1. Kaufman HE, Nesburn AB, Maloney ED: IDU therapy of herpes simplex.
Arch Ophthalmol 67:483, 1962.
2. Kaufman HE, Martola E, Dohlman C: Use of 5-iodo-2-deoxyuridine (IDU)
in treatment of herpes simplex keratitis. Arch Ophthalmol 68:235, 1972.
3. Patterson A, Fox AD, Davies G et al: Controlled studies of IDU in the
treatment of herpetic keratitis. Trans Ophthalmol Soc UK 83:583, 1963.
4. Burns RP: A double-blind study of IDU in human herpes simplex keratitis. Arch Ophthalmol 70:381, 1963. 5. Laibson PR, Leopold IH: An evaluation of double-blind IDU therapy in 100 cases of herpetic
keratitis. Trans Am Acad Ophthalmol Otolaryngol 68:22, 1964. 6. Maxwell E: Treatment of herpes keratitis with 5-iodo-2-deoxyuridine (IDU): A
clinical evaluation of 1500 cases. Am J Ophthalmol 56:571, 1963. 6a. DeClerq E, Descamps J, Verhelst G et al: Comparative efficacy of antiherpes drugs against different strains of herpes
simplex virus. J Infect Dis 141:563, 1980. 7. Prusoff WH: Idoxuridine or how it all began. In DeClerq E (ed): Clinical Use of Antiviral Drugs, pp 15–24. Norwell, MA, Martinus Nijoff, 1988. 8. Prusoff WH, Chang PK: 5-Iodo-2'-deoxyuridine 5'-triphosphate, an
allosteric inhibitor of deoxycytidilate deaminase. J Biol Chem 243:223, 1968. 9. Boston Interhospital Viral Study Group: Failure of high-dose 5-iodo-2'-deoxyuridine in the therapy of herpes
simplex virus encephalitis: Evidence of unacceptable toxicity. N Engl J Med 292:599, 1975. 10. Kaufman HE, Maloney ED: Virus chemotherapy with antimetabolites. Pharmakotherapia 1:43, 1963. 11. Maloney ED, Kaufman HE: Antagonism and toxicity of IDU by its degradation products. Invest Ophthalmol 2:55, 1963. 12. Foster CS, Pavan-Langston D: Corneal wound healing and antiviral medication. Arch Ophthalmol 95:2062, 1977. 13. Pavan-Langston D, Langston RHS: Recent advances in antiviral therapy. Int Ophthalmol Clin 15:89, 1975. 14. Stern GA, Killingworth DW: Complications of topical antimicrobial agents. Int Ophthalmol Clin 29:137, 1989. 15. Wilson FM II: Adverse external ocular effects of topical ocular medications. Surv Ophthalmol 24:57, 1979. 16. Kaufman HE: Treatment of viral diseases of the cornea and external eye. Prog Retinal Eye Res 19:69, 2000. 17. Itoi M, Getter JW, Kaneko N et al: Teratogenicities of ophthalmic drugs: Antiviral ophthalmic drugs. Arch Ophthalmol 93:46, 1975. 18. Wellings PC, Awdry PN, Bors FH et al: Clinical evaluation of trifluorothymidine in the treatment of herpes simplex
corneal ulcers. Am J Ophthalmol 73:932, 1972 19. Pavan-Langston D, Foster CS: Trifluorothymidine and idoxuridine therapy of ocular herpes. Am J Ophthalmol 84:818, 1977. 20. Laibson PR, Arentsen JJ, Mazzanti WD et al: Double controlled comparison of IDU and trifluorothymidine in thirty-three
patients with superficial herpetic keratitis. Trans Am Ophthalmol Soc 75:316, 1977. 21. McGill J, Holt-Wilson AD, McKinnon JR et al: Some aspects of the clinical use of trifluorothymidine in the treatment
of herpetic ulceration of the cornea. Trans Ophthalmol Soc UK 94:342, 1974. 22. Pavan-Langston D, Dohlman CH: A double-blind clinical study of adenine arabinoside therapy of
viral keratoconjunctivitis. Am J Ophthalmol 74:81, 1972. 23. Dresner AJ, Seamans ML: Evidence of the safety and efficacy of adenine arabinoside in the treatment
of herpes simplex epithelial keratitis. In Pavan-Langston D, Buchanon RA, Alford CA Jr (eds): Adenine Arabinoside: An Antiviral Agent, pp 381–391. New York, Raven Press, 1975. 24. Pavan-Langston D, Buchanon RA: Vidarabine therapy of simple and IDU-complicated herpetic keratitis. Trans Am Acad Ophthalmol Otolaryngol 81:813, 1976. 24a. Wilhelmus KR: The treatment of herpes simplex virus epithelial keratitis. Tr Am Ophthalmol Soc 98:505, 2000. 25. Coleman VR, Tsu E, Jawetz E: Treatment resistance to idoxuridine in herpetic keratitis. Proc Soc Exp Biol Med 129:761, 1968. 26. Dubbs DR, Kit S: Mutant strains of herpes simplex deficient in thymidine kinase-inducing
activity. Virology 22:493, 1964. 27. Gottschling H, Heidelberger C: Fluorinated pyrimidines: XIX. Some biological effects of 5-trifluoromethyl-2'-deoxyuridine on Escherichia coli and bacteriophage T4B. J Mol Biol 7:541, 1963. 28. Prusoff WB, Zucker M, Mancini WR et al:. Basic biochemical and pharmacological aspects of antiviral agents.. Antiviral Res 5:1, 1985. 29. Kaufman HE, Heidelberger C: Therapeutic antiviral action of 5-trifluoromethyl-2'-deoxyuridine. Science 145:585, 1964. 30. Hyndiuk RA, Kaufman HE: Newer compounds in the therapy of herpes simplex keratitis. Arch Ophthalmol 78:600, 1967. 31. Collins P, Bauer D: Relative potencies of antiherpes compounds. Ann N Y Acad Sci 26:49, 1977. 32. Hyndiuk RA, Seidman S, Leibsohn JM: Treatment of vaccinial keratitis with trifluorothymidine. Arch Ophthalmol 94:1785, 1976. 33. Spector SA, Tyndall M, Kelley E: Inhibition of human cytomegalovirus by trifluorothymidine. Antimicrob Agents Chemother 23:113, 1983. 34. Lennette DA, Eiferman RA: Inhibition of adenovirus replication in vitro by trifluoridine. Arch Ophthalmol 96:1662, 1978. 35. Pavan-Langston D, Nelson DJ: Intraocular penetration of trifluridine. Am J Ophthalmol 87:814, 1979. 36. Obrien W, Edelhauser H: The corneal penetration of trifluorothymidine, adenine arabinoside, and
idoxuridine: A comparative study. Invest Ophthalmol Vis Sci 16:1093, 1977. 37. Poirer RH, Kingham JD, DeMiranda P et al: Intraocular antiviral penetration. Arch Ophthalmol 100:1964, 1982. 38. Carmine AA, Brogden RN, Heel RC et al: Trifluoridine: A review of its antiviral activity and therapeutic use in
the topical treatment of viral eye infections. Drugs 23:329, 1982. 39. Coster DJ, McKinnon JR, McGill JI et al: Clinical evaluation of adenine arabinoside and trifluorothymidine in the
treatment of corneal disease caused by herpes simplex virus. J Infect Dis 133:A173, 1976. 39a. Udell IJ: Trifluridine-associated conjunctival cicatrization. Am J Ophthalmol 99:363, 1985. 40. Shearer DR, Bourne WM: Severe ocular anterior segment ischemia after long-term trifluridine
treatment for presumed herpetic keratitis. Am J Ophthalmol 109:346, 1990. 40a. Maudgal PC, Van Damme B, Missotten L: Corneal epithelial dysplasia after trifluridine use. Graefes Arch Clin Exp Ophthalmol 220:6, 1983. 41. Pang MP, Peyman GA, Nikoleit J et al: Intravitreal trifluorothymidine and retinal toxicity. Retina 6:260, 1986. 42. Ansfield FJ, Ramirez G: Phase I and II studies of 2-deoxy-5-(trifluoromethyl)-uridine (NSC-75520). Cancer Chemother Rep 55:205, 1971. 43. Kury G, Crosby RJ: The teratogenic effect of 5-trifluoromethyl-2'-deoxyuridine
in chicken embryos. Toxicol Appl Pharmacol 11:72, 1967, 44. Jones BR, McGill JI, McKinnon JR et al: Preliminary experience with adenine arabinoside in comparison with idoxuridine
and trifluorothymidine in the management of herpetic keratitis. In Pavan-Langston D, Buchanon RA, Alford CA (eds): Adenine Arabinoside: An Antiviral Agent, pp 411–416. New York, Raven Press, 1975. 45. Sugar J, Stark W, Binder PS et al: Trifluorothymidine treatment of herpes simplex epithelial keratitis and
comparison with idoxuridine. Ann Ophthalmol 12:611, 1980. 46. Parlato CJ, Cohen EJ, Sakauye CM et al: Role of débridement and trifluridine (Trifluorothymidine) in
herpes simplex dendritic keratitis. Arch Ophthalmol 103:673, 1985. 47. Van Bijsterveld OP, Post H: Trifluorothymidine versus adenine arabinoside in the treatment of herpes
simplex keratitis. Br J Ophthalmol 64:33, 1980. 48. Travers JP, Patterson A: A controlled trial of adenine arabinoside and trifluorothymidine in herpetic
keratitis. J Int Med Res 6:102, 1978. 49. Coster DJ, Jones BR, McGill JI: Treatment of amoeboid herpetic ulcers with adenine arabinoside or trifluorothymidine. Br J Ophthalmol 63:418, 1979. 50. Hyndiuk RA, Charlin RE, Alpren TVP et al: Trifluoridine in resistant human herpetic keratitis. Arch Ophthalmol 96:1839, 1978. 51. Falcon MG, Jones BR, Williams HP et al: Management of herpetic eye disease. Trans Ophthalmol Soc UK 97:345, 1977. 52. Wilhelmus KR, Coster DJ, Donovan HC et al: Prognostic indicators of herpetic keratitis: Analysis of a five-year
observation period after corneal ulceration. Arch Ophthalmol 99:1578, 1981. 53. McNeil JI, Kaufman HE: Local antivirals in a herpes simplex stromal keratitis model. Arch Ophthalmol 97:727, 1979. 54. Wilhelmus KR, Gee L, Hauk WW et al: Herpetic Eye Disease Study. A controlled trial of topical corticosteroids
for herpes simplex stromal keratitis. Ophthalmology 101:1883, 1994. 55. Little JM, Lorenzetti DWC, Brown DC et al: Studies of adenovirus type III infection treated with methisazone and trifluorothymidine. Proc Soc Exp Biol Med 127:1028, 1968. 55a. Cono J, Casey CG, Bell DM: Smallpox vaccination and adverse reactions. Guidance for clinicians. MMWR Recomm Rep 52(RR-4):1, 2003. 56. Lee WW, Benitez A, Goodman L et al: Potential anticancer agents: XL. Synthesis of the B-isomer of 9-($PI$vD-arabinofuranosyl)-adenine. J Am Chem Soc 82:2648, 1960. 57. Fiala M, Chow AW, Miyasaki K et al: Susceptibility of herpes viruses to three nucleoside analogues and their
combinations and enhancement of the antiviral effect of acid pH. J Infect Dis 129:82, 1974. 58. Miller FA, Dixon GJ, Ehrlich J et al: Antiviral activity of 9-B-d arabinofuranashyladenine: Cell
culture studies. In Hobby GI (ed): Antimicrobial Agents and Chemotherapy—1968, pp 136–147. Bethesda, American Society for Microbiology, 1969. 59. Nicholson KG: Properties of antiviral agents: First of two parts. Lancet 2:503, 1984. 60. Whitley R, Alford C, Hess F, Buchanan R: Vidarabine: A preliminary review of its pharmacological properties and
therapeutic use. Drugs 20:267, 1980. 61. Erlich KS, Mills J, Chatis P et al: Acyclovir-resistant herpes simplex virus infections in patients
with the acquired immunodeficiency syndrome. N Engl J Med 320:293, 1989. 62. Hirsch MS, Schooley RT: Resistance to antiviral drugs: The end of innocence. N Engl J Med 320:313, 1989. 63. Larder BA, Darby Gusceptibility to other antiherpes drugs of pathogenic variants of
herpes simplex virus selected for resistance to acyclovir. Antimicrob Agents Chemother 29:894, 1986. 64. Buchanan RA, Hess F: Vidarabine (Vira A) pharmacology and clinical experience. Pharmacol Ther 8:143, 1980. 65. Poirer RH, Kinkel AW, Ellison AC et al: Intraocular penetration of topical 3% adenine arabinoside. In Pavan-Langston D, Buchanon RA, Alford CA (eds): Adenine Arabinoside: An Antiviral Agent, pp. 313–321. New York, Raven Press, 1975. 66. Buchanan RA, Kinkel AW, Alford CA et al: Plasma levels and urinary excretion of vidarabine after repeated dosing. Clin Pharmacol Ther 27:690, 1980. 67. Abel R, Kaufman HE, Sugar J: Intravenous adenine arabinoside against herpes simplex keratouveitis in
humans. Am J Ophthalmol 79:659, 1975. 68. Langston RHS, Pavan-Langston D, Dohlman CH: Antiviral medication and corneal wound healing. Arch Ophthalmol 92:509, 1974. 69. Ross AH, Julia A, Balakrishnan C: Toxicity of adenine arabinoside in humans. J Infect Dis 133:192, 1976. 70. Van Etta L, Brown J, Mastri A: Fatal vidarabine toxicity in a patient with normal renal function. JAMA 246:1703, 1981. 71. Gizzi M, Rudolph S, Perakis A: Ocular flutter in vidarabine toxicity. Am J Ophthalmol 109:105, 1990. 71a. Sachs SL, Scullard GH, Pollard RB et al: Antiviral treatment of chronic hepatitis B virus infectioPharmacokinetics
and side effects of interferon and adenine arabinoside alone and in
combination. Antimicrob Agents Chemother 21:73, 1982. 72. Friedman HM, Gransela T: Adenine arabinoside and allopurinol: Possible adverse drug reaction. N Engl J Med 304:423, 1981. 73. Schardein JL, Hentz DL, Petrere JA et al: The effect of vidarabine on the development of the offspring of rats, rabbits, and
monkeys. Teratology 15:231, 1977. 74. Whitley RJ, Soong S-J, Dolin et al: Adenine arabinoside therapy of biopsy-proven herpes simplex encephalitis. N Engl J Med 297:289, 1977. 75. Whitley R, Hilty M, Haynes R et al: Vidarabine therapy of varicella in immunosuppressed patients. J Pediatr 101:125, 1982. 76. Whitley RJ, Soong S-J, Dolin R et al: Early vidarabine therapy to control the complications of herpes zoster
in immunosuppressed patients. N Engl J Med 307:971, 1982. 76a. Whitley RJ, Gnann JW Jr, Hinthorn D et al: Disseminated herpes zoster in the immunocompromised host: A comparative
trial of acyclovir and vidarabine. J Infect Dis 165:450, 1992. 77. Shepp DH, Dandliker PS, Meyers JD: Treatment of varicella-zoster virus infection in severely immunocompromised
patients: A randomized comparison of acyclovir and vidarabine. N Engl J Med 314:208, 1986. 78. Skoldenberg B, Fosgren M, Alestig K et al: Acyclovir versus vidarabine in herpes simplex encephalitis: Randomized
multicenter study in consecutive Swedish patients. Lancet 2:707, 1984. 79. Whitley RJ, Alford CA, Hirsch MS et al: Vidarabine versus acyclovir therapy in herpes simplex encephalitis. N Engl J Med 314:144, 1986. 80. Whitley RJ, Arvin A, Prober C et al: A controlled trial comparing vidarabine with acyclovir in neonatal herpes
simplex virus infection. N Engl J Med 324:444, 1991. 81. Ch'ien LT, Cannon NJ, Whitley R et al: Effect of adenine arabinoside on cytomegalovirus infection. J Infect Dis 130:32, 1974. 82. Marker SC, Howard RJ, Groth KE et al: A trial of vidarabine for cytomegalovirus infection in renal transplant
patients. Arch Intern Med 140:1441, 1980. 83. Rytel MW, Kauffman HM: Clinical efficacy of adenine arabinoside on cytomegalovirus infections
in renal allograft recipients. J Infect Dis 133:202, 1976. 84. Laibson PR, Krachmer JH: Controlled comparison of adenine arabinoside and idoxuridine therapy of
human superficial dendritic keratitis. In Pavan-Langston D, Buchanon RA, Alford CA Jr (eds): Adenine Arabinoside: An Antiviral AgenT, pp 323–330. New York, Raven Press, 1975. 85. Uninsky E, Jampol LM, Kaufman S et al: Disseminated herpes simplex infection with retinitis in a renal allograft
recipient. Ophthalmology 90:175, 1983. 86. Pollard RB, Egbert PR, Gallagher JG et al: Cytomegalovirus retinitis in immunosuppressed hosts: I. Natural history
and effects of treatment with adenine arabinoside. Ann Intern Med 93:655, 1980. 87. Moriyama K, Asano Y, Fujimoto K et al: Successful combination therapy with acyclovir and vidarabine for disseminated
varicella zoster virus infection with retinal involvement in a
patient with B-cell lymphoma and adult T-cell leukemia. Am J Med 85:885, 1988. 88. Dorsky DI, Crumpacker CS: Drugs five years later: Acyclovir. Ann Intern Med 107:859, 1987. 89. Wagstaff AJ, Faulds D, Goa KL: Aciclovir. A reappraisal of its antiviral activity, pharmacokinetic properties, and
therapeutic efficacy. Drugs 47:153, 1994. 90. Griffiths PD, Emery VC: Cytomegalovirus. In Richman DD, Whitley RJ, Hayden FG (eds): Clinical Virology, pp 433–461. Washington, DC, ASM Press, 2002. 91. Schaeffer HJ: Acyclovir chemistry and spectrum of activity. Am J Med 73:4, 1982. 92. Elion GB, Furman PA, Fyfe JA et al: Selectivity of action of an antiherpetic agent 9-(2-hydroxyethoxymethyl) guanine. Proc Natl Acad Sci U S A 74:5716, 1977. 93. Elion GB: Mechanism of action and selectivity of acyclovir. Am J Med 73:7, 1982. 94. Furman PA, St Clair MH, Fyfe JA et al: Inhibition of herpes simplex virus induced DNA polymerase activity and
viral DNA replication by 9-(2-hydroxyethoxymethyl) guanine
and its triphosphate. J Virol 32:72, 1979. 95. Colby BM, Shaw JE, Elion GB et al: Effect of acyclovir [9-(2-hydroxyethoxymethyl) guanine] on
Epstein-Barr virus DNA replication. J Virol 34:560, 1980. 96. Laskin OL: Acyclovir: Pharmacology and clinical experience. Ann Intern Med 144:1241, 1984. 97. Biron KK, Elion GB: In vitro susceptibility of varicellazoster virus to acyclovir. Antimicrob Agents Chemother 18:443, 1980. 98. Balfour HH Jr: Acyclovir. In Peterson PK, Verhoeft J (eds): Antimicrobial Agents Annual 3, p 345. Amsterdam, Elsevier Science, 1988. 99. DeMiranda P, Whitley RJ, Blum MR: Acyclovir kinetics after intravenous infusion. Clin Pharmacol Ther 26:718, 1979. 100. Whitley RJ, Blum MR, Barton N et al: Pharmacokinetics of acyclovir in humans following intravenous administration: A
model for the development of parenteral antivirals. Am J Med 73:165, 1972. 101. Van Dyke RB, Connor JD, Wyborny C et al: Pharmacokinetics of orally administered acyclovir in patients with herpes
progenitalis. Am J Med 73:172, 1982. 102. McKendrick MW, Case C, Burke C et al: Oral acyclovir in herpes zoster. J Antimicrob Chemother 14:661, 1984. 103. McKendrick MW, McGill JI, Bell HM et al: Oral acyclovir for herpes zoster. Lancet 2:925, 1984. 104. McKendrick MW, McGill JI, White JE et al: Oral acyclovir in acute herpes zoster. Br Med J 293:1529, 1986. 105. Collum LMT, Akhtar J, McGettrick P: Oral acyclovir in herpetic keratitis. Trans Ophthalmol Soc UK 104:629, 198. 106. Collum LMT, McGettrick P, Akhtar, et al: Oral acyclovir (Zovirax) in herpes simples dendritic corneal
ulceration. Br J Ophthalmol 70:435, 1986. 107. Hung SO, Patterson A, Rees PJ: Pharmacokinetics of oral acyclovir (Zovirax) in the eye. Br J Ophthalmol 68:192, 1984. 108. Schulman JA, Peyman GA, Fiscella RG et al: Parentally administered acyclovir for viral retinitis associated with AIDSArch Ophthalmol 102:1750, 1984. 109. Schulman J, Peyman GA, Liu J et al: The intraocular penetration of acyclovir after subconjunctival administration. Ophthalmic Surg 18:111, 1987. 110. Grant DM: Acyclovir (Zovirax) ophthalmic ointment: A review of clinical
tolerance. Curr Eye Res 6:231, 1987. 111. McCulley JP, Binder PS, Kaufman HE, at al: A double-blind, multicenter clinical trial of acyclovir vs idoxuridine
for treatment of epithelial herpes simplex keratitis. Ophthalmology 89:1195, 1982. 112. Carney MD, Peyman GA, Goldberg MF et al: Acute retinal necrosis. Retina 6:85, 1986. 112a. Goldberg LH, Kaufman R, Kurtz TO et al: Continuous five-year treatment of patients with frequently recurring
genital herpes simplex virus infection with acyclovir. J Med Virol 41:45, 1993. 113. Bean B, Aeppli D: Adverse effects of high-dose intravenous acyclovir in ambulatory
patients with acute herpes zoster. J Infect Dis 151:362, 1985. 114. Keeney RE, Kirk LE, Bridgen D: Acyclovir tolerance in humans. Am J Med 73(1A):176, 1982. 115. Sylvester RK, Ogden WB, Draxler CA et al: Vesicular eruption: A local complication of concentrated acyclovir infusions. JAMA 255:385, 1986. 116. Balfour HH Jr: Acyclovir therapy for herpes zoster: Advantages and adverse effects. JAMA 255:387, 1986. 117. Bridgen D, Rosling AE, Woods NC: Renal function after acyclovir intravenous injection. Am J Med 73:182, 1982. 118. Eck P, Silver SM, Clark EC: Acute renal failure and coma after a high dose of oral acyclovir. N Engl J Med 325:1178, 1991. 119. Manka RL: Exogenous lactase in the treatment of oral acyclovir intolerance. Am J Ophthalmol 108:733, 1989. 120. Cohen SMZ, Minkove JA, Zebley JWIII et al: Severe but reversible neurotoxicity from acyclovir. Ann Intern Med 100:920, 1984. 121. Fletcher CV, Chinnock BJ, Chase B et al: Pharmacokinetics and safety of high dose oral acyclovir for suppression
of cytomegalovirus disease after renal transplantation. Clin Pharmacol Ther 44:158, 1988. 122. Sirota P, Stoler M, Meshulam B: Major depression with psychotic features associated with acyclovir therapy. Drug Intell Clin Pharm 22:306, 1988. 123. Tomson CR, Goodiship THJ, Rodger RSC: Psychiatric side-effects of acyclovir in patients with chronic renal
failure. Lancet 1:385, 1985. 124. Wade JC, Meyers JD: Neurologic symptoms associated with parenteral acyclovir treatment after
bone marrow transplantation. Ann Intern Med 98:921, 1983. 125. Clive D, Turner NT, Hozier J et al: Preclinical toxicology studies with acyclovir: Genetic toxicity tests. Fundam Appl Toxicol 3:587, 1983. 126. Tucker WE Jr, Krasny HC, DeMiranda P et al: Preclinical toxicology studies with acyclovir. Carcinogenicity bioassays
and chronic toxicity tests. Fundam Appl Toxicol 3:579, 1983. 127. Klug S, Lewandowski C, Blankenburg G et al: Effect of acyclovir on mammalian embryonic development in culture. Arch Toxicol 58:29, 1985. 128. Moore HL Jr, Szczech GM, Rodwell DE et al: Preclinical toxicology studies with acyclovir: Teratologic, reproductive
and neonatal tests. Fundam Appl Toxicol 3:560, 1983. 129. Reiff-Eldridge R, Heffner CR, Ephross SA et al: Monitoring pregnancy outcomes after prenatal drug exposure through prospective
pregnancy registries: A pharmaceutical company commitment. Am J Obstet Gynecol 182:159, 2000. 130. Greffe BS, Dooley SL, Deddish RB et al: Transplacental passage of acyclovir. J Pediatr 108:1020, 1986. 131. Brown ZA, Baker DA: Acyclovir therapy during pregnancy. Obstet Gynecol 73:526, 1989. 132. Van Landingham KE, Marsteller HB, Ross GW et al: Relapse of herpes simplex encephalitis after conventional acyclovir therapy. JAMA 259:1051, 1988. 133. Bryson YJ, Dillon M, Lovett M et al: Treatment of first episodes of genital herpes simplex virus infection with
oral acyclovir: A randomized double-blind controlled trial
in normal subjects. N Engl J Med 308:916, 1983. 134. Corey L, Benedetti J, Critchlow C et al: Treatment of primary first-episode genital herpes simplex virus
infections with acyclovir: Results of topical, intravenous, and oral therapy. J Antimicrob Chemother 12:79, 1983. 135. Mindel A, Adler MW, Sutherland S et al: Intravenous acyclovir treatment for primary genital herpes. Lancet 1:697, 1982. 136. Nilsen AE, Aasen T, Halsos AM et al: Efficacy of oral acyclovir treatment for primary genital herpes. Lancet 1:697, 1982. 137. Mertz GJ, Jones CC, Mills J: Long-term acyclovir suppression of frequently recurring genital
herpes simplex virus infection. JAMA 260:201, 1988. 138. Spruance SL, Stewart JCB, Rowe NH et al: Treatment of recurrent herpes simplex labialis with oral acyclovir. J Infect Dis 161:185, 1990. 138a. Straus SE, Takiff HE, Mindell S et al: Suppression of frequently recurring genital herpes: A placebo-controlled
double blind trial of oral acyclovir. N Engl J Med 310:1545, 1984. 139. Kaplowitz LG, Baker D, Gelb L et al: Prolonged continuous acyclovir treatment of normal adults with frequently
recurring genital herpes simplex virus infection. JAMA 265:745, 1991. 140. Mertz GJ, Eron L, Kaufman R et al: Prolonged continuous versus intermittent oral acyclovir treatment in normal
adults with frequently recurring genital herpes simplex virus infection. Am J Med 85:14, 1988. 140a. Spruance SL, Nett R, Marbury T et al: Acyclovir cream for the treatment of herpes simplex labialis: Results of
two randomized double-blind vehicle-controlled multicenter
clinical trials. Antimicrob Agents Chemother 46:2238, 2002. 140b. Amir J, Harel L, Smetana Z, Varsano I: Treatment of herpes simplex gingivostomatitis with acyclovir in children: a
randomised double blind placebo controlled trial. BMJ 314:1800, 1997. 141. Gold D, Corey L: Acyclovir prophylaxis for herpes simplex virus infection. Antimicrob Agents Chemother 31:361, 1987. 142. Lundgren G, Wilczek H, Lonnquist B et al: Acyclovir prophylaxis in bone-marrow transplant recipients. Scand J Infect Dis 47:137, 1985. 143. Saral R, Ambinder RF, Burns WH et al: Acyclovir prophylaxis against herpes simplex virus infection in patients
with leukemia. Ann Intern Med 99:773, 1983. 144. Saral R, Burns WH, Laskin OH et al: Acyclovir prophylaxis of herpes simplex virus infections: A randomized
double-blind, controlled trial in bone-marrow transplant
recipients. N Engl J Med 305:63, 1981. 145. Shepp DH, Newton B, Dandliker BS et al: Oral acyclovir therapy for mucocutaneous herpes simplex infections in immunocompromised
marrow transplant recipients. Ann Intern Med 102:783, 1985. 146. Strauss SE, Seidlin M, Takiff H et al: Oral acyclovir to suppress recurrent herpes simplex virus infections in
immunodeficient patients. Ann Intern Med 100:522, 1984. 147. Wade JC, Newton B, McLaren C et al: Intravenous acyclovir to treat mucocutaneous herpes simplex infection after
marrow transplantation. Ann Intern Med 96:265, 1982. 148. Balfour HH, Bean B, Laskin OL et al: Acyclovir halts progression of herpes zoster in immunocompromised patients. N Engl J Med 308:1448, 1983. 149. Balfour HH Jr: Intravenous acyclovir therapy for varicella in immunocompromised children. J Pediatr 104:134, 1984. 150. Bean B, Brain C, Balfour HH Jr: Acyclovir therapy for acute herpes zoster. Lancet 2:118, 1982. 151. Feldman S, Lott L: Varicella in children with cancer: Impact of antiviral therapy and prophylaxis. Pediatrics 80:465, 1987. 152. Peterslund NA, Seyer-Hansen K, Ipsen J et al: Acyclovir in herpes zoster. Lancet 2:827, 1981. 153. Huff JC, Bean B, Balfour HH et al: Therapy of herpes zoster with oral acyclovir. Am J Med 85:84, 1988. 154. Buchi ER, Herbort CP, Ruffieux C: Oral acyclovir in the treatment of acute herpes zoster ophthalmicus. Am J Ophthalmol 102:531, 1986. 155. Cobo LM: Reduction of the ocular complications of herpes zoster ophthalmicus by
oral acyclovir. Am J Med 85:90, 1988. 156. Cobo LM, Foulks GN, Liesegang T et al: Oral acyclovir in the therapy of acute herpes zoster ophthalmicus: An interim
report. Ophthalmology 92:1574, 1985. 157. Cobo LM, Foulks GN, Liesegang T et al: Oral acyclovir in the treatment of acute herpes zoster ophthalmicus. Ophthalmology 93:763, 1986. 158. Wood MJ, Ogan PH, McKendrick MW et al: Efficacy of oral acyclovir treatment of acute herpes zoster. Am J Med 85:79, 1988. 158a. Wood MJ, Kay R, Dowrkin RH et al: Oral acyclovir therapy accelerates pain resolution in patients with herpes
zoster: A meta-analysis of placebo-controlled trials. Clin Infect Dis 22:341, 1996. 158b. Gnann JW, Whitley RJ: Herpes Zoster. N Engl J Med 347:340, 2002. 159. Balfour HH Jr, Kelly JM, Suarez CS et al: Acyclovir treatment of varicella in otherwise healthy children. J Pediatr 116:633, 1990. 160. Dunkle LM, Arvin AM, Whitley RJ et al: A controlled trial of acyclovir for chickenpox in normal children. N Engl J Med 325:1539, 1991. 160a. Balfour HH Jr, Rotbart HA, Feldman S et al: Acyclovir treatment of varicella in otherwise healthy adolescents. J Pediatr 120:627, 1992. 161. Wallace MR, Bowler WA, Murray NB et al: Treatment of adult varicella with oral acyclovir: A randomized, placebo-controlled
trialAnn Intern Med 117:358, 1992. 161a. American Academy of Pediatrics Committee on Infectious Diseases: The use
of oral acyclovir in otherwise healthy children with varicella. Pediatrics 91:674, 1993. 161b. Cohen JI, Brunell PA, Strauss SE et al: Recent advances in varicella-zoster virus infection. Ann Intern Med 130:922, 1999. 162. Balfour HH Jr, Chace BA, Stapleton JT et al: A randomized placebo-controlled trial of oral acyclovir for the
prevention of cytomegalovirus disease in recipients of renal allografts. N Engl J Med 320:1381, 1989. 163. Meyers JD, Reed EC, Shepp DH et al: Acyclovir for prevention of cytomegalovirus infection and disease after
allogeneic marrow transplantation. N Engl J Med 318:70, 1988. 164. Balfour HH Jr, Bean B, Mitchell CD et al: Acyclovir in immunocompromised patients with cytomegalovirus disease. Am J Med 73:243, 1982. 165. Verheyden JPH: Evolution of the therapy for cytomegalovirus infection. Rev Infect Dis 10:S477, 1988. 166. Wade JC, Hintz M, McGuffin RW et al: Treatment of cytomegalovirus pneumonia with high dose acyclovir. Am J Med 73:249, 1982. 167. Plotkin SA, Starr SE, Bryan CK: In vitro and in vivo responses of cytomegalovirus to acyclovir. Am J Med 73:257, 1982. 168. Andersson J, Ernberg I: Management of Epstein-Barr virus infections. Am J Med 85:107, 1988. 169. van der Host C, Jonas J, Ahronheim G et al: Lack of effect of peroral acyclovir for the treatment of infectious mononucleosis. J Infect Dis 164:788, 1991. 170. Resnick L, Herbst JS, Ablashi DV et al: Regression of oral hairy leukoplakia after orally administered acyclovir
therapy. JAMA 259:384, 1988. 171. Murakami S, Mizobuchi M, Nakashiro Y et al: Bell's palsy and herpes simplex virus: Identification of viral DNA
in endoneural fluid and muscle. Ann Intern Med 124:27, 1996. 172. Adour KK, Ruboyianes JM, Von Doertsen PG et al: Bell's palsy treatment with acyclovir and prednisone compared with
prednisone alone: A double-blind, randomized, controlled trial. Ann Otol Rhinol Laryngol 105:371, 1996. 172a. Grogan PM, Gronseth GS: Practice parameter: Steroids, acyclovir, and surgery for Bell's palsy (an
evidence-based review). Report of the Quality
Standards Subcommittee of the American Academy of Neurology. Neurology 56:830, 2001. 173. Coster DJ, Wilhelmus KR, Michael R et al: A comparison of acyclovir and idoxuridine as treatment for ulcerative herpetic
keratitis. Br J Ophthalmol 64:763, 1980. 174. Collum LMT, Benedict-Smith A, Hillary IB: Randomised double-blind trial of acyclovir and idoxuridine in dendritic
corneal ulceration. Br J Ophthalmol 64:766, 1980. 175. McGill J, Tormey P, Walter CB: Comparative trial of acyclovir and adenine arabinoside in the treatment
of herpes simplex corneal ulcers. Br J Ophthalmol 65:610, 1981. 176. Young B, Patterson A, Ravenscroft T: A randomised double-blind clinical trial of acyclovir (Zovirax) and
adenine arabinoside in herpes simplex corneal ulceration. Br J Ophthalmol 66:361, 1982. 177. Laibson PR, Pavan-Langston D, Yeakley WR et al: Acyclovir and vidarabine for the treatment of herpes simplex keratitis. Am J Med 73:281, 1982. 178. Collum LMT, Logan P, McAuliffe-Curtin D et al: Randomised double-blind trial of acyclovir (Zovirax) and
adenine arabinoside in herpes simplex amoeboid corneal ulceration. Br J Ophthalmol 69:847, 1985. 179. La Lau C, Oosterhuis JA, Versteeg J et al: Acyclovir and trifluorothymidine in herpetic keratitis: A multicentre trial. Br J Ophthalmol 66:506, 1982. 180. Hovding G: A comparison between acyclovir and trifluorothymidine ophthalmic ointment
in the treatment of epithelial dendritic keratitis: A double blind, randomized
parallel group trial. Acta Ophthalmologica 67:51, 1989. 181. Hung SO, Patterson A, Clark D, Rees PJ: Oral acyclovir in the management of dendritic herpetic corneal ulceration. Br J Ophthalmol 68:398, 1984. 182. Schwab IR: Oral acyclovir in the management of herpes simplex ocular infections. Ophthalmology 95:423, 1988. 183. Gordon, YJ: The evolution of antiviral therapy for external ocular viral infections
over twenty-five years. Cornea 19:673, 2000. 184. Margolis TP, Ostler HB: Treatment of ocular disease in eczema herpeticum. Am J Ophthalmol 110:274, 1990. 184a. Schwartz GS, Holland EJ: Oral acyclovir for the management of herpes simplex virus keratitis in
children. Ophthalmology 107:278, 2000. 185. Beyer CF, Arens MQ, Hill GA et al: Oral acyclovir reduces the incidence of recurrent herpes simplex keratitis
in rabbits after penetrating keratoplasty. Arch Ophthalmol 107:1200, 1989. 186. Green MT, Dunkel EC, Morris BL: Quantitation of herpes simplex virus type 1 shed in pre-ocular tear
film of rabbits treated with acyclovir. Antimicrob Agents Chemother 20:580, 1981. 187. Pavan-Langston D, Park NH, Lass JH: Herpetic ganglionic latency: Acyclovir and vidarabine therapy. Arch Ophthalmol 97:1508, 1979. 188. Barney NP, Foster CS: A prospective randomized trial of oral acyclovir after penetrating keratoplasty
for herpes simplex keratitis. Cornea 13:232, 1994. 189. Tambasco FP, Cohen EJ, Nguyen LH et al: Oral acyclovir after penetrating keratoplasty for herpes simplex. Arch Ophthalmol 117:445, 1999. 190. Simon AL, Pavan-Langston D: Long term oral acyclovir therapy: Effect on recurrent infectious herpes
keratitis in patients with and without grafts. Ophthalmology 103:1399, 1996. 191. Wilhelmus KR: Diagnosis and management of herpes simplex stromal keratitis. Cornea 6:286, 1987. 192. Oh J: Enhancement of virus multiplication and interferon production by cortisone
in ocular herpes virus infections. J Immunol 104:1359, 1970. 192a. Patterson A, Jones R: The management of ocular herpes. Trans Ophthalmol Soc UK 87:59, 1967. 193. Robbins RM, Galin MA: A model of steroid effects in herpes keratitis. Arch Ophthalmol 93:828, 1975. 194. Collum, LMT, Logan P, Ravenscroft T: Acyclovir (Zovirax) in herpetic disciform keratitis. Br J Ophthalmol 67:115, 1983. 195. VanGanswijk R, Oosterhuis JA, Swart-Van Den Berg M et al: Acyclovir treatment in stromal herpetic keratitis. Doc Ophthalmol 55:57, 1983. 196. Sanitato JJ, Asbell PA, Varnell ED et al: Acyclovir in the treatment of herpetic stromal disease. Am J Ophthalmol 98:537, 1984. 197. Barron BA, Gee L, Hauck WW et al: Herpetic Eye Disease Study. A controlled trial of oral acyclovir for herpes
simplex stromal keratitis. Ophthalmology 101:1871, 1994. 198. The Herpetic Eye Disease Study Group: A controlled trial of oral acyclovir
for the prevention of stromal keratitis or iritis in patients with
herpes simplex virus epithelial keratitis. The epithelial keratitis trial. Arch Ophthalmol 115:703, 1997. 199. The Herpetic Eye Disease Study Group: A controlled trial of oral acyclovir
for iridocyclitis caused by herpes simplex virus. Arch Ophthalmol 114:1065, 1996. 200. The Herpetic Eye Disease Study Group: Acyclovir for the prevention of recurrent
herpes simplex virus eye disease. N Engl J Med 339:300, 1998. 201. The Herpetic Eye Disease Study Group: Oral acyclovir for herpes simplex
virus eye disease. Effect on prevention of epithelial keratitis and stromal
keratitis. Arch Ophthalmol 118:1030, 2000. 202. Grutzmacher RD, Henderson D, McDonald PJ et al: Herpes simplex chorioretinitis in a healthy adult. Am J Ophthalmol 96:788, 1983. 203. Nanda M, Curtin VT, Hilliard JK et al: Ocular histopathologic findings in a case of human herpes B virus infection. Arch Ophthalmol 108:713, 1990. 203a. Ragozzino MW, Melton LJIII , Chu CP et al: Population-based study of herpes zoster and its sequelae. Medicine 61:310, 1982. 204. McGill J: Topical acyclovir in herpes zoster ocular involvement. Br J Ophthalmol 65:542, 1981. 205. McGill J, Chapman C: A comparison of topical acyclovir with steroids in the treatment of herpes
zoster keratouveitis. Br J Ophthalmol 76:746, 1983. 206. McGill J: The enigma of herpes stromal disease. Br J Ophthalmol 71:117, 1987. 207. Marsh RJ, Cooper M: Double-masked trial of topical acyclovir and steroids in the treatment
of herpes zoster ocular inflammation. Br J Ophthalmol 75:542, 1991. 208. Zaal MJW, Maudgal PC, Rietveld E et al: Chronic ocular zoster. Curr Eye Res 10:125, 1991. 209. Engstrom RE, Holland GN: Chronic herpes zoster virus keratitis associated with the acquired immunodeficiency
syndrome. Am J Ophthalmol 105:556, 1988. 209a. Chern KC, Conrad D, Holland GN et al: Chronic varicella-zoster virus epithelial keratitis in patients
with acquired immunodeficiency syndrome. Arch Ophthalmol 116:1011–1017, 1998. 210. Harding SP, Porter SM: Oral acyclovir in herpes zoster ophthalmicus. Curr Eye Res 10:177, 1991. 211. Hoang-Xuan T, Büchi ER, Herbort CP et al: Oral acyclovir for herpes zoster ophthalmicus. Ophthalmology 99:1062, 1992. 212. Wood MJ, Johnson RW, McKendrick MW et al: A randomized trial of acyclovir for 7 days or 21 days with and without
prednisolone for treatment of acute herpes zoster. N Engl J Med 330:896, 1994. 213. Wood MJ, Shukla S, Fiddian AP et al: Treatment of acute herpes zoster: Effect of early (<48h) versus
late (48-72h) therapy with acyclovir and valaciclovir
on prolonged pain. J Infect Dis 178:S81, 1998. 214. Aylward GW, Claove CM, Marsh RJ, Yaseem N: Influence of oral acyclovir on ocular complications of herpes zoster ophthalmicus. Eye 8:70, 1994. 215. Pepose JS: The potential impact of the varicella vaccine and new antivirals on ocular
disease related to varicella-zoster virus. Am J Ophthalmol , 123:243, 1997. 216. Liesegang TJ: Varicella zoster viral disease. Mayo Clin Proc 74:983, 1999. 216a. Severson EA, Baratz KH, Hodge DO, Burke JP: Herpes zoster ophthalmicus in Olmsted County, Minnesota. Have systemic
antivirals made a difference?Arch Ophthalmol 121:386, 2003. 217. Whitley RJ, Weiss H, Gnann JW et al: Acyclovir with or without prednisone for the treatment of herpes zoster. A
randomized, placebo-controlled trialAnn Intern Med 125:376, 1996. 218. Harding SP, Lipton JR, Wells JCD: Natural history of herpes zoster ophthalmicus: Predictors of postherpetic
neuralgia and ocular involvement. Br J Ophthalmol 81:353, 1987. 219. Alper BS, Peter RL: Does treatment of acute herpes zoster prevent or shorten postherpetic neuralgia? A
systematic review of the literature. J Fam Prac 49:255, 2000. 220. Esmann V, Kroon S, Peterslund NA et al: Prednisolone does not prevent post-herpetic neuralgia. Lancet 2:126, 1987. 221. Colin J, Prisant O, Cochener B et al: Comparison of the efficacy and safety of valaciclovir and acyclovir for
the treatment of herpes zoster ophthalmicus. Ophthalmology 107:1507, 2000 222. Buetner KR, Friedman DJ, Forszpaniak C et al: Valacyclovir compared with acyclovir for improved therapy for herpes zoster
in immunocompetent adults. Antimicrob Agents Chemother 39:1546, 1995. 223. Tyring S, Engst R, Corriveau C et al: Famciclovir for ophthalmic zoster: A randomized aciclovir controlled study. Br J Ophthalmol 85:576, 2001. 224. Balfour H: Varicella zoster virus infections in immunocompromised hosts: A review
of the natural history and management. Am J Med 85:68, 1988. 225. Cole EL, Meisler DM, Calabrese LH et al: Herpes zoster ophthalmicus and acquired immunodeficiency syndrome. Arch Ophthalmol 102:1027, 1984. 226. Sandor EV, Millman A, Croxson TS et al: Herpes zoster ophthalmicus in patients at risk for the acquired immunodeficiency
syndrome (AIDS). Am J Ophthalmol 101:153, 1986. 226a. Pepose JS, Biron K: Antiviral sensitivities of the acute retinal necrosis syndrome virus. Curr Eye Res 6:201, 1987. 227. Seiff SR, Margolis T, Oraham SH et al: Use of intravenous acyclovir for treatment of herpes zoster ophthalmicus
in patients at risk for AIDS. Ann Ophthalmol 20:480, 1988. 228. Margolis TP, Milner MS, Shama A et al: Herpes zoster ophthalmicus in patients with human immunodeficiency virus
infection. Am J Ophthalmol 125:285, 1998. 229. Jacobson MA, Berger TG, Fikrig S et al: Acyclovir-resistant varicella zoster virus infection after chronic
oral acyclovir therapy in patients with acquired immunodeficiency syndrome (AIDS). Ann Intern Med 112:187, 1990. 230. Jabs DA, Schachat AP, Liss R et al: Presumed varicella zoster retinitis in immunocompromised patients. Retina 7:9, 1987. 231. Chess J, Marcus DM: Zoster-related bilateral acute retinal necrosis syndrome as presenting
sign in AIDS. Ann Ophthalmol 20:431, 1988. 232. Forster DJ, Dugel PU, Fragieh GT et al: Rapidly progressive outer retinal necrosis in the acquired immunodeficiency
syndrome. Am J Ophthalmol 110:341, 1990. 233. Margolis TP, Lowder CY, Holland GN: Varicella-zoster virus retinitis in patients with the acquired immunodeficiency
syndrome. Am J Ophthalmol 112:119, 1991. 233a. Sellitti TP, Huang AJW, Schiffman J et al: Association of herpes zoster ophthalmicus with acquired immunodeficiency
syndrome and acute retinal necrosis. Am J Ophthalmol 116:297, 1993. 234. Duker JS, Blumenkranz MS: Diagnosis and management of the acute retinal necrosis (ARN) syndrome. Surv Ophthalmol 36:327, 1992. 235. Van Gelder RN, Willis JL, Holland GN, Kaplan HJ: Herpes simplex virus type 2 as a cause of acute retinal necrosis syndrome
in young patients. Ophthalmology 108:869, 2001. 236. Pepose JS, Biron K: Antiviral sensitivities of the acute retinal necrosis syndrome virus. Curr Eye Res 6:201, 1987. 237. Blumenkranz MS, Culbertson WW, Clarkson JG et al: Treatment of the acute retinal necrosis syndrome with intravenous acyclovir. Ophthalmology 93:296, 1986. 238. Hirst L, Beyer TL, Waters D et al: Successful management of acute retinal necrosis with intravenous acyclovir. Arch Ophthalmol 19:445, 1987. 239. Matsuo T, Nakayama T, Koyama T et al: A proposed mild type of acute retinal necrosis syndrome. Am J Ophthalmol 105:579, 1988. 240. Crapotta JA, Freeman WR, Feldman RM et al: Visual outcome in acute retinal necrosis. Retina 13:208, 1993. 241. Palay DA, Sternberg P, Davis J et al: Decrease in the risk of bilateral acute retinal necrosis by acyclovir therapy. Am J Ophthalmol 112:250, 1991. 242. Matsuo T, Koyama M, Matsuo N: Acute retinal necrosis as a novel complication of chickenpox in adults. Br J Ophthalmol 74:443, 1990. 243. Astanides IM, DeSouza S, Wong DTW et al: Oral valacyclovir in the treatment of acute retinal necrosis syndrome. Retina 22:352, 2002. 244. Figueroa MS, Garabito I, Gutierrez C, Fortun J: Famciclovir for the treatment of acute retinal necrosis (ARN) syndrome. Am J Ophthalmol 123:255, 1997. 245. Klein JL, Sandy C, Migdal CS et al: Famciclovir in AIDS-related acute retinal necrosis. AIDS 10:1300, 1996. 246. Luu, KKM, Scott IV, Chaudhry NA et al: Intravitreal antiviral injections as adjunctive therapy in the management
of immunocompetent patients with necrotizing herpetic retinopathy. Am J Ophthalmol 129:811, 2000. 247. Ando F, Kato M, Goto S et al: Platelet function in bilateral acute retinal necrosis. Am J Ophthalmol 96:22, 1983. 248. Peyman GA, Goldberg GF, Uninsky E, at al: Vitrectomy and intravitreal antiviral drug therapy in acute retinal necrosis
syndrome: Report of two cases. Arch Ophthalmol 102:1617, 1984. 249. Engstrom RE, Holland GN, Margolis TP et al: The progressive outer retinal necrosis syndrome: A variant of necrotizing
herpetic retinopathy in patients with AIDS. Ophthalmology 101:1488, 1994. 250. Spaide RF, Martin DF, Teich SA et al: Successful treatment of progressive outer retinal necrosis syndrome. Retina 16:479, 1996. 251. Moorthy RS, Weinberg DV, Teich SA et al: Management of varicella zoster virus retinitis in AIDS. Br J Ophthalmol 81:189, 1997. 252. Matoba AY: Ocular disease associated with Epstein-Barr virus infection. Surv Ophthalmol 35:145, 1990. 253. Wilhelmus KR: Ocular involvement in infectious mononucleosis. Am J Ophthalmol 91:117, 1981. 254. Matoba AY, Wilhelmus KR, Jones DB: Epstein-Barr viral stromal keratitis. Ophthalmology 93:746, 1986. 255. Pinnolis M, McCulley JP, Urman JD: Nummular keratitis associated with infectious mononucleosis. Am J Ophthalmol 89:791, 1980. 256. Pflugfelder SG, Huang A, Crouse C: Epstein-Barr virus keratitis after a chemical facial peel. Am J Ophthalmol 110:571, 1990. 257. Wong KW, D'Amico DJ, Hedges TRIII et al: Ocular involvement associated with chronic Epstein-Barr virus disease. Arch Ophthalmol 105:788, 1987. 258. Schooley RT, Carey RW, Miller G et al: Chronic Epstein-Barr virus infection associated with fever and interstitial
pneumonitis: Clinical and serologic features and response
to antiviral chemotherapy. Ann Intern Med 104:636, 1986. 259. Fay MT, Freeman WR, Wiley CA et al: Atypical retinitis in patients with the acquired immunodeficiency syndrome. Am J Ophthalmol 105:483, 1988. 260. Sha BE, Benson CA, Deutsch TA et al: Suppression of cytomegalovirus retinitis in persons with AIDS with high-dose
intravenous acyclovir. J Infect Dis 164:777, 1991. 261. Fife KH, Crumpacker CS, Mertz GJ et al: Recurrence and resistance pattern of herpes simplex virus following cessation
of >6 years of chronic suppression with acyclovir. J Infect Dis 169:1338, 1994. 262. Charles SJ, Gray JJ: Ocular herpes simplex virus infections: Reduced sensitivity to acyclovir
in primary disease. Br J Ophthalmol 74:286, 1990. 263. Menage MJ, de Clerq E, van Lierde A et al: Antiviral drug sensitivity in ocular herpes simplex virus infection. Br J Ophthalmol 74:286, 1990. 264. Englund JA, Zimmerman ME, Swierkosz EM et al: Herpes simplex virus resistant to acyclovir: A study in a tertiary care
center. Ann Intern Med 112:416, 1990. 265. Chatis PA, Miller CH, Schrager LE et al: Successful treatment with foscarnet of an acyclovir-resistant mucocutaneous
infection with herpes simplex virus in a patient with acquired
immunodeficiency syndrome. N Engl J Med 320:297, 1989. 266. Kaplan MH, Sadick N, McNutt NS et al: Dermatologic findings and manifestations of acquired immunodeficiency syndrome (AIDS). J Am Acad Dermatol 16:485, 1987, 267. Marks GL, Nolan, PE, Erlich KS et al: Mucocutaneous dissemination of acyclovir-resistant herpes simplex
virus in a patient with AIDS. Rev Infect Dis 11:474, 1989, 268. Field, HJ, Darby G: Pathogenecity in mice of strains of herpes simplex virus which are resistant
to acyclovir in vitro and in vivo. Antimicrob Agents Chemother 17:209, 1980, 269. Safrin S, Assaykeen T, Follansbee S et al: Foscarnet therapy for acyclovir-resistant mucocutaneous herpes simplex
virus infection in 26 AIDS patients: Preliminary data. J Infect Dis 161:1078, 1990, 270. Youle MM, Hawkins DA, Collins P et al: Acyclovir-resistant herpes in AIDS treated with foscarnet. Lancet 3:341, 1988. 271. Lalezari JP, Drew WL, Glutzer E et al: Treatment with intravenous (s)-1-[3-hydroxy-2-phosphonylmethoxy)propyl]-cytosine
of acyclovir-resistant mucocutaneous infection with herpes
simplex virus in a patient with AIDS. J Infect Dis 170:570, 1994. 272. Engel JP, Englund JA, Fletcher CV et al: Treatment of resistant herpes simplex virus with continuous-infusion
acyclovir. JAMA 263:1662, 1990. 273. Fletcher CV, Englund JA, Bean B et al: Continuous infusion of high dose acyclovir for serious herpes virus infections. Antimicrob Agents Chemother 33:1375, 1989. 274. Safrin S, Berger TG, Gilson I et al: Foscarnet therapy in five patients with AIDS and acyclovir-resistant
varicella-zoster virus infection. Ann Intern Med 115:19, 1991. 275. Perry CM, Faulds D: Valaciclovir. A review of its antiviral activity, pharmacokinetic properties
and therapeutic efficacy in herpes virus infections. Drugs 52:755, 1996. 276. Acosta EP, Fletcher CV: Valacyclovir. Ann Pharmacother 31:185, 1997. 277. Ormrod D, Goa K: Valaciclovir. A review of its use in the management of herpes zoster. Drugs 59:1317, 2000. 278. Weller S, Blum MR, Doucette M et al: Pharmacokinetics of the acyclovir pro-drug, valacyclovir after escalating
single- and multiple-dose administration to normal
volunteers. Clin Pharmacol Ther 54:595, 1993. 279. Steingrimsdottir H, Gruber A, Palm C et al: Bioavailability of aciclovir after oral administration of aciclovir and
its prodrug valaciclovir to patients with leukopenia after chemotherapy. Antimicrob Agents Chemother 44:207, 2000. 280. Ormrod D, Scott LJ, Perry CM: Valaciclovir. A review of its long term utility in the management of genital
herpes simplex virus and cytomegalovirus infections. Drugs 59:839, 2000. 281. Soul-Lawton J, Seaber E, On N et al: Absolute bioavailability and metabolic disposition of valaciclovir, the $PI$vL-valyl
ester of acyclovir following oral administration
to humans. Antimicrob Agents Chemother 39:2759, 1995. 282. Jacobson MA, Gallant J, Wang LH et al: Phase 1 trial of valaciclovir, the $PI$vL-valyl ester of acyclovir, in
patients with advanced human immunodeficiency virus disease. Antimicrob Agents Chemother 38:1534, 1994. 283. Decroix J, Partsch H, Gonzalez R et al: Factors influencing pain outcome in herpes zoster: An observational study
with valaciclovir. J Eur Acad Dermatol Venereol 14:23, 2000. 284. Lowance D, Neumayer H-H, Legendre CM et al: Valacyclovir for the prevention of cytomegalovirus disease after renal
transplantation. N Engl J Med 340:1462, 1999. 285. Feinberg JE, Hurwitz S, Cooper D et al: A randomized, double-blind trial of valaciclovir prophylaxis for
cytomegalovirus disease in patients with advanced human immunodeficiency
virus infection. J Infect Dis 177:48, 1998. 286. Izzedine H, Mercada IL, Aymard G et al: Neurotoxicity of valacyclovir in peritoneal dialysis: A pharmacokinetic
study. Am J Nephrol 21:162, 2001. 287. Chulay JD, Bell AR: International Valaciclovir HSV Study Group: Long-term safety of
valaciclovir for suppression of herpes simplex virus infections. Clin Infect Dis 23:879, 1996. 288. Fobelo MJ, Delgado JEC, Alonso AR et al: Aseptic meningitis related to valacyclovir. Ann Pharmacother 35:128, 2001. 289. Dekker, CL, Prober CG: Pediatric uses of valacyclovir, penciclovir and famciclovir. Pediatr Infect Dis J 20:1079, 2001. 290. Fife KH, Barbarash RA, Rudolph T et al: Valaciclovir versus acyclovir in the treatment of first-episode
genital herpes infection. Results of an international multicenter double
blind, randomized clinical trial. The Valaciclovir International Herpes
Simplex Virus Study Group. Sex Transm Dis 24:481, 1997. 291. Tyring SK, Douglas JM Jr, Corey L et al: A randomized placebo-controlled comparison of oral valacyclovir
and acyclovir in immunocompetent patients with recurrent genital herpes
infections. The Valaciclovir International Study Group. Arch Dermatol 134:185, 1998. 292. Anonymous: Drugs for non-HIV viral infections. The Medical Letter44 :9, 2002. 293. Reitano M, Tyring S, Lang W et al: Valaciclovir for the suppression of recurrent genital herpes simplex virus
infection: A large scale dose range-finding study. J Infect Dis 1278:603, 1998. 294. Anonymous: Valacyclovir (Valtrex) for herpes labialis. The Medical
Letter44:95, 2002. 295. Tyring SK, Buetner KR, Tucker BA et al: Antiviral therapy for herpes zoster. Randomized controlled clinical trial
of valacyclovir and famciclovir therapy in immunocompetent patients 50 years
and older. Arch Fam Med 9:863, 2000. 296. Leblanc RA, Pesnicak L, Godleski M et al: The comparative effects of famciclovir and valacyclovir on herpes simplex
virus type 1 infection, latency, and reactivation in mice. J Infect Dis 180:594, 1999. 297. Harding SP, Rigal D, Easty DL et al: Superior intraocular penetration of aciclovir from valaciclovir in comparison
with oral aciclovir. J Antimicrob Chemother 44:44, 1999. 298. Asbell PA: Valacyclovir for the prevention of recurrent herpes simplex virus eye disease
after excimer laser photokeratectomy. Trans Am Ophthalmol Soc 98:285, 2000. 299. Dhalimal DK, Romanowski EG, Yates KA et al: Valacyclovir inhibition of ocular herpes simplex type 1 after experimental
reactivation by laser in situ keratomileusis. J Cataract Refract Surg 27:1288, 2001. 300. Grant DM, Mauskopf JA, Bell L et al: Comparison of valaciclovir and acyclovir for the treatment of herpes zoster
in immunocompetent patients over 50 years of age: A cost-consequence
model. Pharmacotherapy 17:333, 1997. 301. Perry CM, Wagstaff AJ: Famciclovir. A review of its pharmacological properties and therapeutic
efficacy in herpes virus infections. Drugs 50:396, 1995. 302. Pue MA, Benet LZ: Pharmacokinetics of famciclovir in man. Antiviral Chem Chemother 4:47, 1993. 302a. Boyd MR, Kern ER, Safrin S: Penciclovir: A review of its spectrum of activity, selectivity and cross-resistance
pattern. Antivir Chem Chemother 4:3, 1993. 302b. Vere Hodge RA: Famciclovir and penciclovir: The mode of action of famciclovir including
its conversion to penciclovir. Antivir Chem Chemother 4:67, 1993. 303. Degreef H,: The Famciclvir Herpes Zoster Clinical Study Group: Famciclovir, a new oral
antiherpes drug: Results of the first controlled clinical study demonstrating
its efficacy and safety in the treatment of uncomplicated
herpes zoster in immunocompetent patients. Int J Antimicrob Agents 4:241, 1994. 304. Saltzman R, Jurewicz R, Boon R: Safety of famciclovir in patients with herpes zoster and genital herpes. Antimicrob Agents Chemother 38:2454, 1994. 305. Loveless M, Sacks SL, Harris RJ: Famciclovir in the management of first-episode genital herpes. Infect Dis Clin Pract 6:S12, 1997. 305a. Sacks SL, Aoki FY, Diaz-Mitoma F et al: Patient-initiated, twice-daily oral famciclovir for early
recurrent genital herpes. A randomized, double-blind multicenter
trial. Canadian Famciclovir Study Group. JAMA 276:44, 1996. 305b. Mertz GJ, Loveless MO, Levin MJ et al: Oral famciclovir for suppression of recurrent genital herpes simplex virus
infection in women. A multicenter, double-blind, placebo-controlled
trial. Collaborative Famciclovir Genital Herpes Research
Group. Arch Intern Med 157:343, 1997. 306. Tyring S, Barbarash RA, Nahlik JE et al: Famciclovir for the treatment of acute herpes zoster: Effects on acute
disease and postherpetic neuralgia. A randomized, double-blind, placebo-controlled
trial. Collaborative Famciclovir Herpes Zoster
Study Group. Ann Intern Med 123:89, 1995. 307. Dworkin RH, Boon RJ, Griffin DRJ, Phung D: Postherpetic neuralgia: impact of famciclovir, age, rash, severity, and
acute pain in herpes zoster patients. J Infect Dis 178:S76, 1998. 308. Spruance RL, Rea TL, Thoming C et al: Penciclovir cream for the treatment of herpes simplex labialis. A randomized, multicenter, double-blind, placebo-controlled trial. JAMA 277:1374, 1997. 309. Kaufman HE, Varnell ED, Thompson HW: Trifluridine, cidofovir, and penciclovir in the treatment of experimental
herpetic keratitis. Arch Ophthalmol 116:777, 1998. 310. Loutsch JM, Sainz B Jr, Marquart ME et al: Effect of famciclovir on herpes simplex virus type 1 corneal disease and
establishment of latency in rabbits. Antimicrob Agents Chemother 45:2044, 2001. 311. Spruance SL, Rowe NH, Raborn GW et al: Peroral famciclovir in the treatment of experimental ultraviolet radiation-induced
herpes simplex labialis: A double blind, dose-ranging, placebo-controlled, multicenter trial. J Infect Dis 179:303, 1999. 312. Diaz-Mitoma F, Sibbald RG, Shafran SD et al: Oral famciclovir for the suppression of recurrent genital herpes: A randomized
controlled trial. JAMA 280:887, 1998. 313. Cole NL, Balfour HH: In vitro susceptibility of cytomegalovirus isolates from immunosuppressed
patients to acyclovir and ganciclovir. Diagn Microbiol Infect Dis 6:255, 1987. 314. Mar E-C, Cheng Y-C, Huang E-S: Effect of 9-(1$PI$XC3 dihydroxy-2-propoxymethyl) guanine
on human cytomegalovirus replication in vivo. Antimicrob Agents Chemother 24:518, 1983. 315. Matthews T, Boehme R: Antiviral activity and mechanism of action of ganciclovir. Rev Infect Dis 10:S490, 1988. 316. Smee DF, Martin JC, Verheyden JPH et al: Anti-herpesvirus activity of the acyclic nucleoside 9-(1$PI$XC3-dihydroxy-2-propoxymethyl) guanine. Antimicrob Agents Chemother 23:676, 1983. 317. Tocci MJ, Livelli TJ, Perry HC et al: Effects of the nucleoside analog 2'-nor-2'-deoxyguanosine
on human cytomegalovirus replication. Antimicrob Agents Chemother 25:247, 1984. 318. Crumpacker CS: Ganciclovir. N Engl J Med 355:721, 1996. 319. Noble S, Faulds D: Ganciclovir. An update of its use in the prevention of cytomegalovirus
infection and disease in transplant recipients. Drugs 56:115, 1998. 320. Mar E-C, Chiou J-F, Cheng Y-C et al: Inhibition of cellular DNA polymerase alpha and human cytomegalovirus-induced
DNA polymerase by the triphosphate of 9-(2-hydroxyethoxymethyl) guanine and 9-(1$PI$XC3-dihydroxy-2-propoxymethyl) guanine. J Virol 53:776, 1985. 321. Jacobson MA, DeMiranda P, Cederberg DM et al: Human pharmacokinetics and tolerance of oral ganciclovir. Antimicrob Agents Chemother 31:1251, 1987. 322. Anderson RD, Griffey KG, Jung D et al: Ganciclovir absolute bioavailability and steady-state pharmacokinetics
after oral administration of two 3,000 mg/d dosing regimens in
human immunodeficiency virus- and cytomegalovirus-seropositive
patients. Clin Ther 17:425, 1995. 323. Lalezari JP, Friedberg DN, Bisset J et al: High dose oral ganciclovir treatment for cytomegalovirus retinitis. J Clin Virol 24:67, 2002. 324. Spector SA, Busch DF, Follansbee S et al: Pharmacokinetics, safety, and antiviral profiles of oral ganciclovir in
persons infected with human immunodeficiency virus: A phase I/II study. J Infect Dis 171:1431, 1995. 325. Jacobson MA: Ganciclovir therapy for opportunistic cytomegalovirus disease in AIDS. In Volberding P, Jacobson MA (eds): AIDS Clinical Review 1990. New York, Marcel Dekker, 1990. 326. Laskin OL, Stahl-Bayliss CM, Kalman CM et al: Use of ganciclovir to treat serious cytomegalovirus infections in patients
with AIDS. J Infect Dis 155:323, 1987. 327. Plotkin SA, Drew WL, Felsenstein D et al: Sensitivity of clinical isolates of human cytomegalovirus to 9-(1$PI$XC3-dihydroxy-2-propoxymethyl) guanine. J Infect Dis 152:833, 1985. 328. Sommadossi JP, Bevan R, Ling T et al: Clinical pharmacokinetics of ganciclovir in patients with normal and impaired
renal function. Rev Infect Dis 10:S507, 1988, 329. Jabs DA, Wingard JR, deBustros S et al: BW B759U for cytomegalovirus retinitis: Intraocular drug penetration. Arch Ophthalmol 107:1436, 1986. 330. Arevalo JF, Gonzalez C, Capparelli EV et al: Intravitreous and plasma concentrations of ganciclovir and foscarnet after
intravenous therapy in patients with AIDS and cytomegalovirus retinitis. J Infect Dis 172:951, 1995. 331. Buhles WC Jr, Mastre BJ, Tinker AJ et al: Ganciclovir treatment of life or sight-threatening cytomegalovirus
infection: Experience in 314 immunocompromised patients. Rev Infect Dis 10:S495, 1988. 332. Davis CL, Springmeyer S, Gmerek BJ: Central nervous system side effects of ganciclovir. N Engl J Med 322:933, 1990. 333. Shea BF, Hoffman S, Sesin GP et al: Ganciclovir hepatotoxicity. Pharmacotherapy 1545:223, 1987. 334. Dennehy PJ, Warman R, Flynn JT et al: Ocular manifestations in pediatric patients with acquired immunodeficiency
syndrome. Arch Ophthalmol 107:978, 1989. 335. Gudnason T, Belani KK, Balfour HH Jr: Ganciclovir treatment of cytomegalovirus disease in immunocompromised children. Pediatr Infect Dis J 8:436, 1989. 336. Markham A, Faulds D: Ganciclovir. An update of its therapeutic use in cytomegalovirus infection. Drugs 48:455, 1994. 337. Erice A: Resistance of human cytomegalovirus to antiviral drugs. Clin Microbiol Rev 12:286, 1999. 338. Smith IL, Cherrington JM, Jiles RE et al: High-level resistance of cytomegalovirus to ganciclovir is associated
with alterations in both the UL97 and DNA polymerase genes. J Infect Dis 178:1821, 1998. 339. Dieterich DT, Kotler DP, Busch DF et al: Ganciclovir treatment of cytomegalovirus colitis in AIDS: A randomized, double-blind, placebo-controlled multicenter study. J Infect Dis 167:278, 1993 340. Lastin OL, Cederberg DM, Mills J et al: Ganciclovir for the treatment and suppression of serious infections caused
by cytomegalovirus. Am J Med 83:201, 1987. 341. Fiala M, Cone LA, Cohen, et al: Response to neurologic complications of AIDS to 3'-azido-3'-deoxythymidine and 9-(1$PI$XC3-dihydroxy-2-propoxymethyl) guanine: I. Clinical features. Rev Infect Dis 10:250, 1988. 342. Merigan TC, Renlund DG, Keay S et al: A controlled trial of ganciclovir to prevent cytomegalovirus disease after
heart transplantation. N Engl J Med 326:1182, 1992. 343. Goodrich JM, Mori M, Gleaves CA et al: Early treatment with ganciclovir to prevent cytomegalovirus disease after
allogenic bone marrow transplantation. N Engl J Med 325:1601, 1991. 344. Schmidt GM, Horak DA, Niland JC et al: A randomized, controlled trial of prophylactic ganciclovir for cytomegalovirus
pulmonary infection in recipients of allogeneic bone marrow transplants. N Engl J Med 324:1005, 1991. 345. Shepp DH, Dandliker PS, deMiranda P et al: Activity of 9-[2-hydroxy-1-(hydroxymethyl) ethoxymethyl] guanine in the treatment of cytomegalovirus
pneumonia. Ann Intern Med 103:368, 1985. 346. Reed EC, Dandliker PS, Meyers JD: Treatment of cytomegalovirus pneumonia with 9-[2-hydroxy-1-(hydroxymethyl) ethoxymethyl] guanine
and high dose corticosteroids. Ann Intern Med 105:214, 1986. 347. Emanuel DI, Cunningham K, Jules-Elysee K et al: Cytomegalovirus pneumonia after bone-marrow transplantation successfully
treated with the combination of ganciclovir and high dose intravenous
immune globulin. Ann Intern Med 109:777, 1988. 348. Schmidt GM, Forman SJ, Zaia JA et al: Treatment of cytomegalovirus associated pneumonitis with ganciclovir (DHPG) and
immunoglobulin following allogeneic bone marrow transplantation: Antiviral
and therapeutic response. Clin Res 36:470A, 1988. 349. Reed EC, Bowden RA, Dandliker PS et al: Treatment of cytomegalovirus pneumonia with ganciclovir and intravenous
cytomegalovirus immunoglobulin in patients with bone marrow transplants. Ann Intern Med 109:783, 1988. 350. Erice A, Jordan C, Chace BA et al: Ganciclovir treatment of cytomegalovirus disease in transplant recipients
and other immunocompromised hosts. JAMA 257:3082, 1987. 351. Hecht DW, Snydman DR, Crumpacker CS et al: Ganciclovir for treatment of renal transplant-associated primary
cytomegalovirus pneumonia. J Infect Dis 157:187, 1988. 352. Keay S, Bissett J, Merigan TC: Ganciclovir treatment of cytomegalovirus infection in iatrogenically immunocompromised
patients. J Infect Dis 156:1016, 1987. 353. Couchoud C: Cytomegalovirus prophylaxis with antiviral agents for solid organ transplantation. Cochrane Database Syst Rev CD001320, 2000. 354. Gourishankar S, Wong W, Dorval M: Meta-analysis of prophylaxis of CMV disease in solid organ transplantation: is
Ganciclovir a superior agent to Acyclovir?Transplant Proc 33:1870, 2001. 354a. Koetz AC, Delbruch R, Meyer-Koenig U et al: CMV pp 65 antigen guided preemptive ganciclovir therapy in solid transplant
recipients: A prospective, double-blind and placebo-controlled
study. Transplantation 72:1325, 2001. 355. Muheim C, Vogel G, Seydoux C et al: Determinants of protracted cytomegalovirus infection in solid-organ
transplant patients. Transplantation 74:226, 2002. 355a. Badley AD, Seaberg EC, Porayko MK et al: Prophylaxis of cytomegalovirus infection in liver transplantation: a randomized
trial comparing a combination of ganciclovir and acyclovir to
acyclovir. NIDDK Liver transplantation Database. Transplantation 64:55, 1997. 355b. Grennan DC, Garlock KA, Singer GG et al: Prophylactic oral ganciclovir compared with deferred therapy for control
of cytomegalovirus in renal transplant recipients. Transplantation 64:1843, 1997. 355c. Rubin RH, Kemmerly SA, Conti D et al: Prevention of primary cytomegalovirus disease in organ transplant recipients
with oral ganciclovir or oral acyclovir prophylaxis. Transpl Infect Dis 2:112, 2000. 356. Shuman JS, Orellana J, Friedman AH et al: Acquired immunodeficiency syndrome (AIDS). Surv Ophthalmol 31:384, 1987. 357. Jabs DA: Ocular manifestations of HIV infection. Trans Am Ophthalmol Soc 93:623, 1995. 358. Hoover DR, Peng Y, Saah A et al: Occurrence of cytomegalovirus retinitis after human immunodeficiency virus
immunosuppression. Arch Ophthalmol 114:821, 1996. 359. Jabs DA, Enger C, Bartlett JG: Cytomegalovirus retinitis and acquired immunodeficiency syndrome. Arch Ophthalmol 107:75, 1989. 360. Sison RF, Holland GN, MacArthur LJ et al: Cytomegalovirus retinopathy as the initial manifestation of the acquired
immunodeficiency syndrome. Am J Ophthalmol 112:243, 1991. 361. Spector SA, McKinley C, Lalezari JP et al: Oval ganciclovir for the prevention of cytomegalovirus disease in persons
with AIDS. N Engl J Med 334:1491, 1996. 362. Henderly DE, Freeman WR, Causey DM et al: Cytomegalovirus retinitis and response to therapy with ganciclovir. Ophthalmology 94:425, 1987. 363. Holland GN, Sidikaro Y, Kreiger EA et al: Treatment of cytomegalovirus retinopathy with ganciclovir. Ophthalmology 94:815, 1987. 364. Masur H, Lane HC, Palestine A et al: Effect of 9(1$PI$XC3 dihydroxy-2-propoxymethyl) guanine
on serious cytomegalovirus disease in eight immunosuppressed
homosexual men. Ann Intern Med 104:41, 1986. 365. Orellana J, Teich SA, Friedman AH et al: Combined short- and long-term therapy of cytomegalovirus
retinitis using ganciclovir (BWB759U). Ophthalmology 94:831, 1987. 366. Palestine AG, Stevens G, Lane HC et al: Treatment of cytomegalovirus retinitis with dihydroxy propoxymethyl guanine. Am J Ophthalmol 101:95, 1986. 367. Rosecan LR, Stahl-Bayliss CM, Kalman CM et al: Antiviral therapy for cytomegalovirus retinitis in AIDS with dihydroxy
propoxymethyl guanine. Am J Ophthalmol 101:405, 1986. 368. Holland GN, Buhles WC Jr, Mastre B et al: A controlled retrospective study of ganciclovir treatment for cytomegalovirus
retinopathy: Use of a standardized system for the assessment of
disease outcome. Arch Ophthalmol 107:1759, 1989. 368a. Spector SA, Weingeist T, Pollard RB et al: A randomized, controlled study of intravenous ganciclovir therapy for cytomegalovirus
retinitis in patients with AIDS. AIDS Clinical Trials Group
and Cytomegalovirus Cooperative Study Group. J Infect Dis 168:447, 1993. 369. Gross JG, Bozzette SA, Mathews WC et al: Longitudinal study of cytomegalovirus retinitis in acquired immune deficiency
syndrome. Ophthalmology 97:681, 1990. 370. Studies of the Ocular Complications of AIDS Research Group, in collaboration
with the AIDS Clinical Trials Group: Foscarnet-ganciclovir
cytomegalovirus retinitis trial 4: Visual outcomes. Ophthalmology 101:1250, 1994. 371. Studies of Ocular Complications of AIDS Research Group in collaboration
with the AIDS Clinical Trials Group: Clinical vs photographic assessment
of cytomegalovirus retinitis. Foscarnet-Ganciclovir Cytomegalovirus
Retinitis Trial Report 8. Arch Ophthalmol 144:848, 1996. 372. D'Amico DJ, Talamo JH, Feldstein D et al: Ophthalmoscopic and histologic findings in cytomegalovirus retinitis treated
with BW-B759U. Arch Ophthalmol 104:1788, 1986. 373. Pepose JS, Newman C, Bach MC et al: Pathologic features of cytomegalovirus retinopathy after treatment with
the antiviral agent ganciclovir. Ophthalmology 94:414, 1987. 374. Teich SA, Castle J, Friedman AH et al: Active cytomegalovirus particles in the eyes of an AIDS patient being treated
with 9-[2-hydroxy-1-(hydroxymethyl) ethoxymethyl] guanine (ganciclovir). Br J Ophthalmol 72:293, 1988. 375. Jacobson MA, O'Donnell JJ, Brodie HR et al: Randomized prospective trial of ganciclovir maintenance therapy for cytomegalovirus
retinitis. J Med Virol 25:339, 1988. 375a. Kupperman BD, Quiceno JI, Flores-Aguilar M et al: Intravitreal ganciclovir concentration after intravenous administration
in AIDS patients with cytomegalovirus retinitis: Implications for therapy. J Infect Dis 168:1506, 1993. 376. Drew WL, Miner RC, Bush DF et al: Prevalence of resistance in patients receiving ganciclovir for serious
cytomegalovirus infection. J Infect Dis 163:716, 1991. 377. Jabs DA, Enger C, Dunn JP et al for the Cytomegalovirus Retinitis and Viral Resistance Study Group: Cytomegalovirus resistance and viral resistance: 4. Ganciclovir resistance. J Infect Dis 177:770, 1998. 378. Jabs DA, Martin BK, Forman MS et al: Cytomegalovirus resistance to ganciclovir and clinical outcomes of patients
with cytomegalovirus retinitis. Am J Ophthalmol 135:26, 2003. 379. The Studies of Ocular Complications of AIDS (SOCA) Research Group
in collaboration with the AIDS Clinical Trials Group (ACTG): Rhegmatogenous
retinal detachment in patients with cytomegalovirus
retinitis: The Foscarnet-Ganciclovir Cytomegalovirus Retinitis
Trial. Am J Ophthalmol 124:61, 1997. 380. Kempen JH, Jabs DA, Dunn JP et al: Retinal detachment risk in cytomegalovirus retinitis related to the acquired
immune deficiency syndrome. Arch Ophthalmol 119:33, 2001. 381. Studies of Ocular Complications of AIDS Research Group in collaboration
with the AIDS Clinical Trials Group: Morbidity and toxic effects associated
with ganciclovir or foscarnet in a randomized cytomegalovirus retinitis
trial. Arch Intern Med 155:65, 1995. 382. Michael D, Min Y-I, Holbrook JT et al: Use of filgrastim as adjuvant therapy in patients with AIDS-related
cytomegalovirus retinitis. AIDS 16:757, 2002. 382a. Studies of the Ocular Complications of AIDS (SOCA) Research Group
in Collaboration with the AIDS Clinical Trials Group: Combination
foscarnet and ganciclovir therapy vs. monotherapy for the treatment of
relapsed cytomegalovirus retinitis in patients with AIDS. Arch Ophthalmol 114:23, 1996. 383. Stanley HD, Charlebois E, Harb G et al: Central venous catheter infections in AIDS patients receiving treatment
for cytomegalovirus disease. J Acquir Immune Defic Syndrome 7:272, 1994. 384. Drew WL, Ives D, Lalezari JP et al: Oral ganciclovir as maintenance treatment for cytomegalovirus retinitis
in patients with AIDS. N Engl J Med 333:615, 1995. 385. The Oral Ganciclovir European and Australian Cooperative Study Group: Intravenous
versus oral ganciclovir: European/Australian comparative study
of efficacy and safety in the prevention of cytomegalovirus retinitis
recurrence in patients with AIDS. AIDS 9:471, 1995. 386. Holland GN, Tufail A: New therapies for cytomegalovirus retinitis. N Engl J Med 333:658, 1995. 387. Martin DF, Kuppermann BD, Wolitz RA et al: Oral ganciclovir for patients with cytomegalovirus retinitis treated with
a ganciclovir implant. N Engl J Med 340:1063, 1999 388. Brosgart CL, Louis TA, Hillman DW et al: A randomized, placebo-controlled trial of the safety and efficacy
of oral ganciclovir for prophylaxis of cytomegalovirus disease in HIV-infected
individuals. AIDS 12:269, 1998. 389. Cheung TW, Teich SA: Cytomegalovirus infection in patients with HIV infection. Mount Sinai J Med 66:113, 1999. 390. Spector SA, Wong R, Hsia K et al: Plasma cytomegalovirus DNA load predicts CMV disease and survival in AIDS
patients. J Clin Invest 101:497, 1998. 391. CDC: Guidelines for preventing opportunistic infections among HIV-infected
persons—2002. Recommendations of the U.S. Public Health
Service and the Infectious Diseases Society of America. MMWR Morb Mortal Wkly Rep 51(No. RR-8):1, 2002. 392. Smith TJ, Pearson PA, Blandford DL et al: Intravitreal sustained-release ganciclovir. Arch Ophthalmol 110:255, 1992. 392a. Musch DC, Martin DF, Gordon JF et al: Treatment of AIDS related CMV retinitis with a sustained-release
ganciclovir implant. N Engl J Med 337:83, 1997. 392b. Martin DF, Parks DJ, Mellow SD et al: Treatment of cytomegalovirus retinitis with an intraocular sustained-release
ganciclovir implant. A randomized controlled clinical trial. Arch Ophthalmol 112:1531, 1994. 393. Martin DF, Dunn JP, Davis JL et al: Use of the ganciclovir implant for the treatment of cytomegalovirus retinitis
in the era of potent antiretroviral therapy: recommendations of
the international AIDS society—USA Panel. Am J Ophthalmol 127:329, 1999. 394. Perkins SL, Chang-Hao Y, Ashton PA et al: Pharmacokinetics of the ganciclovir implant in the silicone-filled
eye. Retina 21:10, 2001. 395. Martidis A, Danis RP, Ciulla TA: Treating cytomegalovirus retinitis-related retinal detachment by
combining silicone oil tamponade and ganciclovir implant. Ophthalmic Surg Lasers 33:135, 2002 396. Kuo IC, Imoi Y, Shum C et al: Genotypic analysis of cytomegalovirus retinitis poorly responsive to intravenous
ganciclovir but responsive to the ganciclovir implant. Am J Ophthalmol 135:20, 2003. 397. Marx JL, Kapusta MA, Patel SS et al: Use of the ganciclovir implant in the treatment of recurrent cytomegalovirus
retinitis. Arch Ophthalmol 114:815, 1996. 398. Roth DB, Fever WJ, Blenke AJ et al: Treatment of recurrent cytomegalovirus retinitis with the ganciclovir implant. Am J Ophthalmol 127:276, 1999. 399. Hatton MP, Duker JS, Reichel E et al: Treatment of relapsed cytomegalovirus retinitis with the sustained-release
ganciclovir implant. Retina 18:50, 1998. 400. Davis JL, Homayoun T, Feuer WJ et al: Effect of potent antiretroviral therapy on recurrent cytomegalovirus retinitis
treated with the ganciclovir implant. Am J Ophthalmol 127:283, 1999. 401. Lim JI, Wolitz RA, Dowling AH et al: Visual and anatomic outcomes associated with posterior segment complications
after ganciclovir implant procedures in patients with AIDS and cytomegalovirus
retinitis. Am J Ophthalmol 127:288, 1999. 402. Guembol HO, Krieglsteiner S, Rosencranz C et al: Complications after implantation of intraocular devices in patients with
cytomegalovirus retinitis. Graefes Arch Clin Exp Ophthalmol 237:824, 1999. 403. Charles NC, Freisberg L: Endophthalmitis associated with extrusion of a ganciclovir implant. Am J Ophthalmol 133:273, 2002. 404. Gentile RC, Lewis JM, Puklin JE: Cyclodialysis complicating intravitreal ganciclovir implantation. Arch Ophthalmol 115:1204, 1997. 405. Martin DF, Ferris FL, Brothers RJ et al: Retinal detachment in eyes treated with a ganciclovir implant. Arch Ophthalmol 113:1355, 1995. 406. The Studies of Ocular Complications of AIDS Research Group in collaboration
with the AIDS Clinical Trials Group: The ganciclovir implant plus
oral ganciclovir versus parenteral cidofovir for the treatment of cytomegalovirus
retinitis in patients with acquired immunodeficiency syndrome: the
Ganciclovir Cidofovir Cytomegalovirus Retinitis Trial. Am J Ophthalmol 131:457, 2001. 407. Pepose JS, Holland GN, Nestor MS et al: Acquired immune deficiency syndrome: Pathogenic mechanisms of ocular disease. Ophthalmology 92:472, 1985. 408. Palestine AG, Rodrigues MM, Macher AM et al: Ophthalmic involvement in acquired immune deficiency syndrome. Ophthalmology 91:1092, 1984. 409. Skolnick PR, Pomerantz RJ, de al Monte SM et al: Dual infection of retina with human immunodeficiency virus type I and cytomegalovirus. Am J Ophthalmol 107:361, 1989. 410. Holland GN, Sison RF, Jatulis DE et al: Survival of patients with the acquired immune deficiency syndrome after
development of cytomegalovirus retinopathy. Ophthalmology 92:204, 1990. 411. Studies of Ocular Complications of AIDS Research Group in collaboration
with the AIDS Clinical Trials Group: Mortality in patients with the acquired
immunodeficiency syndrome treated with either foscarnet or ganciclovir
for cytomegalovirus retinitis. N Engl J Med 326:213, 1992. 412. Studies of the Ocular Complications of AIDS Research Group in collaboration
with AIDS Clinical Trials Group: Antiviral effects of foscarnet and
ganciclovir therapy on human immunodeficiency virus p24 antigen in
patients with AIDS and cytomegalovirus retinitis. J Infect Dis 172:613, 1995. 413. Henry K, Cantrill H, Fletcher C et al: Use of intravitreal ganciclovir for CMV retinitis in a patient with AIDS. Am J Ophthalmol 103:17, 1987. 414. Cantrill HL, Henry K, Melroe NH et al: Treatment of cytomegalovirus retinitis with intravitreal ganciclovir: Long-term
results. Ophthalmology 96:367, 1989. 415. Heinemann M-H: Staphylococcus epidermidis endophthalmitis complicating intravitreal antiviral
therapy for cytomegalovirus retinitis. Arch Ophthalmol 107:643, 1989. 416. Heinemann MH: Long-term intravitreal ganciclovir therapy for cytomegalovirus retinopathy. Arch Ophthalmol 107:1767, 1989. 417. Ussery F, Gibson S, Conklin R et al: Intravitreal ganciclovir in the treatment of AIDS associated with cytomegalovirus
retinitis. Ophthalmology 95:640, 1988. 418. Hodge WG, LaLonde RG, Sampalis J et al: Once weekly intraocular injection of ganciclovir for maintenance therapy
of cytomegalovirus retinitis: Clinical and ocular outcome. J Infect Dis 174:393, 1996. 419. Yoshizumi MO, Lee D, Vinci V et al: Ocular toxicity of multiple intravitreal DHPG injections. Graefes Arch Clin Exp Ophthalmol 228:350, 1990. 420. Cochereau-Massin I, LeHoang P, Lautier-Frau M et al: Efficacy and tolerance of intravitreal ganciclovir in cytomegalovirus retinitis
in acquired immune deficiency syndrome. Ophthalmology 98:1348, 1991. 421. Young SH, Morlet N, Heery S et al: High dose intravitreal ganciclovir in the treatment of cytomegalovirus
retinitis. Med J Aust 157:370, 1992. 422. Young S, Morlet N, Besen G et al: High dose (2$PI$XC000-microgram) intravitreous ganciclovir
in the treatment of cytomegalovirus retinitis. Ophthalmology 105:404, 1998. 423. Morlet N, Young S, Naidoo D et al: High dose intravitreal ganciclovir injection provides a prolonged therapeutic
intraocular concentration. Br J Ophthalmol 80:214, 1996. 424. Desatnick HR, Foster RE, Lowder CY: Treatment of clinically resistant cytomegalovirus retinitis with combined
intravitreal injections of ganciclovir and foscarnet. Am J Ophthalmol 122:121, 1996. 425. Velez G, Roy CE, Whitcup SM et al: High-dose intravitreal ganciclovir and foscarnet for cytomegalovirus
retinitis. Am J Ophthalmol 131:396, 2001. 426. Saran BR, Maguire AM: Retinal toxicity of high dose intravitreal ganciclovir. Retina 14:248, 1994. 427. Hoh, HB, Hurley C, Claoue C et al: Randomised trial of ganciclovir and acyclovir in the treatment of herpes
simplex dendritic keratitis: A multicentre study. Br J Ophthalmol 80:140, 1996. 428. Colin J, Hoh HB, Easty DL et al: Ganciclovir ophthalmic gel (Virgan; 0.15%) in the treatment
of herpes simplex keratitis. Cornea 16:393, 1997. 429. Curran M, Noble S: Valganciclovir. Drugs 61:1145, 2001. 430. Jung D, Dorr A: Single dose pharmacokinetics of valganciclovir in HIV- and CMV-seropositive
subjects. J Clin Pharmacol 39:800, 1999. 431. Martin DF, Sierra-Madero J, Walmsley S et al: A controlled trial of valganciclovir as induction therapy for cytomegalovirus
retinitis. N Engl J Med 346:1119, 2002. 432. Brown F, Banken L, Saywell K, Arum I: Pharmacokinetics of valganciclovir and ganciclovir following multiple oral
doses of valganciclovir in HIV- and CMV-seropositive
volunteers. Clin Pharmacokinet 37:167, 1999. 433. Segarra-Newnham M, Salazar MI: Valganciclovir: A new oral alternative for cytomegalovirus retinitis in
human immunodeficiency virus-seropositive individuals. Pharmacother 22:1124, 2002. 434. Peskovitz MD, Rabkin J, Merion RM et al: Valganciclovir results in improved oral absorption of ganciclovir in liver
transplant recipients. Antimicrob Agents Chemother 44:2811, 2000. 435. Lalezari J, Lindley J, Walmsley S et al: A safety study of oral valganciclovir maintenance treatment of cytomegalovirus
retinitis. J Acquir Immune Defic Syndr 30:392, 2002. 435a. Nichols WG, Boeckh M: Recent advances in the therapy and prevention of CMV infections. J Clin Virol 16:25, 2000. 436. Boivin G, Gadreau GC, Greenfield I et al: Rate of emergence of cytomegalovirus (CMV) mutations in leukocytes
of patients with acquired immunodeficiency syndrome who are receiving
valganciclovir as induction and maintenance therapy for CMV retinitis. J Infect Dis 184:1598, 2001. 437. Palestine AG, Polis MA, DeSmet MD et al: A randomized controlled trial of foscarnet in the treatment of cytomegalovirus
retinitis in patients with AIDS. Ann Intern Med 115:665, 1991. 438. Balfour HH, Benson C, Braun J et al: Management of acyclovir-resistant herpes simplex and varicella-zoster
infections. J Acquir Immune Defic Syndr 7:254, 1994. 439. Wagstaff AJ, Bryson HM: Foscarnet. A reappraisal of its antiviral activity, pharmacokinetic properties
and therapeutic use in immunocompromised patients with viral infections. Drugs 48:199, 1994. 440. Jacobson MA, Crowe S, Levy J et al: Effect of foscarnet therapy on infection with human immunodeficiency virus
in patients with AIDS. J Infect Dis 158:862, 1988. 441. Oberg B: Antiviral effects of phosphonoformate (PFA foscarnet sodium). Pharmacol Ther 19:387, 1983. 442. Sandstrom EG, Kaplan JC, Byington RE et al: Inhibitor of human T-cell lymphotropic virus type III in vitro by
phosphonoformate. Lancet 1:1480, 1985. 443. Kedes DH, Ganem D: Sensitivity of Kaposi's sarcoma–associated herpes virus replication
to antiviral drugs: Implications for potential therapy. J Clin Invest 99:2082, 1997. 444. Crumpacker CS: Mechanism of action of foscarnet against viral polymerases. Am J Med 92:3S, 1992. 445. Jacobson MA, Drew WL, Feinberg J et al: Foscarnet therapy for ganciclovir-resistant cytomegalovirus retinitis
in patients with AIDS. J Infect Dis 163:1348, 1991. 446. Erice A, Chou S, Biron KK et al: Progressive disease due to ganciclovir-resistant cytomegalovirus
in immunocompromised patients. N Engl J Med 320:289, 1989. 447. LeHoang P, Girard B, Robinet M et al: Foscarnet in the treatment of cytomegalovirus retinitis in acquired immune
deficiency syndrome. Ophthalmology 96:865, 1989. 448. Jacobson MA, O'Donnell JJ, Mills J: Foscarnet treatment of cytomegalovirus retinitis in patients with the acquired
immunodeficiency syndrome. Antimicrob Agents Chemother 33:736, 1989. 449. Sjovall J, Karksson A, Ogenstad S et al: Pharmacokinetics and absorption of foscarnet after intravenous and oral
administration to patients with human immunodeficiency virus. Clin Pharmacol Ther 44:65, 1988. 450. Sjovall J, Bergdahl S, Movin G et al: Pharmacokinetics of foscarnet and distribution to cerebrospinal fluid after
intravenous infusion in patients with human immunodeficiency virus
infection. Antimicrob Agents Chemother 33:1023, 1989. 451. López-Cortés LF, Ruiz-Valderas R, Lucero-Muñoz MJ et al: Intravitreal, retinal, and central nervous system foscarnet concentrations
after rapid intravenous administration to rabbits. Antimicrob Agents Chemother 44:756, 2000. 452. Aweeka F, Gambertoglio J, Mills J et al: Pharmacokinetics of intermittently administered intravenous foscarnet in
the treatment of acquired immunodeficiency syndrome patients with serious
cytomegalovirus retinitis. Antimicrob Agents Chemother 33:742, 1989. 453. Ringden O, Lonnqvist B, Paulin T et al: Pharmacokinetics, safety and preliminary clinical experiences using foscarnet
in the treatment of cytomegalovirus infection in allograft recipients. Scand J Infect Dis 17:373, 1986. 454. Noormohamed FH, Youle MS, Higgs CJ et al: Pharmacokinetics and absolute bioavailability of oral foscarnet in human
immunodeficiency virus-seropositive patients. Antimicrob Agents Chemother 42:293, 1998. 455. Deray G, Katlama C, Dohen E: Prevention of foscarnet nephrotoxicity. Ann Intern Med 113:332, 1990. 456. Fanning MM, Read SE, Bendon M et al: Foscarnet therapy of cytomegalovirus retinitis in AIDS. J AIDS 3:472, 1990. 457. MacGregor RR, Graziani AL, Weiss R et al: Successful foscarnet therapy for cytomegalovirus retinitis in an AIDS patient
undergoing hemodialysis: Rationale for empiric dosing and plasma
level monitoring. J Infect Dis 164:785, 1991. 458. Seidel EA, Koenig S, Polis MA: A dose escalation study to determine the toxicity and maximally tolerated
dose of foscarnet. AIDS 7:941, 1993. 459. Jacobson MA, Causey D, Polsky B et al: A dose-ranging study of daily maintenance intravenous foscarnet
therapy for cytomegalovirus retinitis in AIDS. J Infect Dis 168:444, 1993. 460. Jayaweera DT: Minimising the dosage-limiting toxicities of foscarnet induction
therapy. Drug Saf 16:258, 1997. 461. Jacobson MA, Gambertoglio JG, Aweeka FT et al: Foscarnet-induced hypocalcemia and effect of foscarnet on calcium
metabolism. J Clin Endocrinol Metab 72:1130, 1991. 462. Youle MS, Clarbour J, Gazzard B et al: Severe hypocalcemia in AIDS patients treated with foscarnet and pentamidine. Lancet 1:1455, 1988. 463. Yusufi ANR, Sczczepanska-Konkel M, Kempson SA et al: Inhibition of human renal epithelial sodium phosphate co-transport
by phosphonoformic acid. Biochem Biophys Res Comm 139:670, 1986. 464. Jacobson MA, Wulfsohn M, Feinberg JE et al: Phase II dose-ranging trial of foscarnet salvage therapy for cytomegalovirus
retinitis in AIDS patients intolerant of or resistant to
ganciclovir (ACTG Protocol 093). AIDS 8:451, 1994. 465. Farthing CI, Dalgleish AG, Clark A et al: Phosphonoformate (foscarnet): A pilot study in AIDS and AIDS
related complex. AIDS 1:21, 1987. 466. Walmsley SL, Chew E, Read SE et al: Treatment of cytomegalovirus retinitis with trisodium phosphonoformate
hexahydrate (Foscarnet). J Infect Dis 157:569, 1988. 467. Farese RV Jr, Schambelan M, Hollander H et al: Nephrogenic diabetes insipidus associated with foscarnet treatment of cytomegalovirus
retinitis. Ann Intern Med 112:955, 1990. 468. Gilquin J, Weiss L, Kazatchkinl MD: Genital and oral erosions induced by foscarnet. Lancet 335:287, 1990. 469. Van der Pijl JW, Frisser PHJ, Reiss P et al: Foscarnet and penile ulceration. Lancet 335:286, 1990. 470. Safrin S, Kemmerly S, Plotkin B et al: Foscarnet-resistant herpes simplex virus infection in patients with
AIDS. J Infect Dis 169:193, 1994. 470a. Jabs DA, Enger C, Forman M, Dunn JP: Incidence of foscarnet resistance and cidofovir resistance in patients
treated for cytomegalovirus retinitis. The Cytomegalovirus Retinitis and
Viral Resistance Study Group. Antimicrob Agents Chemother 42:2240, 1998. 471. Safrin S, Grumpacker C, Chatis P et al: A controlled trial comparing foscarnet with vidarabine for acyclovir-resistant
mucocutaneous herpes simplex in the acquired immunodeficiency
syndrome. N Engl J Med 325:551, 1991, 472. Apperly JF, Marcus RE, Goldman JM et al: Foscarnet for cytomegalovirus pneumonitis [Letter]. Lancet 1:1151, 1985. 473. Klintmalm G, Lonnqvist B, Oberg B et al: Intravenous foscarnet for the treatment of severe cytomegalovirus infection
in allograft recipients. Scand J Infect Dis 17:157, 1985. 474. Nelson MR, Connolly GM, Hawkins DA et al: Foscarnet in the treatment of cytomegalovirus infection of the esophagus
and colon in patients with the acquired immune deficiency syndrome. Am J Gastroenterol 86:876, 1991. 475. Dieterich DT, Poles MA, Dicker M et al: Foscarnet treatment of cytomegalovirus gastrointestinal infections in acquired
immunodeficiency syndrome patients who have failed ganciclovir
induction. Am J Gastroenterol 88:542, 1993. 476. Jacobson MA, Van der Horst C, Causey DM et al: In vivo additive antiretroviral effect of combined zidovudine and foscarnet
therapy for human immunodeficiency virus infection (ACTG Protocol 053). J Infect Dis 163:1219, 1991. 477. Bergdahl S, Jacobsson B, Moberg L et al: Pronounced anti-HIV-1 activity of foscarnet in patients without
cytomegalovirus infection. J Acquir Immune Defic Syndr Hum Retrovirol 18:51, 1998. 478. Glesby MJ, Hoover DR, Weng S et al: Use of antiherpes drugs and the risk of Kaposi's sarcoma: Data from
the Multicenter AIDS Cohort Study. J Infect Dis 173:1477, 1996. 479. Holland GN, Levinson RD, Jacobson MA,The AIDS Clinical Trials Group Protocol 915 Team: Dose-related difference
in progression rates of cytomegalovirus retinopathy during foscarnet
maintenance. Am J Ophthalmol 119:576, 1995. 480. Freitas VR, Fraser-Smith EB, Matthews TR: Increased efficacy of ganciclovir in combination with foscarnet against
cytomegalovirus and herpes simplex virus type 2 in vitro and in vivo. Antiviral Res 12:205, 1989. 481. Manischewitz JF, Quinnan GV Jr, Lane HC et al: Synergistic effect of ganciclovir and foscarnet on cytomegalovirus replication
in vitro. Antimicrob Agents Chemother 34:373, 1990. 482. Diaz-Lopez M, Chipont E, Sanchez S et al: Intravitreal foscarnet for cytomegalovirus retinitis in a patient with
acquired immunodeficiency syndrome. Am J Ophthalmol 114:742, 1992. 483. Diaz-Lopez, España E, Muñoz G et al: High dose intravitreal foscarnet in the treatment of cytomegalovirus retinitis
in AIDS. Br J Ophthalmol 78:120, 1994. 484. Pearson PA, Jaffe GJ, Ashton P: Intravitreal foscarnet for cytomegalovirus retinitis in a patient with
acquired immunodeficiency syndrome [Letter]. Am J Ophthalmol 115:686, 1993. 485. Hitchcock MJM, Jaffe HS, Martin JC et al: Cidofovir, a new agent with potent anti-herpes virus activity. Antiviral Chem Chemother 7:115, 1996. 486. Bronson JJ, Ghazzouli I, Hitchcock MJM et al: Synthesis and antiviral activity of the nucleotide analogue (s)-1-[3-hydroxy-2-(phosphonylmethoxy) propyl]cytosine. J Med Chem 32:1457, 1989. 487. Safrin S, Cherrington JM, Jaffe HS: Clinical uses of cidofovir. Rev Med Virol 7:145, 1997. 488. Ho HT, Woods KL, Bronson JJ et al: Intracellular metabolism of the anti herpes agent (s)-1-[3-hydroxy-2-(phosphonylmethoxy) propyl] cytosine. J Med Chem 32:1457, 1989. 489. Plosker GL, Noble S: Cidofovir. A review of its use in cytomegalovirus retinitis in patients
with AIDS. Drugs 58:325, 1999. 490. Mulato MS, Cherrington JM, Chen MS: Anti-HCMV activity of cidofovir in combination with antiviral compounds
and immunosuppressive agents: in vitro analyses. Antiviral Chem Chemother 7:203, 1996. 491. Kundy KC: Clinical pharmacokinetics of the antiviral nucleotide analogues cidofovir
and adefovir. Clin Pharmacokinet 36:127, 1999. 492. Polis MA, Spooner KM, Baird BF et al: Anticytomegaloviral activity and safety of cidofovir in patients with human
immunodeficiency virus infection and cytomegalovirus viruria. Antimicrob Agents Chemother 39:882, 1995. 493. Lalezari JP, Stagg RJ, Kuppermann BD et al. Intravenous cidofovir for peripheral cytomegalovirus retinitis in patients
with AIDS. Ann Intern Med 126:257,1997. 494. Studies of Ocular Complications of AIDS Research Group in collaboration
with the AIDS Clinical Trials Group: Parenteral cidofovir for cytomegalovirus
retinitis in patients with AIDS: The HPMPC Peripheral Cytomegalovirus
Retinitis Trial. Ann Intern Med 126:264, 1997. 495. LaLezari JP, Holland GN, Kramer F et al: Randomized, controlled study of the safety and efficacy of intravenous
cidofovir for the treatment of relapsing cytomegalovirus retinitis in
patients with AIDS. J Acquir Immune Defic Syndr Hum Retrovirol 17:339, 1998. 496. The Studies of Ocular Complications of AIDS Research Group in collaboration
with the AIDS Clinical Trial Group: Long-term follow-up
of patients with AIDS treated with parenteral cidofovir for cytomegalovirus
retinitis: The HPMPC Peripheral Cytomegalovirus Retinitis Trial. AIDS 14:1671, 2000. 497. Davis JL, Taskintuna I, Freeman WR et al: Iritis and hypotony after treatment with intravenous cidofovir for cytomegalovirus
retinitis. Arch Ophthalmol 115:733, 1997. 498. Akler ME, Johnson DW, Burman WJ et al: Anterior uveitis and hypotony after intravenous cidofovir for the treatment
of cytomegalovirus retinitis. Ophthalmology 105:651, 1998. 499. Ambati J, Wynne KB, Angerame MC et al: Anterior uveitis associated with intravenous cidofovir use in patients
with cytomegalovirus retinitis. Br J Ophthalmol 83:1153, 1999. 500. Kirsch LS, Arevalo JF, Chavez de la Paz E et al: Intravitreal cidofovir treatment of cytomegalovirus retinitis in patients
with acquired immune deficiency syndrome. Ophthalmology 103:533, 1995. 501. Friedberg DN: Hypotony and visual loss with intravenous cidofovir treatment of cytomegalovirus
retinitis. Arch Ophthalmol 115:801, 1997. 502. Banker AS, Arevalo JF, Munguia D et al: Intraocular pressure and aqueous humor dynamics in patients with AIDS treated
with intravitreal cidofovir (HPMPC) for cytomegalovirus
retinitis. Am J Ophthalmol 124:168, 1997. 503. Taskintuna I, Rahhal FM, Rao NA et al: Adverse events and autopsy findings after intravitreous cidofovir (HPMPC) therapy
in patients with acquired immune deficiency syndrome (AIDS). Ophthalmology 104:1827, 1997. 504. Lea AP, Bryson HM: Cidofovir. Drugs 52:225, 1996. 505. Anonymous: Drugs for non-HIV viral infections. Medical Letter 44:9, 2002. 506. Anonymous: Smallpox vaccine. Medical Letter 45:1, 2003. 507. Jacobson MA, Wilson S, Stanley H et al: Phase I Study of combination therapy with intravenous cidofovir and oral
ganciclovir for cytomegalovirus retinitis in patients with AIDS. J Infect Dis 28:528, 1999. 508. The Studies of Ocular Complications of AIDS Research Group in collaboration
with the AIDS Clinical Trials Group: MSL-109 adjuvant therapy
for cytomegalovirus retinitis in patients with acquired immunodeficiency
syndrome. The Monoclonal Antibody Cytomegalovirus Retinitis Trial. Arch Ophthalmol 115:1527, 1997. 509. Rahhal FM, Arevalo JF, Munguia D et al: Intravitreal cidofovir for the maintenance treatment of cytomegalovirus
retinitis. Ophthalmology 103:1078, 1996. 510. Taskintuna I, Rahhal FM, Arevalo JF et al: Low-dose intravitreal cidofovir (HPMPC) therapy of cytomegalovirus
retinitis in patients with acquired immune deficiency syndrome. Ophthalmology 104:1049, 1997. 511. Kupperman BD, Wolitz R, Stagg R et al: A Phase II randomized, double-masked study of intraocular cidofovir
for relapsing cytomegalovirus retinitis (CMV-R) in
patients with AIDS [abstract]. Vitreous Society Annual Meeting, Cancun, Mexico, Dec. 9–12, 1996. 512. Gordon YJ, Romanowski E, Aravullo-Cruz T et al: Inhibitory effect of (s)-HPMPC, (s)-HPMPA, and 2' nor-cyclic GMP on clinical ocular adenoviral
isolates is serotype-dependent in vitro. Antiviral Res 16:11, 1991. 513. Hillenkamp J, Reinhard T, Ross RS et al: The effects of cidofovir 1% with and without cyclosporin A 1% as
a topical treatment of acute adenoviral keratoconjunctivitis. A
controlled clinical pilot study. Ophthalmology 109:845, 2002. 514. Hillenkamp J, Reinhard T, Ross RS et al: Topical treatment of acute adenoviral keratoconjunctivitis with 0.2% cidofovir
and 1% cyclosporine. A controlled clinical pilot
study. Arch Ophthalmol 119:1487, 2001. 515. Geary RS, Henry SP, Grillone LR: Fomivirsen. Clinical pharmacology and potential drug interactions. Clin Pharmacol 41:255, 2002. 516. Perry CM, Balfour JAB: Fomivirsen. Drugs 57:375, 1999. 517. Freeman WR: Retinal toxic effects associated with intravitreal fomivirsen. Arch Ophthalmol 119:458, 2001. 518. Jabs DA, Griffiths PD: Fomivirsen for the treatment of cytomegalovirus retinitis. Am J Ophthalmol 133:552, 2002. 519. Azad RF, Driver VB, Tanaka K: Antiviral activity of a phosphorothioate oligonucleotide complementary
to RNA of the human cytomegalovirus major immediate-early region. Antimicrob Agents Chemother 37:1945, 1993. 520. Anderson KP, Fox, MC, Brown-Driver V et al: Inhibition of human cytomegalovirus immediate-early gene expression
by an antisense oligonucleotide complementary to immediate-early
RNA. Antimicrob Agents Chemother 40:2004, 1996. 521. Azad R, Brown-Driver V, Buckheit RW Jr et al: Antiviral activity of a phosphorothioate oligonucleotide complementary
to human cytomegalovirus RNA when used in combination with antiviral nucleoside
analogs. Antiviral Res 28:101, 1995. 522. Flores-Aguilar M, Besen G, Vyong C et al: Evaluation of retinal toxicity and efficacy of anti-cytomegalovirus
and anti-herpes simplex virus antiviral phosphorothioate oligonucleotides
ISIS 2922 and ISIS 4015. J Infect Dis 6:1308, 1997. 523. Benjamin M, Lieberman RM, Goldstein DA et al: A pharmacokinetic study of intravitreal fomivirsen (Vitravene™) in
patients with CMVR. ARVO Annual Meeting, May 9–14, 1999. Fort Lauderdale: 10VS 40:5874, 1999. 524. Leeds JM, Henry SP, Truong L et al: Pharmacokinetics of a potential human cytomegalovirus therapeutic, a phosphorothoriate
oligonucleotide after intravitreal injection in the rabbit. Drug Metab Dispos 25:921, 1997. 525. The Vitravene Study Group: The safety of intravitreous fomivirsen for treatment
of cytomegalovirus retinitis in patients with AIDS. Am J Ophthalmol 133:484, 2002. 526. Amin HI, Ai E, McDonald HR: Retinal toxic effects associated with intravitreal fomivirsen. Arch Ophthalmol 118:426, 2000. 527. Uwaydat SH, Li HK: Pigmentary retinopathy associated with intravitreal fomivirsen. Arch Ophthalmol 120:854, 2002. 528. Zambarakji HJ, Mitchell SM, Lightman S et al: Electrophysiological abnormalities following intravitreal vitravene (ISIS 2922) in
two patients with CMV retinitis. Br J Ophthalmol 85:1142, 2001. 529. Stone TW, Jaffe GJ: Reversible bull's-eye maculopathy associated with intravitreal
fomivirsen therapy for cytomegalovirus retinitis. Am J Ophthalmol 130:242, 2000. 530. The Vitravene Study Group: A randomized controlled trial of intravenous
fomivirsen for treatment of newly diagnosed peripheral cytomegalovirus
retinitis in patients with AIDS. Am J Ophthalmol 133:467, 2002. 531. The Vitravene Study Group: Randomized dose-comparison studies of
intravitreous fomivirsen for treatment of cytomegalovirus retinitis that
has reactivated or is persistently active despite other therapies
in patients with AIDS. Am J Ophthalmol 133:475, 2002. 532. Palella FJ Jr, Delaney KM, Moorman AC et al: Declining morbidity and mortality among patients with advanced human immunodeficiency
virus infection. N Engl J Med 338:853, 1998. 533. Jabs DA, Bartlett JG: AIDS and ophthalmology: A period of transition. Am J Ophthalmol 124:227, 1997. 534. Jacobson MA, Stanley H, Holtzer C et al: Natural history and outcome of new AIDS-related cytomegalovirus
retinitis diagnosed in the era of highly active antiretroviral therapy. Clin Infect Dis 30:231, 2000. 535. Jabs DA, Van Natta ML, Kempen JH et al: Characteristics of patients with cytomegalovirus retinitis in the era of
highly active antiretroviral therapy. Am J Ophthalmol 133:48, 2002. 536. Whitcup SM, Fortin E, Nussenblatt RB et al: Therapeutic effect of combination antiretroviral therapy on cytomegalovirus
retinitis. JAMA 277:1519, 1997. 537. Whitcup SM, Fortin E, Lindblad S et al: Discontinuation of anticytomegalovirus therapy in patients with HIV infection
and cytomegalovirus retinitis. JAMA 282:1633, 1999. 538. Macdonald JC, Torriani FJ, Morse LS et al: Lack of reactivation of cytomegalovirus (CMV) retinitis after
stopping CMV maintenance therapy in AIDS patients with sustained elevations
in CD4 T-cells in response to highly active antiretroviral
therapy. J Infect Dis 177:1182, 1998. 539. Jabs, DA, Bolton SG, Dunn JP et al: Discontinuing anticytomegalovirus therapy in patients with immune reconstitution
after combination antiretroviral therapy. Am J Opthalmol 126:817, 1998. 540. Jovan MA, Saves JM, Tubiana R et al: Discontinuation of maintenance therapy for cytomegalovirus retinitis in
HIV-infected patients receiving highly active antiretroviral therapy. AIDS 15:23, 2001. 541. Whitcup SM, Cunningham ET Jr, Polis MA et al: Spontaneous and sustained resolution of CMV retinitis in patients receiving
highly active antiretroviral therapy. Br J Ophthalmol 82:845, 1998. 542. Reed JB, Schwab IR, Gordon J et al: Regression of cytomegalovirus retinitis associated with protease-inhibitor
treatment in patients with AIDS. Am J Ophthalmol 124:199, 1997. 543. Vrabec TR, Baldassano VF, Whitcup SM: Discontinuation of maintenance therapy in patients with quiescent cytomegalovirus
retinitis and elevated CD4+ counts. Ophthalmology 105:1259, 1998. 544. Michelet C, Arvieux C, Francois C et al: Opportunistic infections occurring during highly active antiretroviral
treatment. AIDS 12:1815, 1998. 545. Jacobson MA, Zegans M, Pavan PR et al: Cytomegalovirus retinitis after initiation of highly active antiretroviral
therapy. Lancet 349:1443, 1997. 546. Song MK, Karavellas MP, Macdonald JC et al: Characterization of reactivation of cytomegalovirus retinitis in patients
healed after treatment with highly active antiretroviral therapy. Retina 20:151, 2000. 547. Lin DY, Warren JF, Lazzeroni LC et al: Cytomegalovirus retinitis after initiation of highly active antiretroviral
therapy in HIV-infected patients Natural history and clinical
predictors. Retina 22:268, 2002. 548. Torriani FJ, Freeman WR, Macdonald JC et al: CMV retinitis recurs after stopping treatment in virological and immunological
failure of potent antiretroviral therapy. AIDS 14:173, 2000. 549. Song MK, Schrier RD, Smith IL et al: Paradoxical activity of CMV retinitis in patients receiving highly active
antiretroviral therapy. Retina 22:262, 2002. 550. Jacobson MA, Schrier R, McCune JM et al: Cytomegalovirus (CMV)-specific CD4+ T lymphocyte
immune function in long-term survivors of AIDS-related CMV
end-organ disease who are receiving potent antiretroviral therapy. J Infect Dis 1399, 2001. 551. Kuppermann BD, Holland GN: Immune Recovery Uveitis. Am J Ophthalmol 130:103, 2000. 552. Karavellas MP, Plummer DJ, Macdonald JC et al: Incidence of immune recovery vitritis in cytomegalovirus retinitis patients
following institution of successful highly active antiretroviral
therapy. J Infect Dis 179:697, 1999. 553. Robinson MR, Reed G, Csaky KG et al: Immune-recovery uveitis in patients with cytomegalovirus retinitis
taking highly active antiretroviral therapy. Am J Ophthalmol 130:49, 2000. 554. Nguyen QD, Kempen JH, Bolton SG et al: Immune recovery uveitis in patients with AIDS and cytomegalovirus retinitis
after highly active antiretroviral therapy. Am J Ophthalmol 129:634, 2000. 555. Karavellas MP, Azen SP, Macdonald JC et al: Immune recovery vitritis and uveitis in AIDS. Clinical predictors, sequelae, and
treatment outcomes. Retina 21:1, 2001. 556. Henderson HW, Mitchell SM: Treatment of immune recovery vitritis with local steroids. Br J Ophthalmol 83:540, 1999. 557. Panda A, Das GK, Khokhar S et al: Efficacy of four antiviral agents in the treatment of uncomplicated herpetic
keratitis. Can J Ophthalmol 30:256, 1995. 558. Wutzler P: Antiviral therapy of herpes simplex and varicella-zoster virus infections. Intervirology 40:343, 1997. 559. Gnann JW Jr, Crumpacker CS, Lalezari JP et al: Sorivudine versus acyclovir for treatment of dermatomal herpes zoster in
human immunodeficiency virus–infected patients: Results from a
randomized, controlled clinical trial. Collaborative Antiviral Study
Group/AIDS Clinical Trials Group, Herpes Zoster Study Group. Antimicrob Agents Chemother 42:1139, 1998. 560. Genovesi EV, Lamb L, Medine I et al: Antiviral efficacy of lobucavir (BMS-180194), a cyclobutyl-guanosine
nucleoside analogue, in the woodchuck (Marmota
monax) model of chronic hepatitis B virus (HBV) infection. Antiviral Res 48:197, 2000. 561. Tenney DJ, Yamanaka G, Voss SM et al: Lobucavir is phosphorylated in human cytomegalovirus-infected and -uninfected
cells and inhibits the viral DNA polymerase. Antimicrob Agents Chemother 41:2680, 1997. 562. Lalezari J, Drew WL, Jordan C: In vivo anti-CMV activity and safety of oral lobucavir in HIV-infected
patients (protocol AI 459-007 [Abstract 301]. 9th
Conference on Retroviruses and Opportunistic Infections, 1997. 563. Wilber R, Buffington D, Tyring S: Efficacy and safety of oral lobucavir (LBV) in patients with
recurrent genital herpes. The 38th Interscience Conference on Antimicrobial
Agents and Chemotherapy, Abs H-86, 339, 1998. 564. Reefschlaeger J, Bender W, Hallenberger S et al: Novel non-nucleoside inhibitors of cytomegaloviruses (BAY38-4766): In vitro and in vivo antiviral activity and mechanism of action. J Antimicrob Chemother 757, 2001. 565. McSharry JJ, McDonough A, Olson B et al: Susceptibilities of human cytomegalovirus clinical isolates to BAY38-4766, BAY 43-9695, and ganciclovir. Antimicrob Agents Chemother 45:2925, 2001. 566. Evers DL, Komazin G, Shin D et al: Interactions among antiviral drugs acting late in the replication cycle
of human cytomegalovirus. Antiviral Res 56:61, 2002. 567. Lalezari JP, Aberg JA, Wang LH et al: Phase I dose escalation trial evaluating the pharmacokinetics, anti-human
cytomegalovirus (HCMV) activity, and safety of 1263W94 in
human immunodeficiency virus–infected men with asymptomatic
HCMV shedding. Antimicrob Agents Chemother 46:2969, 2002. | 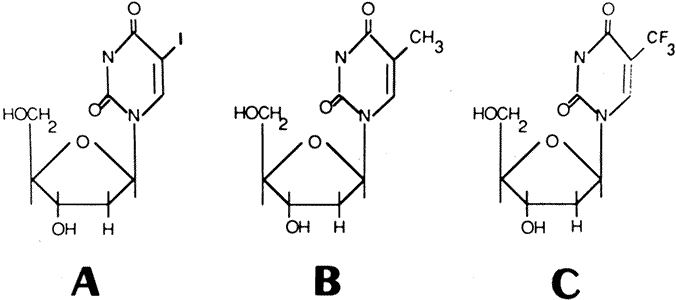
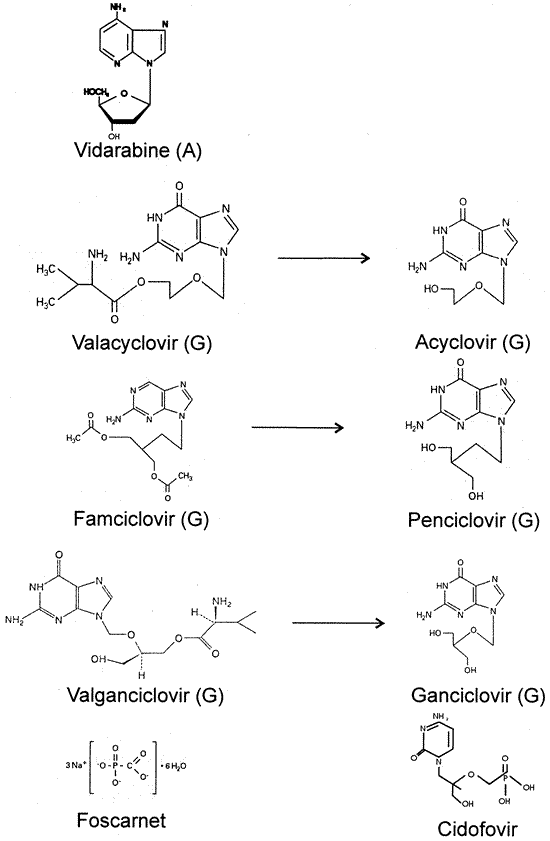
 6 months) increase in CD4 to greater than 100 to 150 cells/μL in
response to HAART, after taking into consideration the magnitude and
duration of CD4 increase, the anatomic location of the retinitis, vision
in the contralateral eye, and the feasibility of regular ophthalmologic
monitoring. Frequent monitoring of immune status is necessary if
anti-CMV therapy is withdrawn, and re-initiation of therapy
may be advisable if CD4 counts fall to less than 100 to150 cells/μl.
6 months) increase in CD4 to greater than 100 to 150 cells/μL in
response to HAART, after taking into consideration the magnitude and
duration of CD4 increase, the anatomic location of the retinitis, vision
in the contralateral eye, and the feasibility of regular ophthalmologic
monitoring. Frequent monitoring of immune status is necessary if
anti-CMV therapy is withdrawn, and re-initiation of therapy
may be advisable if CD4 counts fall to less than 100 to150 cells/μl.