for this provocative action is that mydriasis thickens the iris base and occludes the angle. However, the results of testing are erratic. For example, Sugar712 instilled 4% homatropine into the eyes of four subjects who had had prior attacks of closed-angle glaucoma but who had not received iridectomies. Two did not have an increase in ocular pressure. The other two had marked elevations of more than 15 mmHg. In four narrow-angle glaucoma patients whose eyes were dilated with 10% phenylephrine, an adrenergic agonist, two had no increases in ocular pressure, one had an increase of 32 mmHg 30 minutes after instillation, and one had a latency of more than 5 hours before the pressure rose by 29 mmHg.
There appears to be little relation between the intrinsic muscarinic blocking potency of a drug and its ability to elicit a positive provocative test. Twenty subjects with prior histories of closed-angle glaucoma were tested. In 13 subjects, the drops were placed in the contralateral eyes (i.e., the eyes that had no histories of angle-closure attacks). In 7 subjects, the drops were placed in the diseased eyes.713 Eucatropine (2%) did not elevate the ocular pressure of five eyes but did elevate the ocular pressure of five other eyes by a mean of 21 mmHg. Three of the five unresponsive eyes remained normotensive after 2% homatropine. The two eyes that did respond had ocular pressure increases of 36 and 37 mmHg. Eucatropine elevated the intraocular pressure in one eye that did not respond to homatropine. One of the five eyes unresponsive to eucatropine developed a marked elevation, of 42 mmHg, after 1% hydroxyamphetamine, an adrenergic agonist.
A decrease in tonographic outflow facility may occur more often than an increase in intraocular pressure.714 Tonography was performed before and 1 hour after 5% eucatropine eyedrops were placed in 58 normotensive eyes with narrow angles. Twenty-eight percent had ocular pressure elevations of at least 8 mmHg, and 67% had outflow facility decreases of at least 25%. Of these 58 subjects, 55% eventually had a closed-angle attack. In retrospect, 44% had had a positive pressure provocative test and 88% had had a positive tonography provocative test.
CILIARY BLOCK GLAUCOMA. Ciliary block glaucoma occurs when the ciliary processes or ciliary muscle adhere to the back of the iris. This can occur spontaneously or after intraocular surgery (especially glaucoma filtering procedures). Muscarinic antagonists, such as atropine, can be of therapeutic value by paralyzing and flattening the ciliary muscle, thereby pulling away the adhering tissues.715 The effectiveness of muscarinic antagonists is variable; chronic use may be necessary.
OPEN-ANGLE GLAUCOMA. Harris716 observed that 2% of the “normal” open-angle population had a pressure elevation of 6 mmHg or more after topical applications of 1% cyclopentolate. Homatropine (5%) and 1% atropine were as effective as 1% cyclopentolate in provoking this response, but 0.5% tropicamide, 0.2% cyclopentolate, 5% eucatropine, and 10% phenylephrine were not. In approximately 70% of subjects responding to 1% cyclopentolate, 0.25% scopolamine was equally effective. Harris raised the question of whether this 2% of the population was or was not at risk of developing open-angle glaucoma. Subsequently, 58 subjects with a family history of open-angle glaucoma were studied.717 In none did 1% cyclopentolate elevate the intraocular pressure more than 5 mmHg. This seemed to indicate that muscarinic antagonists were of little predictive value. A different approach was then investigated. Dexamethasone (0.1%) was instilled three times a day for 3 weeks by these 58 subjects. In those with a steroid-induced pressure elevation of at least 6 mmHg, 41% had an additional elevation of at least 6 mmHg after topical application of 1% cyclopentolate. In those without a steroid-induced pressure elevation, only 6% had an elevation of this magnitude after cyclopentolate. A positive cyclopentolate test correlated significantly with a steroid-induced reduction in tonographic outflow facility but not with a steroid-induced elevation in intraocular pressure. The importance of these observations with respect to predicting future cases of open-angle glaucoma remains unclear.
A second use of provocative testing has been to anticipate which open-angle glaucoma patients given systemic muscarinic antagonists will experience increases in their intraocular pressures. Medications with muscarinic blocking activity are given for ulcer therapy and psychiatric conditions, for example. Lazenby and associates718,719 studied 24 subjects who had been off glaucoma therapy 3 or more days and 4 subjects who had been continued on medication. They found that topical 1% cyclopentolate caused a rise of 5 mmHg within 2 hours in half of these patients. Neither the cyclopentolate-responding group nor the cyclopentolate-nonresponding group had ocular pressure elevations during short-term oral atropine sulfate therapy. The atropine sulfate was given in two doses of 0.6 mg, 4 hours apart. A second group of 21 patients with open angles and elevated pressures was given oral atropine sulfate, 0.6 mg, three times a day for 7 days. Nine had responded to prior cyclopentolate eyedrops with an ocular pressure increase of 5 mmHg or more. In 4 of these 9, oral atropine was associated with an elevation in ocular pressure of 6 to 14 mmHg. In 1 of the 4, the pressure in one eye was elevated 9 mmHg and the pressure in the other eye was lowered 2 mmHg. In the remaining 5 cyclopentolate responders, the change in ocular pressure ranged from -2 to + 3 mmHg. A consistent relationship between the ocular pressure response and the tonographic outflow facility response was not observed (e.g., in 1 subject whose ocular pressures increased by 14 and 6 mmHg, the outflow facility values decreased from 0.08 μL/min/mmHg to 0.06 μL/min/mmHg in the former and increased from 0.08 μL/min/mmHg to 0.13 μL/min/mmHg in the latter). The 12 subjects who did not respond to cyclopentolate had little response to oral atropine. Their mean ocular pressures and outflow facilities before and after atropine were, respectively, 22.6 and 22.5 mmHg and 0.18 μL/min/mmHg and 0.19 μL/min/mmHg. These authors concluded that pretesting open-angle glaucoma patients with 1% cyclopentolate eyedrops often identified those at risk of developing further elevations in their ocular pressures from oral anticholinergic medications.
Myopia. It has been suggested that myopia results from accommodation (i.e., ciliary muscle contraction produces elongation of the eye). If so, then muscarinic antagonists, such as atropine, would prevent myopia by putting the ciliary muscle to rest. The evidence is, at best, suggestive. Dyer720 instilled 1% atropine once each day bilaterally in 86 children for 2 to 8 years. Nineteen percent had an increase in myopia of 1 D or more. In another group of 86 children given their full corrections, 84% had an increase of 1 D or more. Bedrossian721 reported that children using 1% atropine for 1 year in one eye had a mean decrease in myopia of 0.21 D in that eye compared with an increase of 0.82 D in the control eye. However, the comparison was made by refracting the control eye with tropicamide. The data may only represent a difference in the cycloplegic potency of atropine versus tropicamide. Sampson722 found that atropine drops seemed to prevent the progression of myopia, but on discontinuing the drops only 12% of subjects maintained their improvement for more than 6 months.
If muscarinic antagonists prevent myopia, then muscarinic agonists, by facilitating accommodation, might induce myopia. However, there are no reports of rapidly developing myopia in children with accommodative esotropia treated with DFP or echothiophate.
Perhaps atropine prevents myopia in a manner that is unrelated to the drug's effect on accommodation. Oral doses of 0.6 mg atropine abolish sleep-associated growth hormone secretion.723 Two 50-μL eyedrops of 1% atropine contain 1 mg atropine, an amount sufficient to have systemic effects.
Amblyopia. As an alternative to occlusive patching of the normal eye, disuse amblyopia can be treated with muscarinic antagonists. The blurring produced by cycloplegia penalizes the eye with normal acuity and encourages use of the amblyopic eye. This form of therapy will succeed only if the reduction in acuity of the normal eye exceeds that of the amblyopic eye. In cases of latent nystagmus, pharmacologic penalization may be the preferred treatment. In patients with eccentric fixation, penalization has been said to be contraindicated.724 However, others have reported dramatic success with this condition.725 Johnson and Antuna726 preferred using atropine in the ointment form because they believed any additional blurring from this viscous material was beneficial.
Single daily instillations of 1% atropine may be inadequate, and multiple drops may have to be given.727 Von Noorden and Milam725 used only one drop, given each morning, and reported good results. Successful therapy, defined as an improvement of two lines or more in visual acuity, occurred in 10 of 17 amblyopic eyes. Pharmacologic penalization not only improved vision but was also used to maintain vision. In 10 of 13 patients whose vision deteriorated when patching was discontinued, vision was maintained within one line of the best acuity by the use of atropine.
Penalization during that period of visual development can result in the initially normal seeing eye developing amblyopia.728 This would occur after the initially amblyopic eye had improved to the point that its acuity surpassed that of the contralateral atropine-blurred eye. Ikeda and Tremain729 studied this phenomenon in experimental animals. The eyes of kittens were chronically blurred with atropine drops, unilaterally or bilaterally. When these cats reached maturity, single-cell recordings were made from their lateral geniculate bodies and visual cortices. Lateral geniculate body recordings were abnormal in unilaterally and bilaterally treated cats. Visual cortex recordings were abnormal only in unilaterally treated cats.
OCULAR HYPERTENSION FROM TOPICAL THERAPY. Muscarinic antagonists given topically or systemically can elevate intraocular pressure. This occurs in normotensive subjects and glaucoma patients whether the angles are open or narrow. The mechanisms involved are varied and not completely understood.
Pigment Release. Mobility of the iris is associated with release of uveal pigment and a transient block of the trabecular meshwork. Mitsui and Takagi730 removed aqueous humor from subjects diagnosed clinically as releasing pigment; the material was identified as being compatible with uveal melanin. They challenged subjects with 5% phenylephrine, 1% atropine, 1% homatropine, 0.5% physostigmine, and 1% pilocarpine. Phenylephrine was by far the most effective in releasing pigment. The other drugs were about equal in effectiveness, with the exception of pilocarpine, which elicited pigment release in only 1 of 16 subjects. Calhoun731 dilated the eyes of subjects having Krukenberg spindles with 2% homatropine plus 10% phenylephrine. The presence or absence of an increase in intraocular pressure correlated with the amount of pigment released into the aqueous humor. Valle732 studied 21 eyes that had or were suspected of having untreated open-angle glaucoma and that responded to a drop of 1% cyclopentolate with intraocular pressure elevations of 8 to 20 mmHg. These pressure responses were maximum 1 to 4 hours after instillation of cyclopentolate. By 6 hours, they approached normal values. In one third of the eyes, pigment was liberated into the anterior chamber. The degree of pressure elevation correlated with the amount of pigment released.
Pseudoexfoliation. A 1% cyclopentolate provocative test was performed on Scandinavian patients suspected of having open-angle glaucoma.733 An increase of 8 mmHg in intraocular pressure was considered positive. Of the 8% of patients having a positive test, half had pseudoexfoliation of the lens capsule.
Open-Angle Glaucoma. The response of eyes with open-angle glaucoma to muscarinic antagonists is not entirely predictable. Some eyes develop elevations of intraocular pressure and some do not. Of 15 eyes with open-angle glaucoma that were dilated with two drops of 5% homatropine given 10 minutes apart, 8 had intraocular pressure elevations of 6 to 13 mmHg, 4 had elevations of less than 6 mmHg, 2 did not change, and 1 had a decrease in ocular pressure of 3 mmHg.734 In a comparison of 5% homatropine, 2% hydroxyamphetamine, and a solution containing both, the mean ocular pressure rise from the combination was the largest. It occurred 3 hours after instillation and averaged 5.6 mmHg. The maximum homatropine and hydroxyamphetamine responses were at 2 hours and averaged 4.5 and 3 mmHg, respectively. Sugar712 instilled 4% homatropine in one eye and 10% phenylephrine in the other eye of 51 open-angle glaucoma subjects who had not taken their medications that day. The changes in intraocular pressures were determined 30 and 60 minutes later. The range of responses to homatropine was -4 to + 8 mmHg and the range of responses to phenylephrine was -8 to + 8 mmHg. Galin735 dilated bilaterally the eyes of three subjects with open-angle glaucoma off medication and six normotensive subjects with open angles; he used 1% cyclopentolate, every 15 minutes for four doses. In seven of the subjects, an increase in intraocular pressure of 8 mmHg or more occurred, but only unilaterally.
Harris and Galin736 studied 69 open-angle glaucoma patients receiving either bilateral pilocarpine or bilateral echothiophate therapy. Each subject was challenged with three drops of 1% cyclopentolate unilaterally. The drops were given 15 minutes apart. The other eye served as control. Forty-one percent of 53 pilocarpine-treated eyes and 50% of 16 echothiophate-treated eyes responded to cyclopentolate with pressure elevations of 6 mmHg or more. It was not unexpected that muscarinic antagonists would reverse the hypotensive effects of muscarinic agonists, but it was surprising that the reversal did not occur in all eyes. Thirty-eight of these patients were then taken off muscarinic therapy for 2 weeks and rechallenged. Twenty-four percent had an increase in intraocular pressure of 6 mmHg or more. This incidence was lower than that while on therapy, probably because the pressures were already near their peak before they were challenged. Combining these results with those of prior studies, the authors stated that the overall incidence of a 6-mmHg or greater ocular pressure elevation after challenge by cyclopentolate drops was 33% for open-angle glaucoma subjects on muscarinic agonist therapy.
Valle737 compared the cyclopentolate response of glaucoma suspects with intraocular pressures of 22 to 24 mmHg with that of untreated open-angle glaucoma patients. Two drops of cyclopentolate were given, each 15 minutes apart, and the ocular pressures were followed for a minimum of 3 hours. In the suspect group, the mean increase in intraocular pressure was 0.4 ± 2.5 mmHg. This was significantly less than that of the glaucoma group (2.5 ± 3.1 mmHg). Only 2% of the suspect group and 9% of the glaucoma group had elevations in pressure of 8 mmHg or more. There was no correlation between the initial ocular pressure and the magnitude of the pressure response.
The eyes of patients with primary open-angle glaucoma were dilated with 1% tropicamide plus phenylephrine.738 The mean ± SD intraocular pressure before dilation was 22.9 ± 6.1 mmHg. One hour after the drops, the mean intraocular pressure was 25.8 ± 7.7 mmHg. Individual changes in ocular pressure ranged from -6 mmHg to + 22 mmHg. An intraocular pressure increase occurred in 60% of subjects and a decrease in 30%. Increases of 10 mmHg or more occurred in 12% of subjects, and increases of 20 mmHg or more occurred in 2%. The only significant risk factor for an elevation was the therapeutic use of muscarinic agonists: 41% of these subjects had an elevation of 5 mmHg or more and 19% had an elevation of 10 mmHg or more. There was no significant correlation between the intraocular pressure rise after dilation and the intraocular pressure rise after laser trabeculoplasty.
The tonographic outflow facility responses to muscarinic antagonists may be more consistent than the ocular pressure responses. Galin735 gave multiple drops of cyclopentolate to 18 eyes. Nine developed elevations in intraocular pressure of 8 mmHg or more and 11 developed decreases in outflow facility of 25% or more. Schimek and Lieberman739 found that all 17 eyes of open-angle glaucoma subjects receiving cyclopentolate had decreases in outflow facility, whereas only 15 eyes had increases in intraocular pressure. Christensen and Pearce740 gave two drops of 5% homatropine unilaterally to 35 chronic simple glaucoma patients. The mean increase in ocular pressure was 3 mmHg, with a range of -2 to + 19 mmHg. The mean decrease in tonographic outflow facility was 28%.
Normotensive Subjects With Open Angles. One eye of each of 70 normotensive subjects, ranging in age from 22 to 80 years, was dilated with 5% homatropine. The ocular pressure change 45 minutes later was similar to that of the control eye, ranging from -6 mmHg to + 3.5 mmHg.740 Tonographic outflow facilities were reduced in the challenged eyes 9% to 10% more than in the control eyes. However, Harris716 found a 2% incidence of a 6-mmHg or greater pressure elevation in 550 normotensive open-angle subjects after 1% cyclopentolate, one drop every 15 minutes for three doses. Homatropine (5%) and 1% atropine were as effective as cyclopentolate in elevating the ocular pressure.
Chronic Narrow-Angle Glaucoma. Lowe741 found that many subjects with chronic narrow-angle glaucoma and open peripheral iridectomies developed angle closure, increased intraocular pressure, and decreased outflow facility after their eyes were dilated with homatropine. Phenylephrine (10%) dilatation did not produce these changes. The explanation offered was that the pull of the sphincter muscle against the phenylephrine-stimulated dilator muscle produced a vector of force pulling the iris base away from the angle. This, presumably, did not occur after homatropine. Harris and Galin742 found similar results. Dilating 26 eyes successfully treated with peripheral iridectomies (i.e., none had residual chronic closed-angle glaucoma) did not produce elevations in ocular pressures. However, in seven of eight eyes with chronic narrow-angle glaucoma, 1% cyclopentolate, but not 10% phenylephrine, produced pressure elevations of 6 mmHg or more. They did not confirm Lowe's741 explanation because the filtration angles, as shown by gonioscopy, remained open in the seven eyes developing elevated intraocular pressures after homatropine.
Acute Closed-Angle Glaucoma. Only 1 of 250 primary care physicians routinely dilates pupils, even when examining diabetics.743 The stated excuse is the fear of provoking acute narrow-angle glaucoma.744 However, if patients with a history of glaucoma or shallow anterior chambers on penlight examination are excluded, there is less than a 0.3% chance of inadvertently dilating the eyes of a subject with potentially occludable filtration angles, as determined by gonioscopy.745
The mechanisms by which muscarinic antagonists produce acute closed-angle glaucoma were stated earlier. However, the incidence of pharmacologically induced angle closure in eyes without previous attacks is unknown. Therefore, a controversy exists. Some ophthalmologists will not perform a dilated intraocular examination on subjects with narrow angles for fear of provoking an acute attack of glaucoma. Other ophthalmologists routinely instill muscarinic antagonists because they believe that the incidence of acute angle closure is lower than the incidences of other types of ocular pathology (e.g., retinal tears, retinal detachments, and melanomas) that would otherwise escape detection. It can also be argued that pharmacologically provoking an attack of angle closure is in the patient's best interest. The ophthalmologist can make the correct diagnosis and provide adequate treatment. Spontaneous attacks that occur without medical supervision are more likely to lead to severe visual loss and intractable glaucoma.
OCULAR HYPERTENSION FROM SYSTEMIC THERAPY. Lund Karlsen192 gave guinea pigs intraperitoneal injections of atropine at a concentration that was lethal in humans (i.e., 0.225 mg/kg). This produced iris concentrations that were only 3% of those achieved by eyedrops containing 0.01 mg atropine. This demonstrates why systemic muscarinic antagonists do not commonly affect intraocular structures (i.e., relatively small amounts enter the eye). The ensuing discussion deals with the ocular effects of systemically administered muscarinic antagonists. Normotensive, open-angle glaucoma, and narrow-angle glaucoma subjects are considered separately. The responses are variable within all three groups. Most subjects demonstrate little or no effect. However, a minority do respond significantly. As a result, the ophthalmologist cannot dismiss a priori systemic anticholinergic medications as being of little consequence.
Normotensive, open-angle subjects were given intramuscular injections of 0.6 to 0.8 mg atropine or 0.4 to 0.6 mg scopolamine. Three of the eight receiving atropine developed 0.5 to 1.5 mm mydriasis, and seven of the eight receiving scopolamine developed 0.5 to 2 mm mydriasis.590 There was a mean increase of 8 mm in the near point of accommodation after atropine and a mean increase of 27 mm after scopolamine. When 100 normotensive subjects ranging in age from 16 to 40 years were given atropine 0.016 mg/kg, 13 developed pupillary dilations of 2 mm or less and 9 developed ocular pressure elevations of 3 mmHg or less.746 Similar results were obtained after intramuscular injection of scopolamine 0.01 mg/kg.747 Cozanitis and co-workers748 reported that atropine 0.01 mg/kg did not affect the pupillary diameter.
A single intramuscular injection of atropine sulfate, 2 mg, in 13 male normotensive subjects produced a mean maximum pupil dilation of 1.25 mm at 2 hours postinjection.749 By 5 hours postinjection this effect was gone. Bye and colleagues750 used pupillography to study the effects of oral atropine sulfate, 0.02 mg/kg. During the 3 hours after ingestion, the mean pupillary diameter increased significantly, going from 7.26 mm to 7.49 mm. The amplitude of the light reflex was significantly diminished. Other anticholinergic drugs that have been studied include oral propantheline751 and intramuscular glycopyrrolate.748 Propantheline, 15 mg every 6 hours, was given for 24 hours. There was a 16% incidence of an ocular pressure elevation of at least 4 mmHg and a 12% incidence of an ocular pressure depression of at least 4 mmHg. The incidences of pupillary dilation or constriction of at least 1 mm were similar. Glycopyrrolate (glycopyrronium), 0.04 mg/kg, did not affect either the pupillary diameters or the ocular pressures of the 10 subjects who were tested.
Eight open-angle glaucoma patients off therapy for 48 hours were given either 0.5 to 0.6 mg atropine or 0.4 to 1 mg scopolamine intramuscularly. Usually the intraocular pressures fell or were unchanged. In one eye there was a pressure increase of 3 mmHg after atropine, and in two eyes there were pressure increases of less than 2 mmHg after scopolamine.752 Thirty-four eyes with open-angle glaucoma were maintained on therapy, and atropine 0.01 mg/kg was injected intramuscularly. Pupillary diameters, ocular pressures, and accommodation were followed for 2 hours.753 The intraocular pressures of 22 eyes were unchanged or were reduced. In the remaining 12 eyes, the ocular pressures increased by 2 to 5 mmHg. Overall, neither the ocular pressure changes nor the pupil diameter changes were significant. Ten glaucoma subjects were given scopolamine 0.01 mg/kg. There were no significant changes in ocular pressure or pupil size.747 The ocular pressures of 94 eyes with untreated open-angle glaucoma were monitored while propantheline, 15 mg every 6 hours, was administered for 24 hours. In 21% of these eyes there was an increase in ocular pressure of 4 mmHg or more, and in 10% there was a decrease of 4 mmHg or more. In one eye, the ocular pressure elevation was 16 mmHg. Pupillary dilations were always less than 1 mm.751
Narrow-angle glaucoma patients were studied by Schwartz and associates.752 Six subjects, normotensive at the time of injection, were given 0.5 to 0.6 mg atropine or 0.4 to 1 mg scopolamine intramuscularly. Five of the six had unilateral iridectomies. The mean elevation in intraocular pressure was less than 0.5 mmHg. Six subjects with previous histories of closed-angle attacks received intramuscular injections of atropine 0.01 mg/kg.753 The ocular pressures remained unchanged or decreased. When seven narrow-angle glaucoma patients were given propantheline, 15 mg every 6 hours for 24 hours, 21% of the 14 eyes had increases in ocular pressure of 4 mmHg or more. In two eyes, the pressure elevations were 16 and 17 mmHg. In none of the 14 eyes did the pupils dilate more than 1 mm.751
SYSTEMIC TOXICITY FROM TOPICAL THERAPY. Toxicities have been reported after topical muscarinic antagonist therapy and from the ingestion of eyedrop solutions.754 All the commonly used blocking agents have been implicated.
Eyedrops produce measurable drug levels in the blood. A single 40-μL drop of 1% atropine given unilaterally to recumbent patients produced a mean ± SD peak plasma concentration of 860 ± 402 pg/mL within 8 minutes.755 A single 35-μL drop of cyclopentolate given unilaterally to supine children whose mean age was 9 ± 3 years produced a maximum plasma level of 2.2 ± 1.6 ng/mL at 5 minutes.756 Two 30-μL drops of 1% cyclopentolate produced a mean peak plasma concentration of approximately 3 ng/mL within 30 minutes; in some subjects a second peak occurred at 2 hours, presumably from gastrointestinal tract absorption.757 When the cyclopentolate drops were separated by 5 minutes, the peak concentration, 8.3 ± 0.5 ng/mL, was reached 10 ± 5 minutes after the second drop189; the second peak occurred at 45 to 60 minutes. The mean half-life in plasma of both atropine and cyclopentolate is just under 2 hours.758 Two 40-μL drops of 0.5% tropicamide instilled in one eye of women whose mean age was 70.4 ± 9.3 years produced a peak concentration of 2.8 ± 1.7 ng/mL in 5 minutes.759 By 5 hours, the plasma level was less than 0.25 ng/mL.
Death occurred in a 3-year-old white girl with a corneal perforation who was given six drops of 1% atropine topically, 0.1 mg atropine subcutaneously, and, at the conclusion of surgery, 1.5 g of 1% atropine ointment on the lids.760 The patient died within 24 hours of the first application of atropine. A 3-year-old white retarded girl developed a fever, convulsed, and died after receiving only four drops of 1% atropine; no postmortem examination was performed. Scopolamine,754 homatropine,761 and cyclopentolate762–765 have produced toxicity. The central nervous system manifestations include cerebellar signs, ataxia, dysarthria, and inappropriate behavior such as hallucinations, incoherent speech, and restlessness. Patients with prior histories of convulsions may develop seizures more readily.766 Dry, flushed skin and fever are often but not always present.761,767 Toxic signs may occur within a few minutes of administration.763 Recovery occurs within 24 hours. Two studies stand out as being especially well documented. In one, an 8-year-old black girl developed toxicity after two drops of 1% tropicamide and four drops of 1% cyclopentolate.767 The child was subsequently readmitted and given 10 drops of 1% tropicamide within 30 minutes. Toxicity did not develop. Five days later, she was challenged with six drops of 1% cyclopentolate within a 15-minute period. Toxicity, including hallucinations, returned. The other report was by Binkhorst and co-workers.768 It was a masked prospective study of drug toxicity. Thirty-five children had psychiatric evaluations before and after receiving four drops bilaterally of 1% cyclopentolate, 2% cyclopentolate, 1% homatropine, or 1% hydroxyamphetamine. Each of the four sets of drops was instilled 10 minutes apart. Only the 2% cyclopentolate solution evoked significant abnormal behavior.
Newborns are especially susceptible to systemic intoxication. Reports have associated increased abdominal distention and necrotizing enterocolitis with the use of muscarinic antagonist eyedrops for ophthalmic examination.765,769 Isenberg and colleagues770 found that 0.25% cyclopentolate had no significant effect on gastric function but that 0.5% cyclopentolate did. Interestingly, these workers reported that, as would be expected, 0.5% cyclopentolate reduced gastric volume output, whereas Hermansen and Sullivan769 reported that 0.5% cyclopentolate was associated with large gastric aspirates.
Systemic toxicity from muscarinic antagonists has been treated with direct and indirect muscarinic agonists. Pilocarpine, 6 mg subcutaneously, has caused death in a 3-year-old child.760 Forrer and Miller771 reversed the delirium and coma that followed the ingestion of 200 mg or more of atropine by injecting 1 to 4 mg physostigmine. The reversal was temporary, and after 30 to 60 minutes additional physostigmine had to be given. Duvoisin and Katz772 and Young and colleagues754 suggested using an initial subcutaneous dose of 0.25 mg physostigmine in children and 1 to 2 mg physostigmine in adults. This initial dose would be supplemented as needed every 15 minutes by doses of similar magnitude.