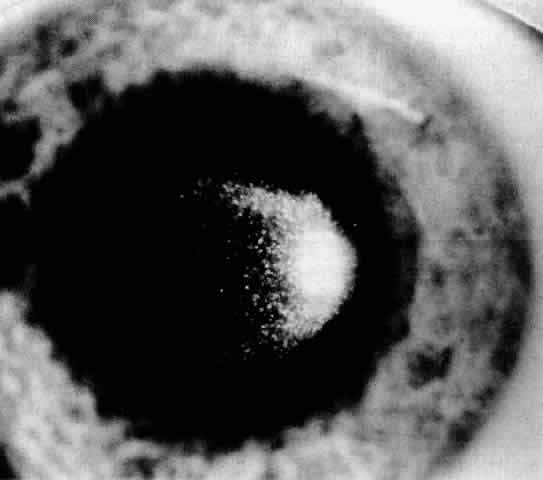
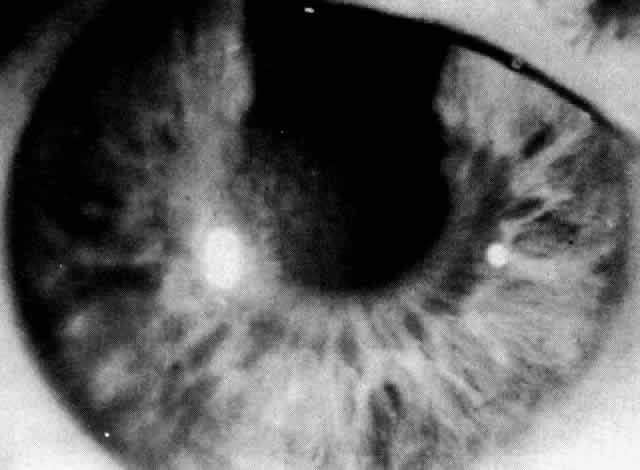
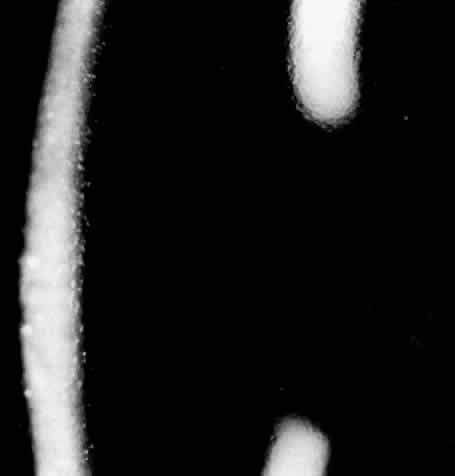
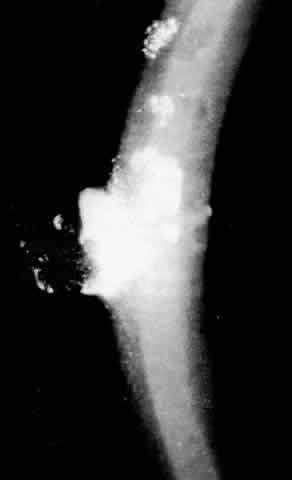
1. Guerry D: Fingerprint lines in the cornea. Am J Ophthalmol 33:724, 1950 2. Chandler PA: Recurrent erosion of the cornea. Am J Ophthalmol 28:355, 1945 3. Spicer WTH, Greeves RA: On superficial linear keratitis together with an account of the pathological
examination of two affected eyes. Ophthalmology 14:116, 1916 4. DeVoe AG: Certain abnormalities of Bowman's membrane with particular reference
to fingerprint lines in the cornea. Trans Am Ophthalmol Soc 60: 195, 1962 5. Cogan DG, Donaldson DD, Kuwabara T, Marshall D: Microcystic dystrophy of the corneal epithelium. Trans Am Ophthalmol Soc 62:213, 1964 6. Guerry D: Observations on Cogan's microcystic dystrophy of the corneal epithelium. Trans Am Ophthalmol Soc 63:320, 1965 7. Wolter JR, Fralick FB: Microcystic dystrophy of the corneal epithelium. Arch Ophthalmol 75:380, 1966 8. Trobe JD, Laibson PR: Dystrophic changes in the anterior cornea. Arch Ophthalmol 87:378, 1972 9. Bron AJ, Tripathi RC: Cystic disorders of the corneal epithelium: I. Clinical aspects. Br J Ophthalmol 57:361, 1973 10. Poohkay ME, Hassard DT: Fingerprint dystrophy of the cornea. Can J Ophthalmol 8:54, 1973 11. Luxenberg MN. Freidland BR, Holder JM: Superficial microcystic corneal dystrophy. Arch Ophthalmol 93: 107, 1975 12. Rodrigues MM, Fine B, Laibson P, Zimmerman L: Disorders of the corneal epithelium: A clinicopathologic study of dot, geographic, and
fingerprint patterns. Arch Ophthalmol 92:475, 1974 13. Laibson PR, Krachmer JH: Familial occurrence of dot (microcystic), map, fingerprinting dystrophy
of the cornea. Invest Ophthalmol 14:397, 1975 14. Rodrigues MM, Laibson PR: Recurrent corneal erosions. Trans PA Acad Ophthalmol Otolaryngol 29: 171, 1976 15. Brown NA, Bron AJ: Superficial lines and associated disorders of the cornea. Am J Ophthalmol 81:34, 1976 16. Brown NA, Bron AJ: Recurrent erosion of the cornea. Br J Ophthalmol 60:84, 1976 17. Franceschetti A et al: Clinical and social aspects of heredity in ophthalmology. In: Acta
XVI Concilium Ophthalmologicum, vol 1, pp 157–299. London, British
Medical Association, 1951 18. Laibson PR: Microcystic corneal dystrophy. Trans Am Ophthalmol Soc 74:488, 1976 19. Werblin TP, Hirst LW, Stark WJ, Maumenee HH: Prevalence of map dot fingerprint change in the cornea. Br J Ophthalmol 65:401, 1981 20. Foulks GN: Treatment of recurrent corneal erosions and corneal edema with topical
osmotic colloidal solution. Ophthalmology 88:801, 1981 21. Buxton JN, Constad WH: Superficial epithelial keratectomy in the treatment of epithelial basement
membrane dystrophy. Ann Ophthalmol 19:92, 1987 22. Wood TO, Griffith ME: Surgery for corneal epithelial basement membrane
dystrophy. Ophthalmic Surg 19:20. 1988 23. Pameijer JK: Ueber eine Fremdartige familiare oberflach liche Hornhautverandering. Klin Monatsbl Augenheilkd 95:516, 1935 24. Burns RP: Meesmann's corneal dystrophy. Trans Am Ophthalmol Soc 66:530, 1968 25. Meesmann A: Über eine bisher nicht Beschriebene, dominant verebte Dystrophia epithelialis
corneae. Bet Dtsch Ophthalmol Ges 52: 154, 1938 26. Meesmann A, Wilke F: Klinische and anatomische Untersuchungen uber eine bisher unbekannte dominant
vererbte epitheldystrophie der Hornhaul. Klin Monatsbl Augenheilkd 103:361, 1939 27. Bron AJ, Tripathi RC: Corneal disorders. In Goldberg MF (ed): Genetic and
Metabolic Eye Disease, pp 281–323. Boston, Little, Brown & Co, 1974 28. Kuwabara T, Ciccarelli EC: Meesmann's corneal dystrophy: A pathological study. Arch Ophthalmol 71:676, 1964 29. Wittebol-Post D, van Bijstervedt OP, Delleman JW: Meesmann's epithelial dystrOphy of the cornea. Ophthalmologica 194:44, 1987 30. Reis H: Familiare, flechige Hornhautartung. Dtsch Med Wochenschr 93:575, 1917 31. Bucklers M: Ueber eine weirere familiare Hornhautdystrophyae (Reis). KIln Monatsbl Augenheilkd 114:386, 1949 32. Grayson M, Wilbrandt H: Dystrophy of the anterior limiting membrane of the cornea (Reis-Bucklers' type). Am J Ophthalmol 61:345, 1966 33. Rice NCS, Ashton N, Jay B, Blach RK: Reis-Bucklers' dystrophy: A clinicopathological study. Br J Ophthalmol 52:577, 1968 34. Griffith DG, Fine BS: Light and electron microscopic observations in a superficial corneal dystrophy: Probable
early Reis-Bucklers' type. Am J Ophthalmol 63:1659, 1967 35. Jones ST, Stouffer LK: Reis-Bucklers' corneal dystrophy. A clinicopathological study. Trans Am Acad Ophthalmol Otolaryngol 74:417, 1970 36. Akiya S, Brown SI: The ultrastructure of Reis-Bucklers' dystrophy. Am J Ophthalmol 72:549, 1971 37. Waring GO, Rodrigues MM, Laibson PR: Corneal dystrophies: I. Dystrophies of the epithelium, Bowman's layer
and stroma. Surv Ophthalmol 23:71, 1978 38. Perry HD, Fine BS, Caldwell DR: Reis-Bucklers' dystrophy. Arch Ophthalmol 97:664, 1979 39. Caldwell DR: Post operative recurrence of Reis-Bucklers dystrophy. Am J
Ophthalmol 85:567. 1978 40. Wood TO, Fleming JC, Dotson RS, Cotten MS: Treatment of Reis-Bucklers' corneal dystrophy by removal of subepithelial
fibrous tissue. Am J Ophthalmol 85:360, 1978 41. Olson RJ, Kaufman H: Recurrence of Reis-Bucklers' corneal dystrophy in a graft. Am J Ophthalmol 85:349, 1978 42. Yamaguchi T, Polack FM, Valenti J: Electron microscopic study of recurrent Reis-Bucklers' corneal dystrophy. Am J Ophthalmol 90:95, 1980 43. Thiel H-J, Behnke H: Eine bisher unbekannte subepitheliale hereditäre Hornhautdystrophie. Klin Monatsbl Augenheilkd 150:862, 1967 44. Yamaguchi T, Polack FM, Rowsy JJ: Honeycomb-shaped corneal dystrophy: A variation of Reis-Bucklers' dystrophy. Cornea 1:71, 1982 45. Wittebol-Post D, Van Bijstervel OP, Delleman JW: The honeycomb type of Reis-Bucklers' dystrophy of the cornea: Biometrics
and an interpretation. Ophthalmologica 194:65, 1987 46. Wittebol-Post D, Van Schoeneveld MJ, Pels E: The corneal dystrophy of Waardenburg and jonkers. Ophthalmic Paediatr Genet 10:249, 1989 47. Vogt A: Lehrlauch und Atlas der Spaltlampen: Mikroskopie des Levenden, Vol 1, Auges. Berlin, Springer, 1930 48. Bron AJ, Tripathi RC: Corneal disorderS. In Golberg MF (ed): Genetic and
Metabolic Eye Disease, pp 281–323. Boston, Little, Brown & Co, 1974 49. Kopsa M, Marusic K: Contribution à l'étude de la dégénérescence
en mosaique de la membrane de Bowman de la cornée. Ophthalmologica 136:83, 1958 50. Valerio M: Due forme rare di degenerazione giovanile della cornea: Degenerazione a
mosaico della cornea. Un nuovo tipo di degenerazione cristalloide della
cornea. Boil Ocul 18:659, 1939 51. Boles-Carenini B: Juvenile familial mosaic degenerations of the cornea associated with megaloCornea. Br J Ophthalmol 45:64, 1961 52. Collier M: Un noveaue cas unilateral de dégénerescence en mosaique de
la membrane de Bowman, associé à une dégénérescence
en bandelette sur oeil malforme. Arch Ophthalmol (Paris) 26:253, 1966 53. Franceschetti A, Forni S: The heredofamilial degenerations of the cornea: Clinical
aspects. In: XVI Concilium Ophthalmologicum, vol 1, p 193. London, British
Medical Association, 1951 54. Velhagen: Über die primäre bandförmige Hornhauttrübung. Klin
Monatsbl Augenheilkd 42:428, 1904 55. Fuchs A: Ueber primaere quertelfoermige Hornhauttruebung. Klin Monatsbl Augenheilkd 103:300, 1939 56. Streiff EB, Zwahlen P: Une famille avec dégénérescence en bandelette de la
cornea. Ophthalmologica 111:129, 1946 57. Groenouw A: Knötchenförmige Hornhauttrübungen (noduli corneae). Arch Augenheilkd 21:281, 1890 58. Groenouw A: Knötchenförmige Hornhauttrübungen vererbte durch drei generationen. Klin Monatsbl Augenheilkd 58:411, 1917 59. Groenouw A: Knötchenf6rmige Hornhauttrübungen vererbte durch vier generationen. Klin Monatsbl Augenheilkd 90:577, 1933 60. Bucklers M: Die erblichen Hornhautdystrophien. Dystrophiae corneae hereditariae. In: Bucherei
des Augenarztes. Stuttgart, Ferdinand Enke, 1938 61. Freiberger M: Corneal dystrophy in three generations. Arch Ophthalmol 16:257, 1936 62. Schutz S: Hereditary corneal dystrophy: History of the condition and presentation
of a pedigree. Arch Ophthalmol 29:523, 1943 63. Moller H: Granular corneal dystrophy Groenouw type I: 115 Danish patients: An epidemiological
and genetic population study. Acta Ophthalmol 68:297, 1990 64. Moller H, Eiberg H, Kruse T: Linkage relations of the locus for granular corneal dystrophy Groenouw
type 1 with 35 polymorphic systems. Acta Ophthalmol 67:721, 1989 65. Pinckers A, Otto A, Van Den Heuvel J: A family pedigree with corneal dystrophy and tapetoretinal degeneration
and albinism. Acta Ophthalmol 51:445, 1973 66. Folberg R, Croxatto O, Dreizen N: Clinically atypical granular corneal dystrophy with pathologic features
of lattice-like amyloid deposits. Ophthalmology 95:46, 1988 67. Smith G, Rabinowitz Y, Sassani J, Smith R: Keratoconus and lattice and granular corneal dystrophies in the same eye. Am J Ophthalmol 108:608, 1989 68. Moller H, Ridgway A: Granular corneal dystrophy Groenouw type I: A report of a probable homozygous
patient. Acta Ophthalmol 68:97, 1990 69. Jones S, Zimmerman L: Histopathologic differentiation of granular, macular, and lattice dystrophies
of the cornea. Am J Ophthalmol 51:394, 1961 70. Sornson E: Granular dystrophy of the cornea: An electron microscopic study. Am J Ophthalmol 59: 1001, 1965 71. Akiya S, Brown S: Granular dystrophy of the cornea: Characteristic electron microscopic lesion. Arch Ophthalmol 84: 179, 1970 72. Rodrigues M, Streeten B, Krachmer J et al: Microfibrillar protein and phospholipid in granular corneal dystrophy. Arch Ophthalmol 101:802, 1983 73. Rodrigues M, Krachmer J: Recent advances in corneal stromal dystrophies. Cornea 7: 19, 1988 74. Miller C, Krachmer J: Corneal diseases. In Renie WA (ed): Goldberg's
Genetic and Metabolic Eye Disease, pp 297–367. Boston, Little. Brown & Co, 1986 75. Brownstein S, Fine B, Sherman M. Zimmerman L: Granular dystrophy of the cornea: Light and electron microscopic confirmation
of recurrence in a graft. Am J Ophthalmol 77:701, 1974 76. Rodrigues M. McGavic J: Recurrent corneal granular dystrophy: A clinicopathologic study. Trans Am Ophthalmol Soc 73:306, 1975 77. Stuart J. Mund M, Iwamoto T et al: Recurrent corneal granular dystrophy. Am
J Ophthalmol 79: 18. 1975 78. Lempert S, Jenkins M et al: A simple technique for removal of recurring
granular dystrophy in corneal grafts. Am J Ophthalmol 86:89. 1978 79. Haddad R, Font R, Fine B: Unusual superficial variant of granular dystrophy of the cornea. Am J Ophthalmol 83:213, 1977 80. Rodrigues M, Gaster R, Pratt M: Unusual superficial form of granular corneal
dystrophy. Ophthalmology 90:1507. 1983 81. Bucklers M: Die erblichen Hornhautdystrophien. Klin Monatsbl Augenheilkd 3(suppl):l, 1938 82. Moller H: Granular corneal dystrophy Groenouw type 1 and Reis-Bucklers' corneal dystrophy: One
entity? Acta Ophthalmol 67:678, 1989 83. Winkelman J, Wittebol-Post D, Delleman J: Ein Beitrag zur Hornhautdystrophie Reis-Bucklers. Klin Monatsbl Augenheilkd 188: 143, 1986 84. Thiel H, Müller-Hermelink K: Zur pathogenese der wa-benformigen hornhaut dystrophie. Ber Dtsch Ophthalmol Ges 78:415, 1981 85. Kompf J et al: Linkage analysis in granular corneal dystrophy (Groenouw
I), Schnyder's crystalline corneal dystrophy, and Reis-Bucklers' corneal
dystrophy. Graefe's Arch Clin Exp Ophthalmol 227:538. 1989 86. Waardenburg P, Jonkers G: A specific type of dominant progressive dystrophy of the cornea developing
after birth. Acta Ophthalmol 39:919, 1961 87. Miller C, Krachmer J: Epithelial and stromal dystrophies.In: The Cornea, pp 383–424. New
York, Churchill Livingstone, 1988 88. Winkelman J, Wittebol-Post D, Delleman J: Ein Beitrag zur hornhautdystrophie Reis-Bucklers. KIln Monatsbl Augenheilkd 188: 143, 1986 89. Biber H: Über einige seltene Hornhauterkrankungen Inaug Dissert, pp 35–42. Zurich, A. Diggelmann, 1890 90. Haab O: Die gitterige keratitis. Zeitr Augenheilkd 2:235, 1899 91. Dimmer F: Ueber oberflachliche gittrige Hornhauttrubungen. Z Augenheilkd 2:353, 1899 92. Franceschetti A, Babel S: II. The heredofamilial degenerations of the cornea: B. Pathological
anatomy. Acta XVI Can Ophthalmol 1951, pp 245–283 93. Seitelberger F, Nemetz V: Beitrag zur frage der gitttrigen Hornhautdystrophie. Graefes Arch Clin Exp Ophthalmol 164:102, 1961 94. Klintworth G: Lattice corneal dystrophy, an inherited variety of amyloidosis restricted
to the cornea. Am J Pathol 50:371, 1967 95. Stansbury F: Lattice type of hereditary corneal degeneration. Arch Ophthalmol 40: 189, 1948 96. Dark A, Thompson D: Lattice dystrophy of the cornea: A clinical and microscopic study. Br J Ophthalmol 44:257, 1960 97. Mehta R: Unilateral lattice dystrophy of the cornea. Br J Ophthalmol 64:53, 1980 98. Malbran E, Meijide R, Croxatto J: Atypical corneal dystrophy with stromal amyloid deposits. Cornea 7:210, 1988 99. Meretoja J: Familial systemic paramyloidosis with lattice dystrophy of the cornea, progressive
cranial neuropathy, skin changes and various internal symptoms. Ann Clin Res 1:314, 1969 100. Meretoja J: Comparative histopathological and clinical findings in eyes with lattice
corneal dystrophy of two different types. Ophthalmologica 165:15, 1972 101. Darras B et al: Familial amyloidosis with cranial neuropathy and corneal
lattice dystrophy. Neurology 36:432.1986 102. Purcell J, Rodrigues M et al: Lattice corneal dystrophy associated with familial systemic amyloidosis (Meretoja's
syndrome). Ophthalmology 90:1512, 1983 103. Hida T, Tsubota K et al: Clinical features of a newly recognized type of lattice corneal dystrophy. Am J Ophthalmol 104:241, 1987 104. Hida T, Proia A et al: Histopathologic and immunochemical features of lattice corneal dystrophy
type 111. Am J Ophthalmol 104:249, 1987 105. Garner A: Amyloidosis of the cornea. Br J Ophthalmol 53:73, 1969 106. Rodrigues M, Zimmerman L: Secondary amyloidosis in ocular leprosy. Arch Ophthalmol 85:277, 1971 107. Stock E, Kielar R: Primary familial amyloidosis of the cornea. Am J Ophthalmol 82:266, 1976 108. Mannis M, Krachmer J et al: Polymorphic amyloid degeneration of the cornea. Arch Ophthalmol 99: 1217, 1981 109. Franceschetti A, Streiff E: Hereditary and constitutional dystrophies of
the cornea. In Ridley F, Sorsby A (eds): Modern Trends in Ophthalmology, pp 414–427. London, Butterworth, 1940 110. Moro F, Amidei B: Distrofia corneale screziata (o di Fehr) associata a sordita e balbuzie. Ann Ottalmol Clin Ocul 83:30, 1957 111. Klintworth GK: Corneal dystrophies, In Nicholson DH (ed): Ocular Pathology
Update. New York, Masson, 1980 112. Donnenfeld E, Cohen EJ, Ingraham H, Poleski S et al: Corneal thinning in macular corneal dystrophy. Am J Ophthalmol 101:112, 1986 113. Ehlers N, Bramsen T: Central thickness in corneal disorders, Acta Ophthalmol 56:412, 1978 114. Quantock A et al: Macular corneal dystrophy: Reduction in both corneal thickness and collagen
interfibrillar sparing. Curr Eye Res 9:393, 1990 115. Hassell J, Hascall V, Ledbetter S et al: Corneal proteoglycan biosynthesis
and macular corneal dystrophy. In Sheffield J, Hilfer S (eds): Cell
and Developmental Biology of the Eye: Heredity and Visual Development, p 101 . New
York, Springer-Verlag, 1984 116. Klintworth G, Meyer R, Dennis R et al: Macular corneal dystrophy: Lack of keratan sulfate in serum and cornea. Ophthalmic Paediatr Genet 7: 139, 1986 117. Thonar E, Meyer R, Dennis R et al: Absence of normal keratan sulfate in the blood of patients with macular
corneal dystrophy. Am J Ophthalmol 102:561, 1986 118. Edward D, Thonar E, Srinivasan M et al: Macular dystrophy of the cornea: A systemic disorder of keratan sulfate
metabolism. Ophthalmology 97:1194, 1990 119. Yang C, Sundaraj N et al: Immunohistochemical evidence of heterogeneity in macular corneal dystrophy. Am J Ophthalmol 106:65, 1988 120. Edward DP, Yue B, Sugar J et al: Heterogeneity in macular corneal dystrophy. Arch Ophthalmol 106: 1579, 1988 121. Klintworth G, Reed J, Stainer G et al: Recurrence of macular corneal dystrophy within grafts. Am J Ophthalmol 95:60, 1983 122. Herman S, Hughes W: Recurrence of hereditary corneal dystrophy following keratoplasty. Am J Ophthalmol 75:689, 1973 123. Van Went J, Wibaut F: Ned T Geneesk 68:2996, 1924 124. Schnyder W: Ein bisher nicht bekannter: Typus von familiärer, praseniler, subkapsularer
schalenkatarakt und gleichzeitiges familiäres vorkommen
von gaskorperveran-derungen. Schweiz Med Wochenschr 57:403, 1927 125. Schnyder W: Mitteilung uber einen neuen Typus von familiärer Hornhauterkrankung. Schweiz Med Wochenschr 59:559, 1929 126. Schnyder W: Scheibenformige kristalleinlagerungen in der hornhaut mitre als Erbleiden. KIln Monatsbl Augenheilkd 103:494, 1939 127. Franceschetti A, Forni D: La degenerayione cristallinea ereditoria del la cornea ed I suoi rapporte
con le distrofie corneali eredofamiliari. Boll Ocul 31:3, 1952 128. Sysi R: Xanthoma cornea as hereditary dystrophy. Br J Ophthalmol 34:369, 1950 129. Ehlers N, Matthiessen M: Hereditary crystalline corneal dystrophy of Schnyder. Acta Ophthalmol 51: 316, 1973 130. Lisch W, Weidle E, Lisch C et al: Schnyder's dystrophy, progression and metabolism. Opbthalmic Paediatr Genet 7:45, 1986 131. Kompf J et al: Linkage analysis in granular corneal dystrophy (Groenouw 1), Schnyder's
crystalline corneal dystrophy, and Reis-Bucklers corneal dystrophy. Graefes Arch Clin Exp Ophthalmol 227:538, 1989 132. Delleman J, Winkelman J: Degeneratio corneae cristal linea hereditaria: A clinical, genetical and
histological study. Ophthalmologica 155:409, 1968 133. Fry W, Pickett W: Crystalline dystrophy of the cornea. Trans Am Ophthalmol Soc 48:220, 1950 134. Luxenberg M: Hereditary crystalline dystrophy of the cornea. Am J Ophthalmol 63:507, 1967 135. Bron A, Williams H, Carruthers M et al: Hereditary crystalline stromal dystrophy of Schnyder: Clinical features
of a family with hyperlipoproteinaemia. Br J Ophthalmol 56:383, 1972 136. Bron A, Williams H: Lipaemia of the limbal vessels. Br J Ophthalmol 56:343, 1972 137. Grop K: Clinical and histological findings in crystalline corneal dystrophy. Acta Ophthalmol 120:52, 1973 138. Rodrigues M, Kruth H, Krachmer J, Willis R: Unesterified cholesterol in Schnyder's corneal crystalline dystrophy. Am J Ophthalmol 104:157, 1987 139. François J: Une nouvelle dystrophie hérédo-familiale de la cornée. J Genet Hum 5:189, 1956 140. Strachan 1M: Cloudy central corneal dystrophy of François. Br J
Ophthalmol 53: 192, 1969 141. Bramsen T, Ehlers N, Baggesen LH: Central cloudy corneal dystrophy of François. Acta Ophthalmol 54:221, 1976 142. Collier M: Dystrophie mouchetée du parenchyme cornéen avec dystrophie
nuageuse centrale. Bull Soc Ophtalmol Fr 64:608, 1964 143. Collier M: Elastorrhexie systematisée et dystrophies cornéennes
chez deux soeurs. Bull Soc Ophtalmol Fr 65:301. 1965 144. François J, Neetens A: Nouvelle dystrophie hérédo-familiale du parenchyme cornéen ( hérédo-dystrophie mouchetée). Bull Soc Beige Ophtalmol 114:641, 1957 145. Bietti G: Contribution h la connaissance des dégénéres-censes
cornéenes seniles. Arch Ophthalmol 25:37, 1965 146. Vogt A: Corneal Degenerations of Various Etiology. In Blodi FC (trans): Textbook
and Atlas of Slit Lamp Microscopy of the Living Eye, vol 1, p 120. Bonn, JP
Wayenborgh, 1981 147. Goodside V: Posterior crocodile shagreen. Am J Ophthalmol 46:748, 1958 148. Tripathi R, Bron A: Secondary anterior crocodile sha green of Vogt. Br J Ophthalmol 59:59, 1975 149. Ansons A, Atkinson P: Corneal mosaic patterns: Morphology and epidemiology. Eye 3:811, 1989 150. Krachmer JH, Dubord PJ, Rodrigues MM, Mannis MJ: Corneal posterior crocodile shagreen and polymorphic amyloid degeneration: A
histopathologic study. Arch Ophthalmol 101:54, 1983 151. Thomsitt J, Bron A J: Polymorphic stromal dystrophy. Br J Ophthalmol 59: 125, 1975 152. Strachan IM: Pre-Descemetic corneal dystrophy. Br J Ophthalmol 52:716, 1968 153. Goldberg MF, Krimmer B, Sugar J et al: Variable expression in flecked (speckled) dystrophy of the cornea. Ann Ophthalmol 9:899, 1977 154. Purcell JJ Jr, Krachmer JH, Weingeist TA: Fleck corneal dystrophy. Arch Ophthalmol 95:440, 1977 155. Patten JT, Hyndiuk RA, Donaldson DD et al: Fleck ( mouchetée) dystrophy of the cornea. Ann Ophthalmol 8:25, 1976 156. Aracena T: Hereditary fleck dystrophy of the cornea: Report of a family. J Pediatr Ophthalmol 12:223, 1975 157. Birndorf LA, Ginsberg SP: Hereditary fleck dystrophy associated with decreased corneal sensitivity. Am J Ophthalmol 73:670, 1972 158. Kiskaddon BM, Campbell RJ, Waller RR, Bourne WM: Fleck dystrophy of the cornea: Case report. Ann Ophthalmol 12:700, 1980 159. Nicholson DH, Green WR, Cross HE et al: A clinical and histopathological study of François-Neetens speckled
corneal dystrophy. Am J Ophthalmol 83:554, 1977 160. Streeten BW, Falls HF: Hereditary fleck dystrophy of the cornea. Am J Ophthalmol 51:275, 1961 161. Gillespie F, Covelli B: Fleck ( mouchetée) dystrophy of the cornea: Report of a family. South Med J 56: 1265, 1963 162. Grayson M, Wilbrandt H: Pre-Descemet dystrophy. Am J Ophthalmol 64:276, 1967 163. Curran RE, Kenyon KR, Green WR: Pre-Descemet's membrane corneal dystrophy. Am J Ophthalmol 77:711, 1974 164. Sever RJ, Frost P, Weinstein G: Eye changes in ichthyosis. JAMA 206:2283, 1968 165. Dodwell DG, Freeman K, Shoch D: Juvenile asteroid hyalosis and pre-Descemet's dystrophy. Am J Ophthalmol 106:504, 1988 166. Witschel H, Fine BS, Grutzner P, McTigue JW: Congenital hereditary stromal dystrophy of the cornea. Arch Ophthalmol 96: 1043, 1978 167. Carpel EF, Sigelman R J, Doughman DJ: Posterior amorphous corneal dystrophy. Am J Ophthalmol 83:629, 1977 168. Dunn SP, Krachmer JH, Ching SS: New findings in posterior amorphous dystrophy. Arch Ophthalmol 102:236, 1984 169. Nakaizumi K: A rare case of corneal dystrophy. Nippon Ganka Gakkai Zasshi 18:949, 1914 170. Weber FL, Babel J: Gelatinous drop-like dystrophy. Arch Ophthalmol 98: 144, 1980 171. Gartry DS, Falcon MG, Cox RW: Primary gelatinous drop-like keratopathy. Br J Ophthalmol 73:661, 1989 172. Kirk HQ, Rabb M, Hatenhauer J, Smith R: Primary familial amyloidosis of the cornea. Trans Am Acad Ophthalmol Otolaryngol 77:411, 1973 173. Ramsey MS, Fine BS, Cohen SW: Localized corneal amyloidosis: Case report with electron microscopic observations. Am J Ophthalmol 73:560, 1972 174. Lewkojewa EF: Weber Einen Fall Primary Degeneration: amyloidose der Kornea. Klin Monatsbl Augenheilkd 83(suppl):117, 1930 175. Pouliquen Y, Dhermy P, Savoldelli M: Dystrophic gelatineuse en goutte de
la cornée (English abstract). J Fr Ophtalmol 4:349. 1981 176. Kanai A, Kaufman HE: Electron microscopic studies of primary band-shaped keratopathy and gelatinous, droplike
corneal dystrophy in two brothers. Ann Ophthalmol 14:535, 1982 177. Kaku K, Suda K: Primary band-shaped and gelatinous drop-shaped degeneration
of the cornea of a sister and brother. Jpn J Clin Ophthalmol 18: 19. 1964 178. Nagataki S, Tanishima T, Sakomoto T: A case of primary gelatinous drop-like corneal dystrophy. Jpn J Ophthalmol 16:107, 1972 179. Matsui M, Ito K, Akiya S: Histochemical and electron microscopic examinations on so-called gelatinous
droplike dystrophy of the cornea. Folia Ophthalmol Jpn 23:466, 1972 180. Krachmer JJ, Purcell JJ, Young CW, Bucher KD: Corneal endothelial dystrophy: A study of sixty-four families. Arch Ophthalmol 96:2036, 1978 181. Hogan MJ, Wood I, Fine M: Fuchs' endothelial dystrophy of the cornea. Am J Ophthalmol 78:363, 1974 182. Krachmer JH, Bucher KD, Purcell JJ, Young CW: Inheritance of endothelial dystrophy of the cornea. Ophthalmo1ogica 181:301–313, 1980 183. Korenzetti DWC, Uotila MH, Parkh N, Kaufman HE: Central corneal guttata: Incidence in the general population. Am J Ophthalmol 64:1155, 1969 184. Rodrigues MM, Krachmer JH, Hackett J et al: Fuchs' corneal dystrophy: A clinicopathologic study of the variation in
corneal edema. Ophthalmology 93:789, 1986 185. Rosenthal WN, Blitzer M, Inslar MS: Aqueous amino acid levels in Fuchs' corneal dystrophy. Am J Ophthalmol 102:570, 1986 186. Roth SI, Stock EL, Jutabba R: Endothelial viral inclusions in Fuchs' corneal
dystrophy. Hum Pathol 18:338. 1987 187. Stocker FW, Irish A: Fate of successful cornea graft in Fuchs' endothelial dystrophy. Am J Ophthalmol 68:820, 1969 188. Arentsen JJ, Laibson PR: Penetrating keratoplasty and cataract extraction: Combined vs nonsimultaneous
surgery. Arch Ophthalmol 96:75, 1978 189. Staz L: An unusual condition of the posterior surface of the cornea (posterior
herpes of the cornea). Br J Ophthalmol 23:622, 1939 190. Tripathi RC, Casey TA, Wise EG: Hereditary posterior polymorphous dystrophy: An ultrastructural and clinical
report. Trans Ophthalmol Soc UK 94:211, 1974 191. Grayson M: The nature of hereditary deep polymorphous dystrophy of the cornea: its
association with iris and anterior chamber dysgenesis. Trans Am Ophthalmol Soc 72:516, 1974 192. McGee HB, Falls HF: Hereditary polymorphous deep degeneration of the cornea. Arch Ophthalmol 50:462, 1953 193. Strachan IM, MacLean H: Posterior polymorphous dystrophy of the cornea. Br J Ophthalmol 52:270, 1968 194. Cibis GW, Krachmer JA, Phelps CD, Weingeist TA: Iridocorneal adhesions in posterior polymorphous dystrophy. Trans Am Acad Ophthalmol Otolaryngol 81:770, 1976 195. Rodrigues MM, Phelps CD, Krachmer JH et al: Glaucoma due to endotheliaziation of the anterior chamber angle. Arch Ophthalmol 98:688, 1980 196. Cibis GW, Krachmer JA, Phelps CD, Weingeist TA: The clinical spectrum of posterior polymorphous dystrophy. Arch Ophthalmol 95: 1529, 1977 197. Boruchoff SA, Kuwabara I: Electron microscopy of posterior polymorphous degeneration. Am J Ophthalmol 72:879, 1971 198. Rodrigues MM, Sun T, Krachmer J, Newsome D: Epithelization of the corneal
endothelium in posterior polymorphous dystrophy. Invest Ophthalmol
Vis Sci 19:832. 1980 199. Boruchoff SA, Weiner MJ, Albert DM: Recurrence of posterior polymorphous corneal dystrophy after penetrating
keratoplasty. Am J Ophthalmol 109:323, 1990 200. Pearce WG, Tripathi RC, Morgan G: Congenital endothelial corneal dystrophy: Clinical, pathological and genetic
study. Br J Ophthalmol 53:577, 1969 201. Maumenee AE: Congenital hereditary corneal dystrophy. Am J Ophthalmol 50:1114, 1960 202. Waardenburg PJ, Franceschetti A, Klein D: Genetics and Ophthalmology, vol 1, pp 458–562. Oxford, Blackwell, 1961 203. François J: Heredity in Ophthalmology, pp 300–302. St. Louis, CV
Mosby, 1961 204. Goldberg MF: Genetic and Metabolic Eye Disease, pp 305–306. Boston, Little, Brown & Co, 1974 205. Harboyen G, Mamo J, Der Kaloustian V, Karam F: Congenital corneal dystrophy: Progressive sensorineural deafness in a family. Arch Ophthalmol 85:27, 1971 206. Judisch GF, Maumenee IH: Clinical differentiation of recessive congenital hereditary endothelial
dystrophy. Am J Ophthalmol 85:606, 1978 207. Pedersen O, Rushoud A, Olsen EG: Anterior mesenchymal dysgenesis of the eye. Acta Ophthalmol 67:470, 1989 |