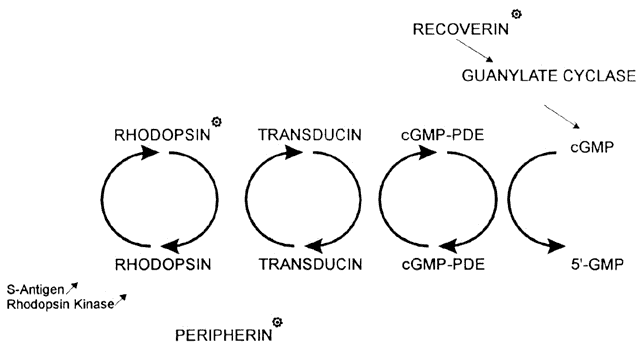
1. Rommens JR, Iannuzzi MC, Kerem BS et al: Identification of the cystic fibrosis gene: chromosomal walking and jumping. Science 245:1059, 1989 2. Hall JM, Friedman L, Guenther C et al: Closing in on a breast cancer gene on chromosome 17q. Am J Hum Genet 50:1235, 1992 3. Easton DF, Bishop DT, Ford D et al: The Breast Cancer Linkage Consortium: Genetic linkage analysis in a familial
breast and ovarian cancer: results from 214 families. Am J Hum Genet 52:678, 1993 4. Fishel R, Loscoe MK, Rao MRS et al: The human mutator gene homolog MSH2 and its association with hereditary
polyposis colon cancer. Cell 75:1027, 1993 5. The Huntington's Disease Collaborative Research Group: A novel gene
containing a trinucleotide repeat that is expanded and unstable on Huntington's
disease chromosomes. Cell 72:971, 1993 6. Saunders AM, Strittmatter WJ, Schmechel D et al: Association of apolipoprotein E allele e4 with late-onset familial and
sporadic Alzheimer's disease. Neurology 43:1467, 1993 7. Frezal J, Abule MS, De Fougerolle T: Gene atlas: A catalogue of mapped
genes and other markers, 2nd ed, p 1013. Paris, Inserm/John Libbey, 1991 8. Frezal J, Kaplan J, Dolifus H: Mapping the eye diseases. Ophthalmic Paediatr Genet 13:37, 1992 9. Musarella MA: Gene mapping of ocular diseases. Surv Ophthalmol 36:285, 1992 10. Jay B, Jay M: Molecular genetics in clinical ophthalmology. In Davidson
SI, Jany B (eds): Recent Advances in Ophthalmology, Vol 8, pp 185–206. New
York, Churchill Livingstone, 1992 11. Wang MX, Jenkins JJ, Cu-Unjieng AB et al: Eye tumors. In Parham DM (ed): Pediatric
Neoplasia: Morphology and Biology, pp 405–422. Philadelphia, LippincottRaven, 1996 12. Petrash JM: Applications of molecular biological techniques to the understanding of
visual system disorders. Am J Ophthalmol 113:573, 1992 13. MacDonald IM, Sasi R: Molecular genetics of inherited eye disorders. Clin Invest Med 17:474, 1994 14. Lindsay S, Ingelharn CF, Curtis A et al: Molecular genetics of inherited retinal degenerations. Curr Opin Genet Dev 2:459, 1992 15. Deeb SS: Genetic determinants of visual functions. Curr Opin Neurobiol 3:506, 1993 16. Huber A: Genetic disease of vision. Curr Opin Neurol 7:65, 1994 17. Wiggs JL: Molecular genetics and ocular disease. Int Ophthalmol Clin 33(2):1, 1993 18. Zhang K, Wang MX, Munier F et al: Molecular genetics of retinoblastoma. Int Ophthalmol Clin 33(3):53, 1993 19. Rosenfeld PJ, McKusick VA, Amberger JS et al: Recent advances in the gene map of inherited eye disorders: Primary hereditary
disease of the retina, choroid, and vitreous. J Med Genet 31:903, 1994 20. Wang MX, Donoso LA: Gene research and the eye. Curr Opin Ophthalmol 4(suppl III):102, 1993 21. Small KW: Application of molecular genetics to ocular disease. J Fla Med Assoc 81:264, 1994 22. Keith CG: Molecular biology in ocular disorders. Med J Aust 158:615, 1993 23. Griffin JR: Genetics review: Relation to ocular disease. J Optom Vis Sci 71:164, 1994 24. Wang MX, Sandos R, Crandal A, Donoso LA: Recent advances in the molecular genetics of retinitis pigmentosa. Curr Opin Ophthalmol 6(III):1, 1995 25. Wiggs JL: Genetics of glaucoma. In Wiggs JL (ed): Molecular Genetics of
Ocular Disease. New York, Wiley-Liss, 1995 26. Stephens JC, Cavanaugh ML, Gradie MI et al: Mapping the human genome: current status. Science 250:237, 1990 27. Thompson MW, McInnes RR, Willard HF: Genetics in medicine, 5th ed. Philadelphia, WB
Saunders, 1991 28. Sakai T, Ohtani N, McGee TL et al: Oncogenic germline mutations in Sp1 and ATF sites in the human retinoblastoma
gene. Nature 353:83, 1991 29. Onadim X, Hogg A, Baird PN et al: Oncogenic point mutations in exon 20 of the RB1 gene in families showing
incomplete penetrance and mild expression of the retinoblastoma phenotype. Proc Natl Acad Sci USA 89:6177, 1992 30. Munier F, Wang MX, Taonney F et al: Pseudo low penetrance: fortuitous familial aggregation of sporadic retinoblastomas
caused by independently-derived mutations in two large pedigrees. Arch Ophthalmol 111:1507, 1993 31. Dryja TP, Rapaport J, McGee TL et al: Molecular etiology of low-penetrance retinoblastoma in two pedigrees. Am J Hum Genet 52:1122, 1993 32. NIH/CEPH Collaborative Mapping Group: A comprehensive genetic linkage map
of the human genome. Science 258:67, 1992 33. Weissenbach J, Gyapay G, Dib C et al: A second-generation linkage map of the human genome. Nature 359:794, 1992 34. Saiki RK, Gelfand DH, Stoffel S et al: Primer-directed enzymatic amplification of DNA with a thermostable DNA
polymerase. Science 239:487, 1988 35. Southern EM: Detection of specific sequence among DNA fragments separated by gel electrophoresis. J Mol Biol 98:503, 1975 36. Botstein D, White RL, Skolnick M, Davis RW: Construction of a genetic linkage map using restriction fragment length
polymorphisms. Am J Hum Genet 32:314, 1980 37. Wiggs JL, Nordenskjold M, Yandell D et al: Prediction of the risk of hereditary retinoblastoma, using DNA polymorphisms
within the retinoblastoma gene. N Engl J Med 318:151, 1988 38. Wyman AR, White R: A highly polymorphic locus in human DNA. Proc Natl Acad Sci USA 77:6754, 1980 39. Kidd KK, Bowcock AM, Schmidtke J et al: Report of the DNA committee and catalogs of cloned and mapped genes and
DNA polymorphisms. Human Gene Mapping 10: Tenth International Workshop
on Human Gene Mapping. Cytogenet Cell Genet 51:622, 1989 40. Williamson R, Bowcock A, Kidd KK et al: Report of the DNA committee and catalogues of cloned and mapped genes and
DNA polymorphisms. Human Gene Mapping 10.5: Update to the Tenth International
Workshop on Human Gene Mapping. Cytogenet Cell Genet 55:457, 1990 41. Orita M, Iwahana H, Kanazawa et al: Detection of polymorphism of human DNA by electrophoresis as single-strand
conformation polymorphism. Proc Natl Acad Sci USA 86:2766, 1989 42. Sanger F, Nicklen S, Coulson AR: DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci USA 74:5463, 1977 43. Maxam AM, Gilbert W: A new method of sequencing DNA. Proc Natl Acad Sci USA 74:560, 1977 44. Glaser T, Walton DS, Cai J et al: PAX6 gene mutations in aniridia. In Wiggs
JL (ed): Molecular Genetics of Ocular Disease, pp 51–82. New
York, Wiley-Liss, 1995 45. Franckle U, Holmes LB, Atkins L et al: Aniridia-Wilms' tumor association: evidence for specific deletions of 11p13. Cytogenet Cell Genet 24:185, 1979 46. Gessler M, Thomas GH, Couillin P et al: A deletion map of the WAGR region on chromosome 11. Am J Hum Genet 44:486, 1989 47. Dryja TP, McGee TL, Reichel E et al: A point mutation of the rhodopsin gene in one form of retinitis pigmentosa. Nature 343:364, 1990 48. Rosenfeld PJ, Cowley GS, McGee TL et al: A null mutation in the rhodopsin gene causes rod photoreceptor dysfunction
and autosomal recessive retinitis pigmentosa. Nature Genet 1:209, 1992 49. Horowitz JM, Yandell DW, Park SH et al: Point mutation inactivation of the retinoblastoma antioncogene. Science 243:937, 1989 50. Riordan JR, Rommens JR, Kerem RS et al: Identification of the cystic fibrosis gene: Cloning and characterization
of complementary DNA. Science 245:1066, 1989 51. Kerem BS, Rommens JR, Buchanan JA et al: Identification of the cystic fibrosis gene: Genetic analysis. Science 245:1073, 1989 52. Kerem E, Corey M, Kerem BS et al: The relationship between genotype and phenotype in cystic fibrosis: Analysis
of the most common mutation (508). N Engl J Med 323:1517, 1990 53. Berson EL: Retinitis pigmentosa. Invest Ophthalmol Vis Sci 34:1659, 1993 54. Chen Z, Battinelli EM, Fielder A et al: A mutation in the Norrie disease gene (NDP) associated with Xlinked familial
exudative vitreoretinopathy. Nature Genet 5:180, 1993 55. Cavenee WK, Murphree AL, Shull MM: Prediction of familial predisposition to retinoblastoma. N Engl J Med 314:1201, 1986 56. Naumova A, Hanser M, Strong L et al: Concordance between parental origin of chromosome 13q loss and 6p amplification
in sporadic retinoblastoma. Am J Hum Genet 54:274, 1994 57. Naumova AK, Bird L, Slamka C et al: Transmission-ratio distortion of X chromosomes among male offspring of
females with skewed X-inactivation. Dev Genet 17:198, 1995 58. Scharf SJ, Bowcock AM, McClure G et al: Amplification and characterization of the retinoblastoma gene VNTR by PCR. Am J Hum Genet 50:371, 1992 59. Goddard AD, Phillips RA, Greger V et al: Use of the RB1 cDNA and a diagnostic probe in retinoblastoma families. Clin Genet 37:117, 1990 60. Onadim ZO, Mitchell CD, Rutland PC et al: Application of intragenic DNA probes in prenatal screening for retinoblastoma
gene carriers in the United Kingdom. Arch Dis Child 65:651, 1990 61. Scheffer H, Meerman GJ, Kruize YCM et al: Linkage analysis of families with hereditary retinoblastoma: Nonpenetrance
of mutation, revealed by combined use of markers within and flanking
the RB1 gene. Am J Hum Genet 45:252, 1989 62. McWilliam P, Farrar GJ, Kenna P et al: Autosomal dominant retinitis pigmentosa (ADRP): Localization of an ADRP
gene to the long arm of chromosome 3. Genomics 5:619, 1989 63. Kranich H, Bartkowski S, Denton MJ et al: Autosomal dominant ‘sector’ retinitis pigmentosa due to a point
mutation predicting an Asn-15-Ser substitution of rhodopsin. Hum Mol Genet 2(6):813, 1993 64. Sullivan LJ, Makris GS, Dickinson P et al: A new codon 15 rhodopsin gene mutation in autosomal dominant retinitis
pigmentosis associated with sectorial disease. Arch Ophthalmol 11(11):1512, 1993 65. Sung CH, Davenport CM, Hennessey JC et al: Rhodopsin mutations in autosomal dominant retinitis pigmentosa. Proc Natl Acad Sci USA 88:6481, 1991 66. Dryja TP, Hahn LB, Cowley GS et al: Mutation spectrum of the rhodopsin gene among patients with autosomal dominant
retinitis pigmentosa. Proc Natl Acad Sci USA 88:9370, 1991 67. Kim RY, Al-Maghtheh M, Fitzke FW et al: Dominant retinitis pigmentosa associated with two rhodopsin gene mutations: leu-40-arg
and an insertion disrupting the 5'-splice junction
of exon 5. Arch Ophthalmol 111:1518, 1993 68. Rodriguez JA, Herrena CA, Birch DG et al: A leucine to arginine amino acid substitution at codon 46 of rhodopsin
gene is responsible for a severe form of autosomal dominant retinitis
pigmentosa. Hum Mutat 2:205, 1993 69. Inglehearn CF, Keen TJ, Bashir R et al: A complete screen for mutations of rhodopsin gene in a panel of patients
with autosomal dominant retinitis pigmentosa. Hum Mol Genet 1:41, 1992 70. Dryja TP, McGee TL, Hahn LB et al: Mutations within the rhodopsin gene in patients with autosomal dominant
retinitis pigmentosa. N Engl J Med 323:1302, 1990 71. Macks JP, Davenport CM, Jacobson SG et al: Identification of novel rhodopsin mutations responsible for retinitis pigmentosa: implications
for the structure and function of rhodopsin. Am J Hum Genet 53:80, 1993 72. Keen TJ, Inglehearn CF, Lester DH et al: Autosomal dominant retinitis pigmentosa: four new mutations in rhodopsin, one
of them in the retinal attachment site. Genomics 11:199, 1991 73. Vaithinathan R, Berson EL, Dryja TP: Further screening of the rhodopsin gene in patients with autosomal dominant
retinitis pigmentosa. Genomics 21:461, 1994 74. Sheffield VC, Fishman GA, Beck JS et al: Identification of novel rhodopsin mutations associated with retinitis pigmentosa
using GC-clamped denaturing gradient gel electrophoresis. Am J Hum Genet 49:699, 1991 75. Farrar GJ, Findlay JB, Kumar SR et al: Autosomal dominant retinitis pigmentosa: a novel mutation in the rhodopsin
gene in the original 3q linked family. Hum Mol Genet 1:769, 1992 76. Reig C, Leicha N, Antich J et al: A missense mutation (His211Arg) and a silent (Thr160) mutation within the
rhodopsin gene in a Spanish autosomal dominant retinitis pigmentosa
family. Hum Mol Genet 3:195, 1994 77. Cideciyan AV, Jacobsen SG, Kemp CM et al: Stop codon and splice site rhodopsin mutations causing retinitis pigmentosa: retinal
function phenotypes. Invest Ophthalmol Vis Sci 35:1478, 1994 78. Horn M, Humphries P, Kunisch M et al: Deletions in exon 5 of the human rhodopsin gene causing a shift in the
reading frame and autosomal dominant retinitis pigmentosa. Hum Genet 90:255, 1992 79. Rosenfeld PJ, Dryja TP: Molecular genetics of retinitis pigmentosa and
related degenerations. In Wiggs JL (ed): Molecular Genetics of Ocular
Disease, pp 99–126. New York, Wiley-Liss, 1995 80. Farrar GJ, Kenna P, Jordan SA et al: A three base pair deletion in the peripherin-RDS gene in one form of retinitis
pigmentosa. Nature 354:478, 1991 81. Kajiwara K, Hahn LB, Mukai S et al: Mutations in the human retinal degeneration slow gene in autosomal dominant
retinitis pigmentosa. Nature 354:480, 1991 82. Wells J, Wroblewski J, Keen J et al: Mutations in the human retinal degeneration slow (RDS) gene can cause either
retinitis pigmentosa or macular dystrophy. Nature Genet 3:213, 1993 83. Farrar GJ, Kenna P, Jordan SA et al: Autosomal dominant retinitis pigmentosa: a novel mutation at the peripherin/RDS
locus in the original 6p-linked pedigree. Genomics 14:805, 1992 84. Farrar GJ, Kenna P, Jordan SA et al: Autosomal dominant retinitis pigmentosa: a novel mutation at the peripherin/RDS
locus in the original 6p-linked pedigree (errata). Genomics 15:466, 1992 85. Saga M, Mashima Y, Akeo K et al: A novel cys-214-ser mutation in the peripherin/RDS gene in a Japanese family
with autosomal dominant retinitis pigmentosa. Hum Genet 92:519, 1993 86. Bateman BJ, Klisak I, Kolis T et al: Assignment of the B-subunit of rod photoreceptor cGMP phosphodiesterase
gene (homolog of mouse rd gene) to human chromosome 4p 16. Invest Ophthalmol 32:890, 1991 87. Kajiwara K, Berson EL, Dryja TP: Digenic retinitis pigmentosa due to mutations at the unlinked peripherin/RDS
and ROM I loci. Science 264(5165):1604, 1994 88. Al-Maghtheh M, Kim RY, Hardcastle A et al: A 150 bp insertion in the rhodopsin gene of an autosomal dominant retinitis
pigmentosa family. Hum Mol Genet 3:205, 1994 89. Farber DB, Hechenlively JR, Sparkes RS et al: Molecular genetics of retinitis pigmentosa. West J Med 155:388, 1991 90. Farrar GJ, Kenna P, Redmond R et al: Autosomal dominant retinitis pigmentosa: a mutation in codon 178 of the
rhodopsin gene in two families of Celtic origin. Genomics 11:1170, 1991 91. Goldberg MF: Molecular heterogeneity in retinitis pigmentosa. Ophthalmic Genet 15:47, 1994 92. Al-Maghtheh M, Gregory C, Inglehearn C et al: Rhodopsin mutations in autosomal dominant retinitis pigmentosa. Hum Mutat 2:249, 1993 93. Oetting WS, King RA: Molecular basis of oculocutaneous albinism. J Invest Dermatol 103(suppl):131S, 1994 94. Tomita YT: The molecular genetics of albinism and piebaldism. Arch Dermatol 130:355, 1994 95. Spritz RA: Molecular genetics of oculocutaneous albinism. Semin Dermatol 21:167, 1993 96. Shastry BS: Recent development in certain X-linked genetic eye disorders. Biochim Biophys Acta 1182:119, 1993 97. Bergen AA, Samanns C, Schuurman EJ et al: Multipoint linkage analysis in X-linked ocular albinism of the Nettleship-Falls
type. Hum Genet 88:162, 1991 98. Pillers DA, Towbin JA, Chamberlain JS et al: Deletion mapping of Aland Island eye disease to Xp21 between DXS67 (B24) and
Duchenne muscular dystrophy. Am J Hum Genet 47:795, 1990 99. Giebel LB, Strunk KM, Spritz RA: Organization and nucleotide sequence of the human tyrosinase gene and a
truncated tyrosinase-related segment. Genomics 9:435, 1991 100. Kwon BS, Hag AK, Pomerrantz SH et al: Isolation and sequence of a putative cDNA clone for human tyrosinase that
maps at the mouse. Proc Natl Acad Sci USA 84:7473, 1987 101. Wittbjer A, Dahlback B, Odh G et al: Isolation of human tyrosinase from culture melanoma cells. Acta Derm Venereol (Stockh) 69:125, 1989 102. Barton DE, Kwon BS, Franckle U: Human tyrosinase gene, mapped to chromosome 11 (q14–q21), defines
second region of homology with mouse chromosome 7. Genomics 3:17, 1988 103. Giebel LB, Strunk KM, King RA et al: A frequent tyrosinase gene mutation in classic, tyrosinase-negative (type
IA) oculocutaneous albinism. Proc Natl Acad Sci USA 87:3255, 1990 104. Witkop CJ Jr, Quevedo WC Jr, Fitzpatrick TB et al: Albinism. In Scriver
CR et al (eds): The Metabolic Basis of Inherited Disease, pp 2905–2947. New
York, McGraw-Hill, 1989 105. Silvers W: The Coat Colors of Mice, pp 90–101. New York, Springer-Verlag, 1979 106. Elsas FJ, Maumenee IH, Kenyon KR, Yoder F: Familial aniridia with preserved ocular function. Am J Ophthalmol 83:718, 1977 107. Traboulsi EI et al: Hypoplasia of the iris: the aniridia spectrum. Int Pediatr 5:275, 1990 108. Riccardi VM, Sujansky E, Smith AC et al: Chromosomal imbalance in the aniridia-Wilms' tumor association: 11p interstitial
deletion. Pediatrics 61:604, 1978 109. Flanagan JC, DiGeorge AM: Sporadic aniridia and Wilms' tumor. Am J Ophthalmol 67:558, 1969 110. Lyons LA, Martha A, Mintz-Hitter HA et al: Resolution of the two loci for autosomal dominant aniridia, AN1 and AN2, to
a single locus on chromosome 11p13. Genomics 13:965, 1992 110a. Gessler M, Simola KOJ, Bruns CAP: Cloning breakpoint of a chromosome translocation identifies the AN2 locus. Science 244:1575, 1989 111. Ton CTT et al: Positional cloning and characterization of paired box- and homeobox-containing
gene from the aniridia region. Cell 67:1059, 1991 112. Glaser T, Walton DS, Maas RL: Genomic structure, evolutionary conservation and aniridia mutations in
the human PAX6 gene. Nature Genet 2:232, 1992 113. Walther C et al: PAX: A murine multigene family of paired box-containing genes. Genomics 11:424, 1991 114. Fantes JA et al: Non-radioactive in situ hybridization for the rapid analysis of submicroscopic
deletions at the WAGR locus. Am J Hum Genet 51:1286, 1992 115. Haber DA et al: An internal deletion within 11p13 zinc finger gene contributes to the development
of Wilms' tumor. Cell 61:1257, 1990 116. Martha A, Ferrell RE, Saunder GF. Dinucleotide repeat polymorphism in the
human aniridia (PAX6) gene. Hum Mol Genet 2:1982, 1993 117. Martha A, Mintz-Hittner H et al: Paired box mutations in familial and sporadic aniridia predicts truncated
aniridia protein. Am J Hum Genet 54:801, 1994 118. Chisholm IA, Chudley AE: Autosomal dominant iridogeniodysgenesis with associated somatic anomalies: four-generation
family with Rieger's syndrome. Br J Ophthalmol 67:529, 1983 119. Motegi T, Nakamura K, Terakawa T et al: Deletion of a single chromosome band 4q26 in a malformed girl: exclusion
of Rieger syndrome associated gene(s) from the 4q26 segment. J Med Genet 25:628, 1988 120. Lugutic I, Brecevic L, Petkovic I et al: Interstitial deletion 4q and Rieger syndrome. Clin Genet 20:323, 1981 121. Servile F, Brouset A: Pericentric inversion and partial monosomy 4q associated with congenital
anomalies. Hum Genet 39:239, 1977 122. Mitchell JA, Packman S, Loughman WE: Deletions of different segments of the long arm of chromosome 4. Am J Med Genet 89:73, 1981 123. Vaux C, Sheffield L, Keith CG et al: Evidence that Rieger syndrome maps to 4q25 or 4q27. J Med Genet 29:256, 1992 124. Alward WLM, Murray JC: Axenfeld-Rieger syndrome. In Wiggs JL (eds): Molecular
Genetics of Ocular Disease, pp 31–50. New York, Wiley-Liss, 1995 125. Abbott BD, Pratt RM: EGF receptor expression in the developing tooth is altered by exogenous
retinoic acid and EGF. Dev Biol 128:300, 1988 126. Raymond GM, Jumblatt MM, Barrels SP et al: Rabbit corneal endothelial cells in vitro: effects of EGF. Invest Ophthalmol Vis Sci 27:474, 1986 127. Partanen A, Ekblom P, Theleff I: Epidermal growth factor inhibits morphogenesis and cell differentiation
in culture mouse embryonic teeth. Dev Biol 111:84, 1989 128. Rhodes JA, Fitzgibbon DH, Macchiarulo PA: Epidermal growth factor-induced precocious incisor eruption is associated
with decreased tooth size. Dev Biol 121:247, 1987 129. Kronmiller JE, Upholt WB, Kollar EJ: EGF antisense oligodeoxynucleotides block murine odontogenesis. Dev Biol 147:485, 1991 130. Jumblatt MM, Matkin ED, Neufeld AH: Pharmacological regulation of the morphology and mitosis in culture rabbit
corneal endothelium. Invest Ophthalmol Vis Sci 29:586, 1988 131. Lewis RA, Nussbaum RL, Stambolian D: Mapping X-linked ophthalmic diseases. IV. Provisional assignment of the
locus for x-linked congenital cataracts and microcornea (the Nance-Horan
syndrome) to Xp22.2-p22.3. Ophthalmology 97:110, 1990 132. Nance WE, Warburg M, Bixler D et al: Congenital X-linked cataract, dental anomalies and brachymetacarpalia. Birth Defects 10:285, 1974 133. Horan MB, Billson FA: Aust Paediatr J 10:98, 1974 134. Bixler D, Higgis D, Hartsfield J Jr: The Nance-Horan syndrome: a rare X-linked ocular-dental trait with expression
in heterozygous females. Clin Genet 26:30, 1984 135. Lewis RA, Otterud B, Stauffer D et al: Mapping recessive ophthalmic disease: linkage of the locus for Usher syndrome
type II to a DNA marker on chromosome 1q. Genomics 7:250, 1990 136. Lewis RA: Mapping the gene for X-linked cataracts and microcornea with facial, dental, and
skeletal features to Xp22: an appraisal of the Nance-Horan syndrome. Trans Am Ophthalmol Soc 87:658, 1989 137. Stambolian D, Lewis RA, Buetow K et al: Nance-Horan syndrome: localization within the region Xp21.1–Xp22.3 by
linkage analysis. Am J Hum Genet 47:13, 1990 138. Lubsen NH, Renwick JH, Tsui L-C et al: A locus for a human hereditary cataract is closed linked to the gamma crystallin
gene family. Proc Natl Acad Sci USA 84:489, 1987 139. Eiberg H, Marner E, Rosenberg T et al: (CAM) assigned to chromosome 16: linkage to haptoglobin. Clin Genet 34:272, 1988 140. Marner E: Autosomal dominant congenital cataract on chromosome 16. Clin Genet 36:326, 1989 141. Moross T, Vaithilingham SS, Styles S et al: Autosomal dominant anterior polar cataracts associated with a familial 2;14 translation. J Med Genet 21:52, 1984 142. Warburg M: Letter to the editor: X-linked cataract and X-linked microphthalmos: How
many deletion families? Am J Med Genet 34:451, 1989 143. Alitalo T, Kruss TA, Forsuis H et al: Localization of Aland island eye disease locus to the pericentromeric region
of the X chromosome by linkage analysis. Am J Hum Genet 48:31, 1991 144. Nussbaum RL, Lewis RA, Lesko JG et al: Choroideremia is linked to the restriction fragment length polymorphism
DXYS1 at Xq13. Am J Hum Genet 37:473, 1985 145. Schwartz M, Rosenberg T, Niebuhr E et al: Choroideremia: further evidence for assignment of the locus to Xq13–Xq21. Hum Genet 74:449, 1986 146. Sankila EM, de La Chapelle A, Karna J et al: Choroideremia: close linkage to DXYS1 and DXYS12 demonstrated by segregation
analysis and historical-genealogical evidence. Clin Genet 31:315, 1987 147. Lasko JG, Lewis RA, Nussbaum RL: Multipoint linkage analysis of loci in the proximal long arm of the human
X chromosome: application to mapping the choroideremia locus. Am J Hum Genet 40:303, 1987 148. Cremers PM, van de Pol DJR, Kerkhoff LPM et al: Cloning of a gene that is rearranged in patients with choroideremia. Nature 347:674, 1990 149. Merry DE, Janne PA, Landers JE et al: Isolation of a candidate gene for choroideremia. Proc Natl Acad Sci USA 89:2135, 1992 150. van der Hark JAJM, van de Pol TJR, Molloy CM et al: Detection and characterization of point mutations in the choroideremia
candidate gene by PCR-SSCP analysis and direct DNA sequencing. Am J Hum Genet 50:1195, 1992 151. Sankila EM, Tolvanen R, van den Hurk JAJM et al: Aberrant splicing of the CHM gene is a significant cause of choroideremia. Nature Genet 1:109, 1992 152. Schwartz M, Rosenberg T, van den Hurk JAJM et al: Identification of mutations in Danish choroideremia families. Hum Mutat 2:43, 1993 153. Pascal O, Donnelly P, Fouanon C et al: A new (old) deletion in the choroideremia gene. Hum Mol Genet 1:1489, 1993 154. Seabra MC, Brown MS, Slaughter CA et al: Purification of component A of Rab geranylgeranyl transferase: possible
identity with the choroideremia gene product. Cell 70:1049, 1992 155. Seabra MC, Brown MS, Goldstein JL: Retinal degeneration in choroideremia: deficiency of Rab geranylgeranyl
transferase. Science 259:377, 1993 156. Haynie GD, Mukai S: Genetic basis of color vision. Int Ophthalmol Clin 33:141, 1993 157. Nathans J, Hogness DS: Isolation, sequence analysis, and intro-exon arrangement of the gene encoding
bovine rhodopsin. Cell 34:807, 1983 158. Nathans J, Hogness DS: Isolation and nucleotide sequence of the gene encoding human rhodopsin. Proc Natl Acad Sci USA 81:4851, 1984 159. Nathans J, Piantanida TP, Eddy RL et al: Molecular genetics of inherited variation in human color vision. Science 232:203, 1986 160. Nathans J, Thomas D, Hogness DS: Molecular genetics of human color vision: the genes encoding blue, green, and
red pigments. Science 232:193, 1986 161. Nathans J, Davenport CM, Maumenee IH et al: Molecular genetics of human blue cone monochromasy. Science 245:831, 1989 162. Nathans J, Merbs SL, Sung C-H et al: Molecular genetics of human visual pigments. Annu Rev Genet 26:403, 1992 163. Deeb SS, Motulaky AG: Molecular genetics of red-green color vision defect. In
Wiggs JL (ed): Molecular Genetics of Ocular Disease, pp 161–181. New
York, Wiley-Liss, 1995 164. Neitz J, Jacobs GH: Polymorphism of the longwavelength cone in normal human color vision. Nature 323:623, 1986 165. Mollom JD: Perception: questions of sex and color. Nature 323:578, 1986 166. Piantanida T: The molecular genetics of color vision and color blindness. Trends Genet 4:319, 1988 167. Neitz J, Jacobs GH: Polymorphism in normal human color vision and its mechanism. Vision Res 30:621, 1990 168. Winderickx J, Lindsey DT, Sanocki E et al: Polymorphism in red photopigment underlies variation in color matching. Nature 356:431, 1992 169. Winderickx J, Sanocki E, Lindsay DT et al: Defective color vision associated with a missense mutation in the human
green visual pigment gene. Nature Genet 1:251, 1992 170. Weitz CJ, Miyake Y, Shinzato K et al: Human tritanopia associated with two amino acid substitutions in the blue-sensitive
opsin. Am J Hum Genet 50:498, 1992 171. Krill AE: Congenital stationary night blindness. In Krill AE (ed): Krill's
Hereditary Retinal and Choroidal Diseases, Vol II, pp 391–420. New
York, Harper & Row, 1977 172. Carr RE: Congenital stationary nightblindness. Trans Am Ophthalmol Soc 72:448, 1974 173. Merin S, Rowe H, Auerbach E, Landau J: Syndrome of congenital high myopia with nyctalopia. Report of findings
in 25 families. Am J Ophthalmol 70:541, 1970 174. François J, Verriest G, DeRouck A: A new pedigree of idiopathic congenital night-blindness: transmitted as
a dominant hereditary trait. Am J Ophthalmol 59:621, 1965 175. Musarella MA, Weleber RG, Murphrey WH et al: Assignment of the gene for complete X-linked congenital stationary night
blindness (CSNB1) to chromosome Xp11.3. Genomics 5:727, 1989 176. Aldred MA, Dry KL, Sharp DM et al: Linkage analysis in X-linked congenital stationary night blindness. Genomics 14:99, 1992 177. Bech-Hansen NT, Moore BJ, Pearce WG: Mapping of locus for X-linked congenital stationary night blindness (CSNB1) proximal
to DXS7. Genomics 12:409, 1992 178. Dry KL, Van DD, Aldred MA et al: Linkage analysis in a family with complete type congenital stationary night
blindness with and without myopia. Clin Genet 43:250, 1993 179. Bhattacharya SS, Wright AF, Clayton JF et al: Close genetic linkage between X-linked retinitis pigmentosa and a restriction
fragment length polymorphism identified by recombinant DNA probe
L1.28. Nature 309:253, 1984 180. Clayton JF, Wright AF, Jay M et al: Genetic linkage between X-linked retinitis pigmentosa and DNA probe DXS7 (L1.28): further
linkage data, heterogeneity testing, and risk estimation. Hum Genet 74:168, 1986 181. Farrar GJ, Geraghty MT, Moloney JMB et al: Linkage analysis of X-linked retinitis pigmentosa in the Irish population. J Med Genet 25:222, 1988 182. Meitinger T, Fraser NA, Lorenz B et al: Linkage of X-linked retinitis pigmentosa to the hypervariable DNA marker
M27B (DXS255). Hum Genet 81:283, 1989 183. Wirth B, Denton MJ, Chen J-D et al: Two different genes for X-linked retinitis pigmentosa. Genomics 2:263, 1988 184. Musarella MA: Mapping of the X-linked recessive retinitis pigmentosa gene: a review. Ophthalmic Pediatr Genet 2:77, 1990 185. Musarella MA, Burghes A, Anson-Cartwright L et al: Localization of the gene for X-linked recessive type of retinitis pigmentosa (XLRP) to
Xp21 by linkage analysis. Am J Hum Genet 43:484, 1988 186. Nussbaum RL, Lewis RA, Lesko JG et al: Mapping X-linked ophthalmic diseases: II. Linkage relationship of X-linked
retinitis pigmentosa to X chromosomal short arm markers. Hum Genet 70:45, 1985 187. Traboulsi EI: Ectopia lentis and associated systemic disease. In Wiggs
JL (ed): Molecular Genetics of Ocular Disease, pp 219–233. New York, Wiley-Liss, 1995 188. Dietz HC, Pyeritz RD, Hall BD et al: The Marfan syndrome locus: confirmation of assignment to chromosome 15 and
identification of tightly linked markers at 15q15-q21.3. Genomics 9:355, 1991 189. Kanulainen K, Pulkinen L, Savolainen A et al: Location on chromosome 15 of the gene defect causing Marfan syndrome. N Engl J Med 323:935, 1990 190. Lee B, Godfrey M, Vitale E et al: Linkage of Marfan syndrome and a phenotypically related disorder to two
different fibrillin genes. Nature 352:330, 1991 191. Tsipouras P, Del Mastro R, Sarfarazi M et al: Genetic linkage of the Marfan syndrome, ectopia lentis, and congenital
contractual arachnodactyly to the fibrillin genes on chromosomes 15 and 5. N Engl J Med 326:905, 1992 192. Magenis RE, Maslen CL, Smith L et al: Localization of the fibrillin (FBN) gene to chromosome 15, band q21.1. Genomics 11:346, 1991 193. Dietz HC, Cutting GR, Pyeritz RE et al: Marfan syndrome caused by a recurrent de novo missense mutation in the
fibrillin gene. Nature 352:337, 1991 194. Dietz HC, Saraiva JM, Pyeritz RE et al: Clustering fibrillin (FBN1) mutations in Marfan syndrome patients at cysteine
residues in EGF-like domains. Hum Mutat 1:366, 1992 195. Dietz HC, Valle D, Francomano CA et al: The skipping of constitutive exons in vivo induced by nonsense mutations. Science 259:680, 1993 196. Kainulainen K, Karttunen L, Puhakka L et al: Mutations in the fibrillin gene responsible for dominant ectopia lentis
and neonatal Marfan syndrome. Nature Genet 6:64, 1994 197. Criswick VG, Schepens CL: Familial exudative vitreoretinopathy. Am J Ophthalmol 68:578, 1969 198. van Nouhuys CE: Dominant exudative vitreoretinopathy. Ophthalmic Paediatr Genet 5:31, 1985 199. Feldman EL, Norris JL, Cleasby GW: Autosomal dominant exudative vitreoretinopathy. Arch Ophthalmol 101:1532, 1983 200. Plager DA, Orgel IK, Ellis FD et al: X-linked recessive familial exudative vitreoretinopathy. Am J Ophthalmol 114:145, 1992 201. Tasman W, Augsberger JJ, Shields JA et al: Familial exudative vitreoretinopathy. Trans Am Ophthalmol Soc 79:211, 1981 202. Ohba N, Yamashita T: Primary vitreoretinal dysplasia resembling Norrie's disease in a female: Association
with X:autosome chromosomal translocation. Br J Ophthalmol 70:64, 1986 202a. Ober RR, Bird AC, Hamilton AM et al: Autosomal dominant exudative vitreoretinopathy. Br J Ophthalmol 64:112, 1980 203. Li Y, Fuhrmann C, Schwinger E et al: Letter to the editor: The gene for autosomal dominant familial exudative
vitreoretinopathy (Criswick-Scheperns) on the long arm of chromosome 11. Am J Ophthalmol 113:712, 1992 204. Li Y, Muller B, Fuhrmann C et al: The autosomal dominant familial exudative vitreoretinopathy locus maps
on 11q and is closely linked to D11S533. Am J Hum Genet 51:749, 1992 205. Stone EM, Nichols BE, Streb LM et al: Genetic linkage of vitelliform macular degeneration (Best's disease) to
chromosome 11q13. Nature Genet 1:246, 1992 206. Fullwood P, Jones J, Bundey S et al: X-linked exudative vitreoretinopathy: Clinical features and genetic linkage
analysis. Br J Ophthalmol 77:168, 1993 207. Netland PA, Wiggs JL, Dreyer EB: Inheritance of glaucoma and genetic counseling of glaucoma patients. Int Ophthalmol Clin 33(2):101, 1993 208. Miller SJH: Genetics of glaucoma and family studies. Trans Ophthalmol Soc UK 98:290, 1978 209. Rosenthal AR, Perkins ES: Family studies in glaucoma. Br J Ophthalmol 69:664, 1985 210. Becker B, Kolker AF, Roth FD: Glaucoma family study. Am J Ophthalmol 50:557, 1960 211. Davies TG: Tonographic survey of the close relatives of patients with chronic simple
glaucoma. Br J Ophthalmol 52:32, 1968 212. Paterson G: A nine-year follow-up of studies on first-degree relatives of patients
with glaucoma simplex. Trans Ophthalmol Soc UK 90:515, 1970 213. Jay B, Paterson G: The genetics of simple glaucoma. Trans Ophthalmol Soc UK 90:161, 1970 214. François J, Heintz-De Bree C: Personal research on the heredity of chronic simple (open-angle) glaucoma. Am J Ophthalmol 62:1067, 1966 215. Kellerman L, Posner A: The value of heredity in the detection and study of glaucoma. Am J Ophthalmol 40:681, 1955 216. Tielsch JM, Sommer A, Katz J et al: Racial variations in the prevalence of primary open-angle glaucoma: The
Baltimore eye survey. JAMA 266:369, 1991 217. Lichter PR: Genetic clues to glaucoma's secrets: The L. Edward Jackson Memorial
Lecture, Part 2. Am J Ophthalmol 117:706, 1994 218. Leopold IH: The HLA system and glaucoma. Am J Ophthalmol 87:578, 1979 219. Brooks AM, Gillies WE: Blood groups as genetic markers in glaucoma. Br J Ophthalmol 72:127, 1965 220. Garg MP, Pahwa JM: Primary glaucoma and blood groups. J All India Ophthalmol Soc 13:127, 1965 221. David R, Maier G, Jenkins T: Genetic markers in glaucoma. Br J Ophthalmol 64:227, 1980 222. Ritch R, Podos SM, Henley W et al: Lack of association of histocompatibility antigens with primary open-angle
glaucoma. Arch Ophthalmol 96:2204, 1978 223. Becker B, Morton WR: Phenylthiourea taste testing and glaucoma. Arch Ophthalmol 72:323, 1964 224. Becker B: Diabetes mellitus and primary open-angle glaucoma. Am J Ophthalmol 71:1, 1971 225. Renie WA (ed): Glaucoma. In Renie WA (ed): Goldberg's Genetic and
Metabolic Eye Disease, 2nd ed, pp 285–286. Boston, Little, Brown & Co, 1986 226. Allen TD, Ackerman WG: Hereditary glaucoma in a pedigree of three generations. Arch Ophthalmol 27:139, 1942 227. Courtney RH, Hill E: Hereditary juvenile glaucoma simplex. JAMA 97:1602, 1931 228. Martin JP, Zorab EC: Familial glaucoma in nine generations of South Hampshire family. Br J Ophthalmol 58:536, 1974 229. Stokes WH: Hereditary primary glaucoma. Arch Ophthalmol 24:85, 1940 230. Weatherhill JR, Hart CT: Familial hypoplasia of the iris stroma associated with glaucoma. Br J Ophthalmol 53:433, 1969 231. Sheffield VC, Stone EM, Alward WL et al: Genetic linkage of familial open-angle glaucoma to chromosome 1q21-q31. Nature Genet 4:47, 1993 232. Johnson AT et al: Clinical feature and linkage analysis of a family with autosomal dominant
juvenile glaucoma. Ophthalmology 100:524, 1993 233. Richards JE, Lichter PR, Boehnke M et al: Mapping of a gene for autosomal dominant juvenile-onset open-angle glaucoma
to chromosome 1q. Am J Hum Genet 54:62, 1994 234. Valle D, Kaiser-Kupfer MI, Valle LAD: Gyrate atrophy of the choroid and retina: Deficiency of ornithine aminotransferase
in transformed lymphocytes. Proc Natl Acad Sci USA 74:5159, 1977 235. Inana G, Hotta Y, Zintz C et al: Molecular basis of ornithine aminotransferase defect in gyrate atrophy. Prog Clin Biol Res 362:191, 1991 236. Ramesh V, Gussela JF, Shih VE: Molecular pathology of gyrate atrophy of the choroid and retinal due to
ornithine and aminotransferase deficiency. Mol Biol Med 8:81, 1991 237. Pauling L, Itano HA, Singer SJ et al: Sickle anemia: a molecular disease. Science 110:543, 1949 238. Ramesh V, Eddy R, Bruns GA et al: Localization of the ornithine aminotransferase gene and related sequences
on two human sequences. Hum Genet 76:121, 1987 239. Barrett DJ, Bateman JB, Sparkes RS et al: Chromosomal localization of human ornithine aminotransferase gene sequences
to 10q26 and Xp11.2. Invest Ophthalmol Vis Sci 28:1037, 1987 240. Wu J, Ramesh V, Kidd JR et al: The ornithine aminotransferase (OAT) locus is linked and distal to D10S20 on
the long arm of chromosome 10. Cytogenet Cell Genet 48:126, 1988 241. Ramesh V, McClatchey AI, Ramesh N et al: Molecular basis of ornithine aminotransferase deficiency in B-6-responsive
and nonresponsive forms of gyrate atrophy. Proc Natl Acad Sci USA 85:3777, 1988 242. Mitchell GA, Looney JE, Brody LC et al: Human ornithine-delta aminotransferase: cDNA cloning and analysis of the
structural gene. J Biol Chem 263:14288, 1988 243. Inana G, Hotta Y, Zintz C et al: Expression defect of ornithine aminotransferase gene in gyrate atrophy. Invest Ophthalmol Vis Sci 29:1001, 1988 244. Mitchell GA, Brody LC, Sipila I et al: At least two mutant alleles of ornithine-delta-aminotransferase cause gyrate
atrophy of the choroid and retina in Finns. Proc Natl Acad Sci USA 86:197, 1989 245. Inana G, Chambers C, Hotta Y et al: Point mutation affecting processing of the ornithine aminotransferase precursor
protein in gyrate atrophy. J Biol Chem 264:17432, 1989 246. McClatchey AI, Kaufman DL, Berson E et al: Splicing defect at the ornithine aminotransferase (OAT) locus in gyrate
atrophy. Am J Hum Genet 47:790, 1990 247. Mitchell GA, Labuda D, Fontaine G et al: Splicemediated insertion of an Alu sequence inactivates ornithine delta-aminotransferase: A
role for Alu elements in human mutation. Proc Natl Acad Sci USA 88:815, 1991 248. Akaki Y, Hotta Y, Mashima Y et al: A deletion in the ornithine aminotransferase gene in gyrate atrophy. J Biol Chem 267:12950, 1992 249. Michaud J, Brody LC, Stell G et al: Strand-separating conformational polymorphism analysis: Efficacy of detection
of point mutations in the human ornithine delta-aminotransferase
gene. Genomics 13:389, 1992 250. Mashima Y, Weleber RG, Kennaway NG et al: A single base change at a splice acceptor site in the ornithine aminotransferase
gene causes abnormal RNA slicing in gyrate atrophy. Hum Genet 90:305, 1992 251. Mashima Y, Murakami A, Weleber RG et al: Nonsense codon mutations of the ornithine aminotransferase gene with decreased
levels of mutant mRNA in gyrate atrophy. Am J Hum Genet 51:81, 1992 252. Park JK, Herron BJ, O'Donnell JJ et al: Three novel mutations of the ornithine aminotransferase (OAT) gene in gyrate
atrophy. Genomics 14:553, 1992 253. Park JK, O'Donnell JJ, Shin VE et al: A 15-bp deletion in exon 5 of the ornithine aminotransferase (OAT) locus
associated with gyrate atrophy. Hum Mutat 1:293, 1992 254. Brody LC, Mitchell GA, Obie C et al: Ornithine delta-aminotransferase mutations in gyrate atrophy: Allelic heterogeneity
and function consequences. J Biol Chem 267:3302, 1992 255. Dietz HC, McIntosh I, Sakai LY et al: Four novel FBN1 mutations: significance for mutant transcript level and
EGF-like domain calcium binding in the pathogenesis of the Marfan syndrome. Genomics 17:468, 1993 256. Inana G, Hotta Y, Mashima Y et al: Molecular genetic basis of gyrate atrophy. Invest Ophthalmol Vis Sci 35:1984, 1994 256a. Mitchell G, Brody LA, Looney J et al: An Initiator codon mutation in the ornithine-delta-aminotransferase causing
gyrate atrophy of the choroid and retina. J Clin Invest 81:630, 1988 256b. Ramesh V, Benoit LA, Crawford P et al: The ornithine aminotransferase (OAT) locus: analysis of RFLPs in gyrate
atrophy. Am J Hum Genet 42:365, 1988 257. Leber T: Ueber Hereditare und congenital-angelegte Schnervenleiden. Graefes Arch Ophthalmol 17:249, 1871 258. Riordan-Eva P, Harding AE: Leber's hereditary optic neuropathy: The clinical relevance of different
mitochondrial DNA mutations. J Med Genet 32:81, 1995 259. Singh G, Lott MT, Wallace DC: A mitochondrial DNA mutation as a cause of Leber's hereditary optic
neuropathy. N Engl J Med 320:1300, 1989 260. Brown MD, Voljavec AS, Lott MT et al: Leber's hereditary optic neuropathy: a model for mitochondrial neurodegenerative
diseases. FASCEB J 6:2791, 1992 261. Savontaus ML: mtDNA mutations in Leber's hereditary optic neuropathy. Biochim Biophys Acta 1271:261, 1995 262. Newman NJ: Leber's hereditary optic neuropathy. Arch Neurol 50:540, 1993 263. Vilkki J, Savontaus ML, Kelimo H et al: Mitochondrial DNA polymorphism in Finnish families with Leber's hereditary
optic neuroretinopathy. Hum Genet 82:208, 1989 264. Holt IJ, Miller DH, Harding AE: Genetic heterogeneity and mitochondrial DNA heteroplasmy in Leber's
hereditary optic neuropathy. J Med Genet 26:739, 1989 265. Parker WD Jr, Oley CA, Parks JK: A defect in mitochondrial electron-transport activity (NADH-coenzyme Q
oxidoreductase) in Leber's hereditary optic neuropathy. N Engl J Med 320:1331, 1989 266. Newman NJ, Lott MT, Wallace DC: The clinical characteristics of pedigrees of Leber's hereditary optic
neuropathy with the 11778 mutation. Am J Ophthalmol 111:750, 1991 267. Nikoskelainen EK: Clinical picture of LHON. Clin Neurosci 2:115, 1994 268. Majander A, Huoponen K, Savontaus ML et al: Electron transfer properties of NADH:ubiquinone reductase in the ND1/3460 and
the ND4/11778 mutations of the Leber hereditary optic neuroretinopathy (LHON). FEBS Lett 292:289, 1991 269. Larsson NG, Andersen O, Holme E et al: Leber's hereditary optic neuropathy and complex I deficiency in muscle. Ann Neurol 30:701, 1991 270. Lessel S, Horovitz B: Histochemical study of enzymes of optic nerve of monkey and rat. Am J Ophthalmol 74:118, 1972 271. Bu XD, Rotter JI: X chromosome-linked and mitochondrial gene control of Leber hereditary
optic neuropathy: evidence from segregation analysis for dependence on
X chromosome inactivation. Proc Natl Acad Sci USA 88:8198, 1991 272. Vilkki J, Ott J, Savontaus ML et al: Optic atrophy in Leber hereditary optic neuroretinopathy is probably determined
by an X chromosomal gene closely linked to DXS7. Am J Hum Genet 48:486, 1991 273. Chen JD, Cox I, Denton MJ: Preliminary exclusion of an X-linked gene in Leber optic atrophy by linkage
analysis. Hum Genet 82:203, 1989 274. Sweeney MG, Davis MB, Lashwood A et al: Evidence against an X-linked locus close to DXS7 determine visual loss
susceptibility in British and Italian families with Leber hereditary optic
neuropathy. Am J Hum Genet 51:741, 1992 275. Juvonen V, Vilkki J, Aula P et al: Reevaluation of the linkage of an optic atrophy susceptibility gene to
Xchromosomal markers in Finnish families with Leber hereditary optic neuroretinopathy (LHON). Am J Hum Genet 53:289, 1993 276. Nikoskelainen E, Hoyt WF, Nummelin K: Ophthalmoscopic findings in Leber's hereditary optic neuropathy, II: The
fundus finds in the affected family members. Arch Ophthalmol 101:1059, 1983 277. Nikoskelainen E, Sogg RL, Rosenthal AR et al: The early phase in Leber hereditary optic atrophy. Arch Ophthalmol 95:969, 1977 278. Smith JL, Hoyt WF, Susac JO: Ocular fundus in acute Leber optic neuropathy. Arch Ophthalmol 90:349, 1973 279. Berninger TA, von Meyer L, Siess E: Leber's hereditary optic atrophy: Further evidence for a defect of
cyanide metabolism? Br J Ophthalmol 73:314, 1989 280. Nikoskelainen E: New aspects of the genetic etiologic, and clinical puzzle of Leber's
disease. Neurology 34:1482, 1984 281. Wallace DC, Singh G, Lott MT et al: Mitochondrial DNA mutation associated with Leber's hereditary optic
neuropathy. Science 242:1427, 1988 282. Bressler NM, Bressler SB, Fine SL: Age-related macular degeneration. Surv Ophthalmol 32:375, 1988 283. Segato T, Midenam E, Blarzino MC: Age-related macular degeneration. Aging Clin Exp Res 5:165, 1993 284. Vinding T: Age-related macular degeneration: Macular changes, prevalence and sex ratio. Acta Ophthalmol 67:609, 1989 285. Gass JDM: Drusen and disciform macular detachment and degeneration. Arch Ophthalmol 90:206, 1973 286. Melrose MA, Margargal LE, Lucier AC: Identical twins with subretinal neovascularization complicating senile
macular degeneration. Ophthalmic Surg 16:648, 1985 287. Meyers SM, Zachary AA: Monozygotic twins with age-related macular degeneration. Arch Ophthalmic 106:651, 1988 288. Heinrich P: Senile degeneration of the macula. Klin Monatsbl Augenheilkd 162:3, 1973 289. Small KW: Molecular genetics of macular degeneration. In Wiggs JL (ed): Molecular
Genetics of Ocular Disease. New York, Wiley-Liss, 1995 290. Lefler WH, Wadsworth JAC, Sidbury JB: Hereditary macular degeneration and aminoaciduria. Am J Ophthalmol 71:224, 1971 291. Weber JL, May PE: Abundant class of human polymorphism can be typed using the polymerase
chain reaction. Am J Hum Genet 44:388, 1989 292. Small KW, Weber JL, Roses A et al: North Carolina macular dystrophy is assigned to chromosome 6. Genomics 13:681, 1992 293. Small KW, Weber JL, Hung WY et al: North Carolina macular dystrophy: exclusion mapping using RFLPs and microsatellites. Genomics 11:763, 1991 294. Small KW, Killian J, McLean WC: North Carolina's dominant progressive foveal dystrophy: How progressive
is it? Br J Ophthalmol 75:401, 1991 294a. Small KW, Weber JL, Roses A et al: North Carolina macular dystrophy (MCDR1). A review and refined mapping
to 6q14-q16.2. Ophthalmic Paediatr Genet 14:143, 1994 295. Best F: Uber eine hereditar makula affektion: Beitrage zur Vereburngalehre. Z Augenheilkd 13:199, 1905 296. Forsman K, Graff C, Nordstrom S et al: The gene for Best's macular dystrophy is located 11q13 in a Swedish
family. Clin Genet 42:156, 1992 297. Nichols BE, Bascom R, Litt M et al: Refining the locus for Best vitelliform macular dystrophy and mutation
analysis of the candidate gene ROM1. Am J Hum Genet 54:95, 1994 298. Stone E, Kimura AE, Folk JC et al: Genetic linkage of autosomal dominant neovascular inflammatory vitreoretinopathy
to chromosome 11q13. Hum Mol Genet 1:685, 1992 299. Nichols BE, Drack AV, Vandenburgh K et al: A 2 base pair deletion in the RDS gene associated with butterfly-shaped
pigment dystrophy of the fovea. Hum Mol Genet 2:1347, 1993 300. Bascom RA, Schappert, McInnes RR: Cloning of the human and murine ROM1 genes: genomic organization and sequence
conservation. Hum Mol Genet 2:385, 1993 301. Bascom RA, Lui L, Humphries et al: Polymorphisms and sequence variants at the ROM1 locus. Hum Mol Genet 2:1975, 1993 302. Bascom RA, Garcia-Heras J, Haieh CL et al: Localization of the photoreceptor gene ROM1 to human chromosome 11 and
mouse chromosome 19: sublocation to human 11q13 between PGA and PYGM. Am J Hum Genet 51:1028, 1992 303. Bascom RA, Manara S, Colins L et al: Cloning of the cDNA for a novel photoreceptor membrane protein (rom-1) identified
a disk rim protein family implicated in human retinopathies. Neuron 8:1171, 1992 304. Stargardt K: Uber familiare progressive deneration in dermakulagegend des auges. Graefes Arch Klin Ophthalmol 71:534, 1909 305. Kaplan J, Gerber S, Larget-Piet D et al: A gene for Stargardt's disease (fundus flavimaculatus) maps to the
short arm of chromosome 1. Nature Genet 5:308, 1993 306. Zhang K, Bither PP, Park R et al: A dominant Stargardt's macular dystrophy locus maps to chromosome 13q34. Arch Ophthalmol 112:759, 1994 307. Stone EM, Nichols RE, Kimura AE et al: Clinical features of a Stargardt-like dominant progressive macular dystrophy
with genetic linkage to chromosome 6q. Arch Ophthalmol 112:765, 1994 308. Deutman AF, van Blommerstein JDA, Henkes HE et al: Butterfly-shaped pigment dystrophy of the fovea. Arch Ophthalmol 83:558, 1970 309. Gass JDM: A clinicopathologic study of a peculiar foveomacular dystrophy. Trans Am Ophthalmol Soc 72:139, 1974 310. Hsieh RC, Fine BS, Lyons JS: Patterned dystrophies of the retinal pigment epithelium. Arch Ophthalmol 95:429, 1977 311. Marmor MF, Byers B: Pattern dystrophy of the pigment epithelium. Am J Ophthalmol 84:32, 1971 312. Nichols BE, Sheffield VC, Vanderburgh K et al: Butterfly-shaped pigment dystrophy of the fovea caused by a point mutation
in codon 167 of the RDS gene. Nature Genet 3:202, 1993 313. Welleber RG, Carr RE, Murphey WH et al: Phenotypical variation including retinitis pigmentosa, pattern dystrophy, and
fundus flavimaculatus in a single family with a deletion of codon 153 or 154 of
the peripherin/RDS gene. Arch Ophthalmol 111:1531, 1993 314. Wells J, Wroblewski J, Keen J et al: Mutations in the human retinal degeneration slow (RDS) gene can cause either
retinitis pigmentosa or macular dystrophy. Nature Genet 3:213, 1993 315. Travis GH, Brennan MB, Danielson PE et al: Identification of a photoreceptor-specific mRNA encoded by the gene responsible
for retinal degeneration slow (RDS). Nature 338:70, 1989 316. Farrar JF, Kenna P, Jordan SA et al: A three-based-pair deletion in the peripherin-RDS gene in one form of retinitis
pigmentosa. Nature 365:478, 1990 317. Cawthon RM, Weiss R, Xu GF et al: A major segment of the neurofibromatosis type 1 gene: cDNA sequence, genomic
structure, and point mutations. Cell 62:193, 1990 318. Marchuk DA, Saulino AM, Tavakkol R: cDNA cloning of the type 1 neurofibromatosis gene: Complete sequence of
the NF1 gene product. Genomics 11:931, 1991 319. Wallace MR, Anderson LB, Saulino AM: A de novo insertion mutation causing neurofibromatosis type 1. Nature 353:864, 1991 320. Guttmann DH, Collins FS: Neurofibromatosis type 1. Arch Neurol 50:1185, 1993 321. Viskochil D, White R, Cawthon R: The neurofibromatosis type 1 gene. Ann Rev Neurosci 16:183, 1993 322. Rouleau GA, Merel P, Lutchman M et al: Alteration in a new gene encoding a putative membrane-organizing protein
causes neurofibromatosis type 2. Nature 363:515, 1993 323. Trafatter JA, MacCollin MM, Rutter JL et al: A novel moesinezrin-, radixin-like gene is a candidate for the neurofibromatosis 2 tumor
suppressor. Cell 72:791, 1993 324. Norrie G: Causes of blindness in children. Acta Ophthalmol (Copenh) 5:357, 1927 325. Sims KB, de la Chapelle A, Norio R et al: Monoamine oxidase deficiency in males with an X chromosome deletion. Neuron 2:1069, 1989 326. Warburg M: Norrie's disease. Arch Ophthalmol 66:30, 1961 327. Warburg M: Norrie's disease, a congenital progressive oculo-acoustico-cerebral
degeneration. Acta Ophthalmol 89(suppl):1, 1966 328. Warburg M: Norrie's disease—differential diagnosis and treatment. Acta Ophthalmol 53:217, 1975 329. Sims KB, Lebo RV, Benson G et al: The Norrie disease gene maps to a 150 kb region on chromosome Xp11.3. Hum Mol Genet 1:83, 1992 330. Berger W, Meindl A, van de Pol TJR et al: Isolation of a candidate gene for Norrie disease by positional cloning. Nature Genet 1:199, 1992 331. Chen Z-Y, Sims SB, Coleman M et al: Characterization of a YAC containing part or all of the Norrie disease
locus. Hum Mol Genet 1:161, 1992 332. Meindl A, Berger W, Meitinger T et al: Norrie's disease is caused by mutations in an extracellular protein
resembling C-terminal globular domain of mucins. Hum Mol Genet 2:139, 1992 333. Meitinger T, Mendl A, Bork P: Molecular modelling of the Norrie disease protein predicts a cystine knot
growth factor tertiary structure. Nature Genet 5:376, 1993 334. Black G, Redmond RM: The molecular biology of Norrie's disease. Eye 8:491, 1994 335. de la Chapelle A, Sankila E-M, Lindlof M et al: Norrie's disease caused by a gene deletion allowing carrier detection
and prenatal diagnosis. Clin Genet 28:317, 1985 335a. McLaughlin ME, Sandberg MA, Berson EL et al: Recessive mutations in the gene encoding the beta-subunit of rod phosphodiesterase
in patients with retinitis pigmentosa. Nature Genet 4:130, 1993 336. Olsson JE, Gordon JW, Pawlyk BS et al: Transgenic mice with a rhodopsin mutation (pro23his): a mouse model of
autosomal dominant retinitis pigmentosa. Neuron 9:815, 1992 337. Huang PC, Gaitin AE, Hao Y et al: Cellular interactions in the mechanism of photoreceptor degeneration in
transgenic mice expressing a mutant rhodopsin gene. Proc Natl Acad Sci USA 90:8484, 1993 338. Shields JA, Shields CL: Genetics of retinoblastoma. In Shields JA, Shields
CL (eds): Intraocular Tumors: A Text and Atlas, pp 333–339. Philadelphia, WB
Saunders, 1992 339. Knudson AG: Mutation and cancer: Statistical study of retinoblastoma. Proc Natl Acad Sci USA 68:820, 1971 340. Friend SH, Bernards R, Rodlj S et al: A human DNA segment with properties of the gene that predisposes to retinoblastoma
and osteosarcoma. Nature 323:643, 1986 341. Lee W-H, Bookstein R, Hong F et al: Human retinoblastoma susceptibility gene: Cloning, identification, and
sequence. Science 235:1934, 1987 342. Fung YKT, Murphree AL, T'Ang A et al: Structural evidence for the authenticity of the human retinoblastoma gene. Science 236:1659, 1987 343. McGee TL, Yandell DW, Dryja TP: Structure and partial genomic sequence of the human retinoblastoma susceptibility
gene. Gene 80:119, 1989 344. Bookstein R, Shew JY, Chen PL et al: Suppression of tumorigenicity of human prostate carcinoma cells by replacing
a mutated retinoblastoma gene. Science 247:712, 1990 345. Yandell DW, Campbell TA, Dayton SH et al: Oncogenic point mutations in the human retinoblastoma gene: their application
to genetic counseling. N Engl J Med 321:1689, 1989 346. Horowitz JM, Park S, Bogenmann E et al: Frequent inactivation of the retinoblastoma anti-oncogene is restricted
to a subset of human tumor cells. Proc Natl Acad Sci USA 87:2775, 1990 347. Lohmann D, Horsthemke B, Gillessen-Kaesback G et al: Detection of small RB1 gene deletions in retinoblastoma by multiple PCR
and high resolution gel electrophoresis. Hum Genet 89:49, 1992 348. Weir-Thompson E, Condie A, Leonard RCF et al: Familial RB1 mutation detected by HOT technique is homozygous in a second
primary neoplasm. Oncogene 6:2363, 1991 349. Dunn JM, Phillips RA, Zhu X et al: Mutations in the retinoblastoma 1 gene and their effects on transcription. Mol Cell Biol 9:4596, 1989 350. Bignon YJ, Shew JY, Rappolee D et al: A single Cys(706) to Phe substitution in the retinoblastoma protein causes
the loss of binding to SV40T antigen. Cell Growth Differ 1:647, 1990 351. Scheffner M, Munger K, Byrne JC et al: The state of p53 and retinoblastoma genes in human cervical carcinoma cell
lines. Proc Natl Acad Sci USA 88:5523, 1991 352. Kaye FJ, Kratke RA, Gerster JL et al: A single amino acid substitution results in a retinoblastoma protein defective
in phosphorylation and oncoprotein binding. Proc Natl Acad Sci USA 87:6922, 1990 353. Mori N, Yokota J, Akiyama et al: Variable mutations for the RB gene in small cell carcinoma. Oncogene 5:1713, 1990 354. Murakami Y, Katahira M, Makino R et al: Inactivation of the retinoblastoma gene in a human lung carcinoma cell
line detected by a single-strand conformation polymorphism analysis of
the polymerase chain reaction product of cDNA. Oncogene 6:43, 1991 355. Shew JY, Chen PL, Boostein R et al: Deletion of a splice donor site ablates expression of the following exon
and produces an unphosphorylated RB protein unable to bind SV40 T antigen. Cell Growth Differ 1:17, 1990 356. Harbour JW, Lai SL, Whang PJ et al: Abnormalities in structure and expression of the human retinoblastoma gene
in SCLC. Science 241:353, 1988 357. Friend SH, Horowitz JM, Gerber MR et al: Deletion of a DNA sequence in retinoblastoma and mesenchymal tumors: organization
of the sequence and its encoded protein. Proc Natl Acad Sci USA 84:9059, 1987 358. Lee W-H, Shew J-Y, Hong FD et al: The retinoblastoma susceptibility gene encodes a nuclear phosphoprotein
associated with DNA binding activity. Nature 329:642, 1987 359. Riley DJ, Lee EHP, Lee WH: The retinoblastoma protein: More than a tumor suppressor. Annu Rev Cell Biol 10:1, 1994 360. Schubert EL, Han MF, Strong LC: The retinoblastoma gene and its significance. Ann Med 26:177, 1994 361. Goodrich DW, Wang NP, Qian YW et al: The retinoblastoma gene product regulates progression through the G1 phase
of the cell cycle. Cell 67:293, 1991 362. Nevins JR: E2F: a link between the Rb tumor suppressor protein and viral oncoproteins. Science 258:424, 1992 363. Chellappan SP, Hiebert S, Mundryj M et al: The E2F transcription factor is a cellular target for the RB protein. Cell 65:1053, 1991 364. Whyte P, Buchkovich KJ, Horowitz JM et al: Association between an oncogene and an anti-oncogene: The adenovirus E1A
proteins bind to the retinoblastoma gene product. Nature 334:124, 1988 365. DeCaprio JA, Ludlow JW, Figge J et al: SV40 large tumor antigen forms a specific complex with the product of the
retinoblastoma susceptibility gene. Cell 54:275, 1988 366. Dyson N, Howley PM, Munger K et al: The human papillomavirus-16 E7 oncoprotein is able to bind to the retinoblastoma
gene product. Science 243:934, 1989 367. Szekely L, Selivanova G, Magnusson KP et al: EBNA-5, an Epstein-Barr virus-encoded nuclear antigen, binds to the retinoblastoma
and p53 proteins. Proc Natl Acad Sci USA 90:5455, 1993 368. Jiang WQ, Szekely L, Wendel-Hansen V et al: Colocalization of the retinoblastoma protein and the Epstein-Barr virus-encoded
nuclear antigen EBNA-5. Exp Cell Res 197:314, 1991 369. Ewen ME, Xing YG, Lawrence JB et al: Molecular cloning, chromosomal mapping, and expression of the cDNA for
p107, a retinoblastoma gene product-related protein. Cell 66:1155, 1991 370. Hu QJ, Bautista C, Edwards GM et al: Antibodies specific for the human retinoblastoma protein identify a family
of related polypeptides. Mol Cell Biol 11:5792, 1991 371. Mayol X, Grana W, Baldi A et al: Cloning of a new member of the retinoblastoma gene family (pRb2) which
binds to the E1A transforming domain. Oncogene 8:2561, 1993 372. Lee EY, Change CY, Hu N et al: Mice deficient for Rb are nonviable and show defect in neurogenesis and
hematopoiesis. Nature 359:288, 1992 373. Jacks T, Fazeli A, Schmitt EM et al: Effects of an Rb mutation in the mouse. Nature 359:295, 1992 374. Strohmeyer T, Reissmann P, Cordon-Cardo et al: Correlation between retinoblastoma gene expression and differentiation
in human testicular tumors. Proc Natl Acad Sci USA 88:6662, 1991 375. Wunder JS, Czitron AA, Kandel R et al: Analysis of alterations in the retinoblastoma gene and tumor grade in born
and soft-tissue sarcomas. J Natl Cancer Inst 83:194, 1991 376. Cance WG, Brennan MF, Dudas ME: Altered expression of the retinoblastoma gene product in human sarcomas. N Engl J Med 323:1457, 1990 377. Reissmann PT, Simon MA, Lee WH: Studies of the retinoblastoma gene in human sarcomas. Oncogene 4:839, 1989 378. T'Ang A, Varley JM, Charkraborty et al: Structural rearrangement of the retinoblastoma gene in human breast carcinoma. Science 242:263, 1988 379. Varley JM, Armour J, Swallow JE et al: The retinoblastoma gene is frequently altered leading to loss of expression
in primary breast tumors. Oncogene 4:725, 1989 380. Goodrich DW, Chen Y, Scully P et al: Expression of the retinoblastoma gene product in bladder carcinoma cells
associated with a low frequency of tumor formation. Cancer Res 52:1968, 1992 381. Takahashi R, Hashimoto T, Xu HJ et al: The retinoblastoma gene functions as a growth and tumor suppressor in human
bladder carcinoma cells. Proc Natl Acad Sci USA 88:5257, 1991 382. Xu HJ, Cairns P, Hu SX et al: Loss of RB protein expression in primary bladder cancer correlates with
loss of heterozygosity at the RB locus and tumor progression. Int J Cancer 53:781, 1993 383. Cordon-Cardo C, Wartinger D, Petrylak D et al: Altered expression of the retinoblastoma gene product: Prognostic indicator
in bladder cancer. J Natl Cancer Inst 84:1251, 1992 384. Logothetis CJ, Xu HJ, Ro JY et al: Altered expression of retinoblastoma protein and known prognostic variables
in locally advanced bladder cancer. J Natl Cancer Inst 84:1256, 1992 385. Steeg PS: Suppressor genes in breast cancer: an overview. Cancer Treat Res 61:45, 1992 386. Fung YK, T'Ang A: The role of the retinoblastoma gene in breast cancer development. Cancer Treat Res 61:59, 1992 387. Borg A, Zhang QX, Alm P et al: The retinoblastoma gene in breast cancer: Allele loss is not correlated
with loss of gene protein expression. Cancer Res 52:2991, 1992 388. Birch JM: Epidemiology of sarcomas. Curr Opin Oncol 2:462, 1990 388a. Wang NP, To H, Lee WH et al: Tumor suppressor activity of RB and p53 genes in human breast carcinoma
cells. Oncogene 8:279, 1993 389. Stickler GB, Belau PG, Farrell F et al: Mayo Clin Proc 40:433, 1965 390. Ahmad NN, Ala-Kokko L, Knowlton RG et al: Stop codon in the procollagen II gene (COL2A1) in a family with the Stickler
syndrome (arthro-ophthalmopathy). Proc Natl Acad Sci USA 88:6624, 1991 391. Francomano CA, Liberfarb RM, Hirose T et al: The Stickler syndrome: evidence for close linkage to the structural gene
for type II collagen. Genomics 1:293, 1987 392. Francomano CA, Rowan BG, Liberfarb RM et al: The Stickler and Wagner syndromes: evidence for genetic heterogeneity (abstr). Am J Hum Genet 43:A83, 1988 393. Knowlton RG, Weaver EJ, Struyk AF et al: Genetic linkage analysis of hereditary arthro-ophthalmopathy (Stickler
syndrome) and the type II procollagen gene. Am J Hum Genet 45:681, 1989 394. Fryer AE, Upadhyaya M, Littler M et al: Exclusion of COL2A1 as a candidate gene in a family with Wagner-Stickler
syndrome. J Med Genet 27:91, 1990 395. Ahmad NN, McDonald-McGinn DM, Zackai EH et al: A second mutation in the type II procollagen gene (COL2A1) causing stickler
syndrome (arthro-ophthalmopathy) is also a premature termination
codon. Hum Genet 52:39, 1993 396. Haines JL, Short MP, Kwiatkowski DJ et al: Localization of one gene for tuberous sclerosis within 9q32–9q34, and
further evidence for heterogeneity. Am J Hum Genet 49:764, 1991 397. Kandt RS, Haines JL, Smith M: Linkage of an important gene locus for tuberous sclerosis to a chromosome 16 marker
for polycystic kidney disease. Nature Genet 2:37, 1992 398. Jannsen LA, Povey S, Altwood J: A comparative study of genetic heterogeneity in tuberous sclerosis: evidence
for one gene on 9q34 and a second gene on 1q22-23. Ann NY Acad Sci 615:306, 1991 399. Smith M, Smalley S, Cantor R et al: Mapping of a gene determining tuberous sclerosis to human chromosome 11q14–11q23. Genomics 6:105, 1990 400. Silcock AQ: Hereditary sarcoma of the eyeball. Trans Pathol Soc Lond 43:1401, 1892 401. Parsons JH: Some anomalous sarcomata of the choroid. Trans Ophthalmol Soc UK 25:205, 1905 402. Davenport RC: Family history of choroidal sarcoma. Br J Ophthalmol 11:443, 1927 403. Gutmann G: Casuistischer Bertrag zur Lehre von den Geschweulsten des Augafels. Arch Argenheilkd 31:158, 1895 404. Pfingst AO, Graves S: Melanosarcoma of the choroid occurring in two brothers. Arch Ophthalmol 50:431, 1921 405. Waardenburg PJ: Malanosarcoma van bet vog bij verschillendeleden eener zelfde familie. Ned Tijdschr Geneeskd 84:4718, 1940 405a. Tasman W: Familial intraocular melanoma. Trans Am Acad Ophthalmol Otolaryngol 74(5):955, 1070 406. Lynch HT, Anderson DE, Krush AJ: Heredity and intraocular melanomas. Cancer 21:119, 1968 407. Singh AD, Wang MX, Donoso LA et al: Familial uveal melanoma—III: Is
the occurrence of familial uveal melanoma coincidental? Arch Ophthalmol (in
press) 408. Wang MX, Early JJ, Shields JA et al: An ocular melanoma-associated antigen: molecular characterization. Arch Ophthalmol 110:399, 1992 408a. Wang MX, Shields JA, Donoso LA: Subclinical metastasis of uveal melanoma. Int Ophthalmol Clin 33:119, 1993 409. von Hippel E: Uber sine sehr self seltene erkrankung dernetzhaut Klinische Beobachtungen. Arch Ophthalmol 59:83, 1904 410. Lindau A: Studien ber kleinbirncysten bau: Pathogenese und beziehunger zur angiomatosis
retinae. Acta Radiol Microbiol Scand 1(suppl):1, 1926 411. Latif F, Kalman T, Gnarra J et al: Identification of the von Hippel-Lindau disease tumor suppressor gene. Science 260:1317, 1993 412. Glenn GM, Linehan M, Hosoe S et al: Screening for von Hippel-Lindau disease by DNA polymorphism analysis. JAMA 267:1226, 1992 413. Deutman AF: Vitreoretinal dystrophies. In Krill AE (ed): Krill's Hereditary
Retinal and Choroidal Diseases, Vol II, pp 1043–1100. New
York, Harper & Row, 1977 413a. Miller RF, Dowling JE: Intracellular responses of the Muller (glial) cells of mudpuppy retina: their
relation to b-wave of the electroretinogram. J Neurophysiol 33:323, 1970 413b. Yanoff M, Kertesz Rahn E, Zimmerman LE: Histopathology of juvenile retinoschisis. Arch Ophthalmol 79:49, 1968 413c. Condon GP, Brownstein S, Wang NS et al: Congenital hereditary (juvenile X-linked) retinoschisis. Histopathologic
and ultrastructural findings in three eyes. Arch Ophthalmol 104:576, 1986 413d. Sieving PA, Bingham E, Roth MS et al: Linkage relationship of X-linked juvenile retinoschisis with Xp22.1-p22.3 probes. Am J Hum Genet 47:616, 1990 413e. Alitalo T, Kruse TA, de la Chapelle A: Refined localization of the gene causing X-linked juvenile retinoschisis. Genomics 9:505, 1991 413f. Woo SLC, Kidd VJ, Pam ZK et al: Bandury report 14. In Caskey CT, White
RL (eds): Recombinant DNA Application to Human Disease. Cold Spring Harbor, NY, Cold
Spring Harbor Laboratory, 1983 414. Gibbs RA, Nguen PN, Caskey T: Detection of single DNA base difference by competitive oligonucleotide
priming. Nucleic Acids Res 17:2437, 1989 415. Newton CR, Graham A, Heptinstall LE et al: Analysis of any point mutation in DNA: the amplification refractory mutation
systems (ARMS). Nucleic Acids Res 17:2503, 1989 416. Landegren U, Kaiser R, Sander J et al: A ligase-mediated gene detection technique. Science 241:1077, 1988 417. Gibbs RA, Caskey CT: Identification and localization of mutations at the Lesch-Nyhan locus by
ribonuclease A cleavage. Science 236:303, 1987 418. Sheffield VC, Beck JS, Nichols B et al: Detection of multiallele polymorphism within gene sequences by GC-clamped
denaturing gradient gel electrophoresis. Am J Hum Genet 50:567, 1992 419. Borresen AL, Hovig E, Smith-Borensen B et al: Constant denaturing gel electrophoresis as a rapid screening technique
for p53 mutations. Proc Natl Acad Sci USA 88:8405, 1991 420. Soracher EJ, Huang Z: Diagnosis of genetic disease by primer-specified restriction map modification, with
application to cystic fibrosis and retinitis pigmentosa. Lancet 337:1115, 1991 421. Cotton RGH, Rodrigues NR, Campbell RD: Reactivity of cytosine and thymine in single-base-pair mismatches with
hydroxylamine and osmium tetroxide and its application to the study of
mutations. Proc Natl Acad Sci USA 85:4397, 1988 422. Chen Z-Y, Battinelli EM, Woodruff G et al: Characterization of a mutation within the NDP gene in a family with a manifesting
female carrier. Hum Mol Genet 2:1727, 1993 423. Mange AP, Mange EJ: Genetics: Human Aspects. Sunderland, MA, Sinauer Associates, 1990 424. Wachtel S, Elias S, Price J et al: Fetal cells in the maternal circulation: isolation by multiparameter flow
cytometry and confirmation by polymerase chain reaction. Hum Reprod 6:1466, 1991 425. Bianchi DW, Flint AF, Pizzimenti MF et al: Isolation of fetal DNA from nucleated erythrocytes in maternal blood. Proc Natl Acad Sci USA 87:3279, 1990 426. Omadim Z, Hungerfold J, Cowell JK: Follow-up retinoblastoma patients having prenatal and perinatal predictions
for mutant gene carrier status using intragenic probes for the RB1 gene. Br J Cancer 65:711, 1992 427. Maher ER, Bentley E, Payne SJ: Presymptomatic diagnosis of von Hippel-Lindau disease with flanking DNA
markers. J Med Genet 29:902, 1992 428. Miller AD: Human gene therapy comes of age. Nature 357:455, 1992 429. Thompson L: At age 2, gene therapy enters a growth phase. Science 258:744, 1992 430. Piatigorsky J, Wistow GJ: Enzyme/crystallins: Gene sharing as an evolutionary strategy. Cell 57:197, 1989 431. Kumar R, Zack DJ: Regulation of visual pigment gene expression. In Wiggs
JL (eds): Molecular Genetics of Ocular Diseases. New York, Wiley-Liss, 1995 432. Wang MX, Church GM: A whole genome approach to in vivo DNA-protein interaction in E. coli. Nature 360:606, 1992 433. Wax M, Patil R: A rationale for gene targeting in glaucoma therapy. J Ocular Pharmacol 10:403, 1994 434. Milam AH: Strategies for rescue of retinal photoreceptor cells. Curr Opin Neurol 3:797, 1993 |