DNA
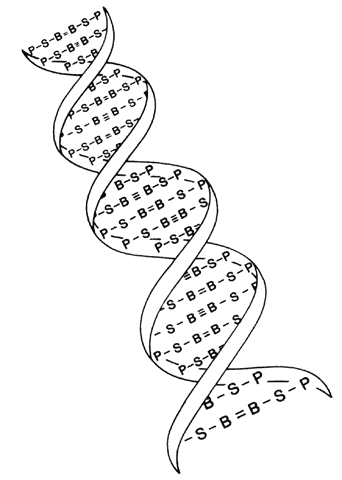
The structure of DNA is a double helix with four bases: adenine, guanine, cytosine, and thymine. These are paired by hydrogen bonds between the two helices like rungs in a ladder (Fig. 1). Each base is associated with a sugar and phosphate forming a nucleotide. The nucleotides are numbered in order, giving the DNA sequence. Nucleotides in the linear polymer chains that make up the two helices are connected by phosphodiester bonds. The sequence of bases (in nucleotide form), in groups of three called codons, makes up the genetic code. Each triplet codes for a specific amino acid. Since 64 different triplets are possible from the four bases, and there are only 20 amino acids, more than one triplet can code for a given amino acid.5 For example, the codon AAA will always and only produce the amino acid phenylalanine in the protein, but other codons may also produce phenylalanine in the protein. Amino acids are ultimately arranged into proteins, the building blocks of living structures.
|
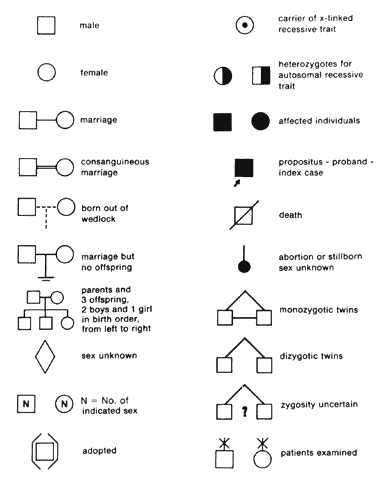
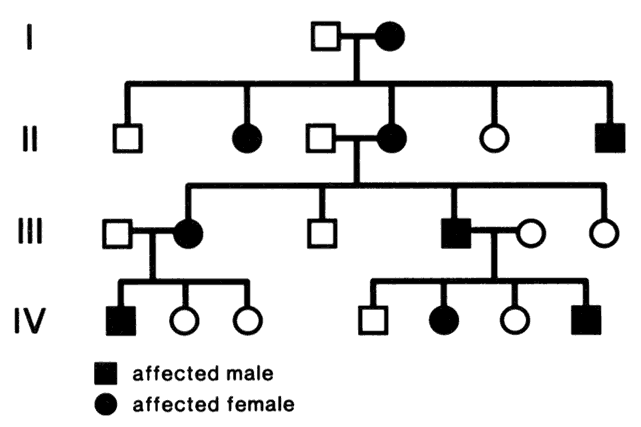
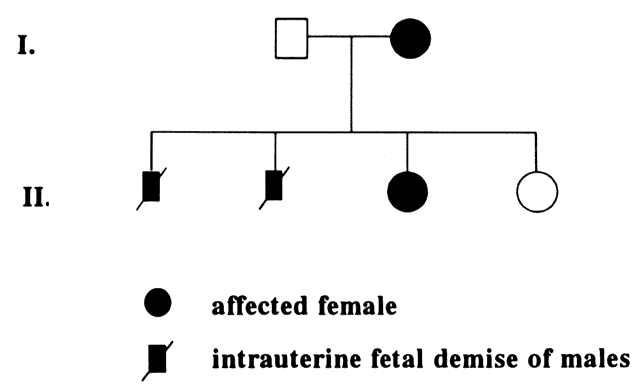
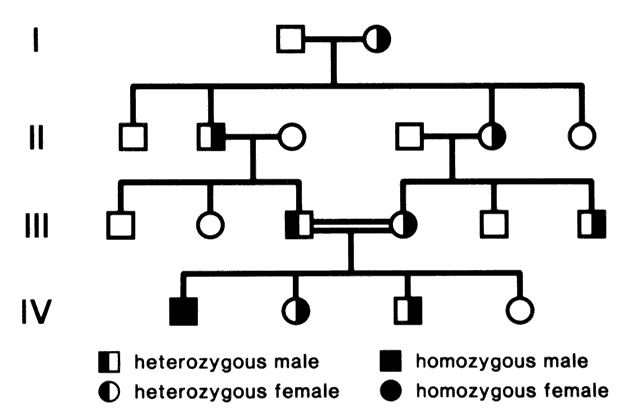
In 1911, Johannsen coined the term “gene” as the basic unit of hereditary characteristics.6 In 1901, the monk Gregor Mendel described the dominant and recessive patterns by which some traits are inherited, leading to the term Mendelian inheritance.7 Garrod developed the concept of human biochemical disease (i.e., inborn errors of metabolism) and its inheritance in a recessive Mendelian fashion in 1908 and8,9
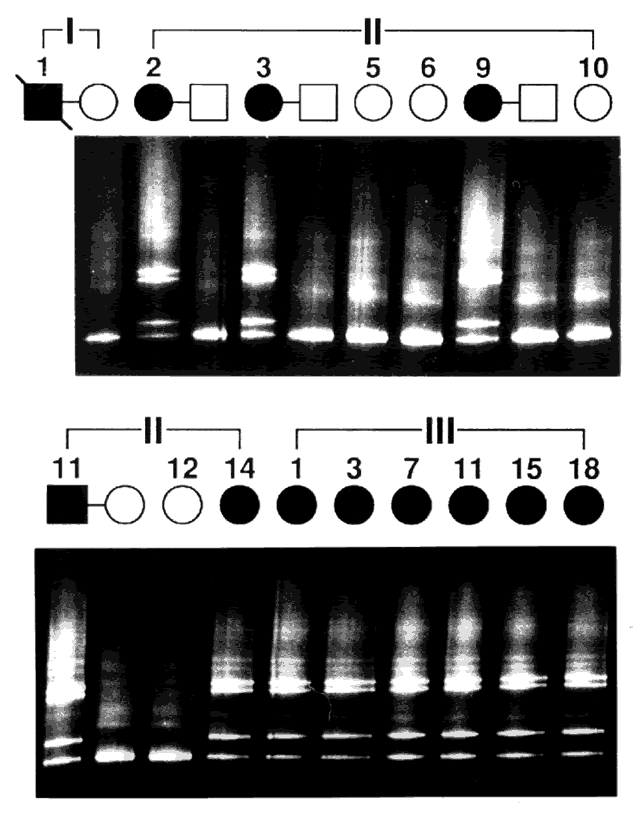
Genes are arranged into coding sequences, called exons, and intervening sequences that do not code, called introns. In addition, there may be separate promoter regions. Only 3% to 5% of human DNA codes for proteins; the rest has no known function.10 Although variation (mutation) in exons is likely to cause disease and is therefore rare, variation in introns does not cause a problem, yet is inherited in a Mendelian fashion. Since these variations are benign, they are also plentiful and vary from family to family. These variations are therefore used as genetic markers (e.g., in linkage analysis).
DNA sequences are written in a 5' to 3' direction (Fig. 2), and the sequence given is for the sense strand. The other strand is complementary and is called the template strand or antisense strand and runs in the opposite direction.10 DNA is transcribed in the following manner:
|
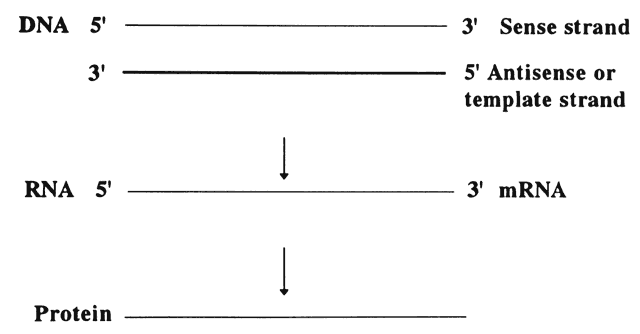
- Near the 5' end of genes, there are promotor sequences that instruct
messenger RNA (mRNA) to start transcription using the DNA antisense
strand as a template. That is, a new RNA strand is made that is complementary
in its base composition to the DNA antisense strand and thus
is the same as the sense strand. Each DNA codon is thereby turned into
a corresponding RNA codon.
- The double helix uncoils, and the appropriate bases are added along the
strand. RNA processing then occurs, cutting out (splicing) the introns, which do not code for anything, and rejoining the edges.
- Translation then occurs. This refers to the new mRNA strands, which include
only coding sequences that move out of the nucleus and into the cytoplasm
where they bind to the ribosomes. The ribosomes assemble a polypeptide
according to the recipe specified by the sequence of codons.
- Various modifications may then occur before the chain is transported to
its functional location within or outside of the cell.
DNA replicates itself in an analogous fashion. The two helical strands separate, and each is copied by a series of enzymes that position a complementary base opposite each base in the strand of DNA. One double helix thereby gives rise to two identical double helices.
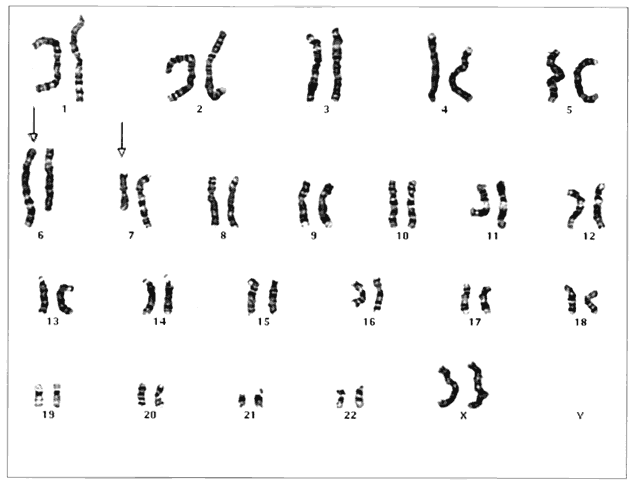
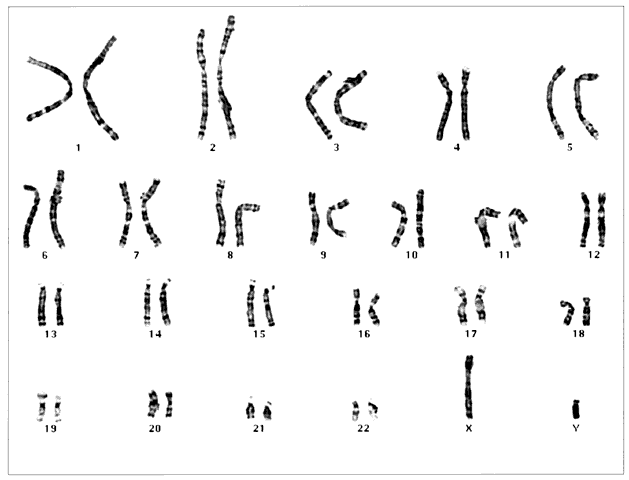
Humans have approximately 2 m of DNA arranged into 46 chromosomes composed of 23 homologous pairs. This includes 44 autosomes, which are the same in males and females, plus either an XY in males (the Y chromosome determines testis development) or an XX in females (Fig. 3). Each member of a homologous pair carries corresponding, although not necessarily identical, genes in the same sequence. One member of each chromosome pair is inherited from the father, the other from the mother. For most of the cell cycle, each chromosome exists as a single chromatid. When a cell is committed to division, however, each chromatid is replicated. This is the form in which we usually see pictures of chromosomes: two identical sister chromatids joined at a centromere, making an “X” shape.
|
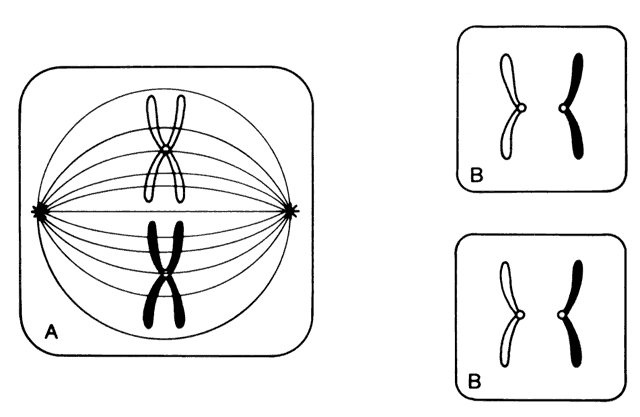
Each somatic cell has 46 chromosomes and divides by mitosis, which results in daughter cells with 46 chromosomes (Fig. 4). Mitosis gives rise to the body cells needed for growth and development. The normal sperm or ova, however, are the result of division by meiosis (Fig. 5) and have 23 chromosomes each, only one from each chromosome pair. In this manner, each parent passes on half of his or her genetic information to each child. Interestingly, the drive toward genetic diversity is so great that it can be seen even within a single sperm or egg. Each parent has two copies of every chromosome and passes on only one. However, the version of each chromosome that is passed on is not identical to either of the copies that the parent possesses. During meiosis, before the two copies of each chromosome separate to go to different eggs or sperm, the homologous chromosomes exchange genetic material in a process called crossing over (see Fig. 5B). At this stage, each homologue is made of two identical sister chromatids joined at the centromere. A crossover involves an exchange of genetic material between two chromatids on two different homologues.
|
|
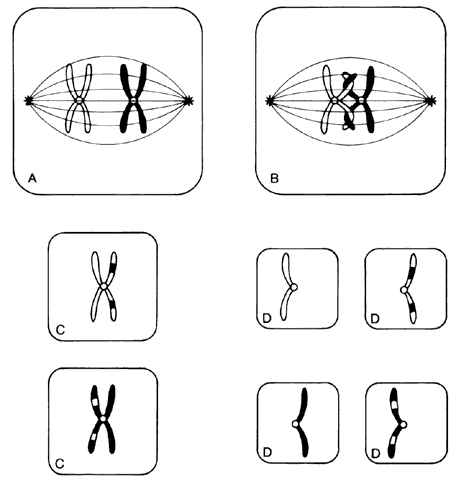
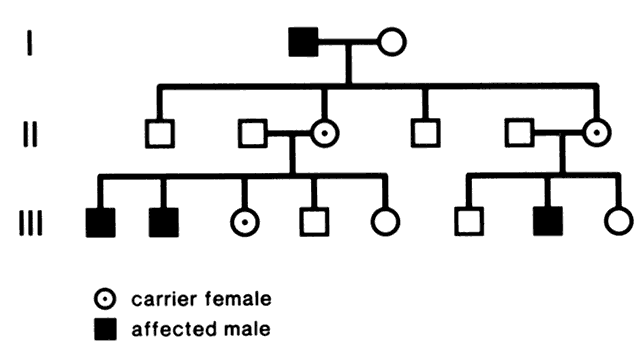
For example, a person has one copy of chromosome 1 from her mother and another copy from her father. Assume that she has brown eyes and is esotropic. Assume hypothetically that an esotropia gene is present on the maternal chromosome 1 as well as a gene for brown eyes. On the paternal chromosome 1 are genes for straight eyes, color blue. We might expect that in meiosis, when one copy of each chromosome goes to each egg, that the resultant eggs would all have either the maternally derived chromosome 1 with brown eyes and esotropia or the paternal straight blue. In fact, however, crossing over exchanges material between this person's two copies of chromosome 1, making unique versions to be passed on: very possibly esotropia with blue eyes or straight brown eyes. The chance that traits will be separated by crossovers increases as their genetic distance from each other increases.
Many crossovers may occur involving one, two, three, or four arms. In human male meiosis, an average of 55 crossovers occurs, with at least 1 crossover per homologous pair. Crossovers in females are even more frequent.10 Thus, the version of chromosome 1 passed on by a parent is actually an amalgam of different pieces from each of his or her two different chromosome 1 copies. This is important to understand because in some families, diseases caused by mutations in genes that are physically close together on the chromosome may occur together in some family members, whereas others may have only one trait or the other. It is also vital to understand crossing over because it plays an important role in understanding linkage analysis.
At conception, a sperm and ovum unite to form a zygote, which reestablishes the 46-chromosome complement. These chromosomes represent a random assortment of the genetic information present in each parent, in a unique combination that allows for advantageous new gene interactions in the offspring. Conversely, it may bring together genes that add to each other in a negative way, producing what we know as human malformation or disease. In addition, with such a complex process it is understandable that “mistakes” sometimes occur: chromosome pairs may not completely separate during meiosis, leading to extra genetic material in the offspring called trisomies or rearrangements called translocations. Trisomies occur with increased frequency as maternal age increases; mutations occur with increased frequency as paternal age increases. Pieces of DNA may be lost (deleted), or one or more bases may not pair up normally, changing a codon (mutations). These occurrences are not rare, and often result in early miscarriages. Although they are genetic in nature, they may or may not be inherited or heritable; it may be a chance occurrence without increased risk of recurrence.
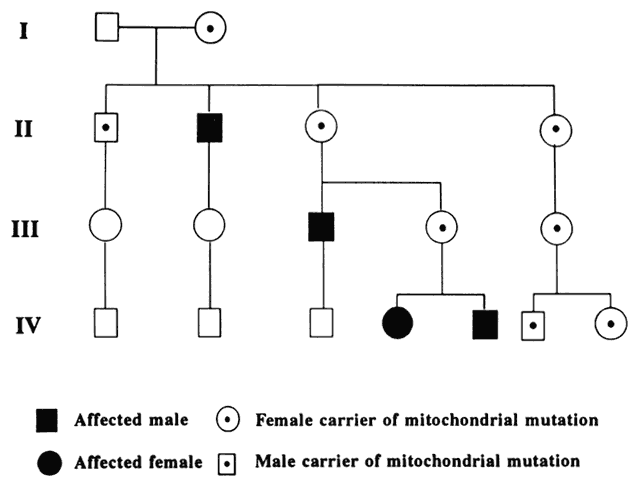
A mutation is a stable, heritable alteration in DNA transmitted from parent to progeny.11 From an evolutionary standpoint, mutations are necessary to generate genetic diversity, which makes adaptation to differing environments possible. While some mutations cause disease, others contribute to success and survival of the fittest. Some mutations do both: when one of a person's two homologous hemoglobin genes carries a sickle mutation, the person is resistant to malaria. Thus, this particular mutation protects persons who live in areas in which malaria is endemic, and it has been passed on to large numbers of progeny in some populations. If a person carries two copies of the sickle mutation, however, sickle cell anemia results, which is associated with high morbidity and mortality.