The glaucomas can be roughly segregated into two major classes, open-angle and closed-angle, with multiple subdivisions for each type. One such subdivision is based on our understanding of the cause of the condition. Primary glaucomas refer to diseases in which we are clinically unable to determine the cause of disease, while secondary glaucomas are those caused by a specific insult, such as inflammation, trauma, or other coexisting eye condition. Of all the forms of glaucoma, the primary open-angle glaucomas are by far the most common among whites and blacks, and primary angle-closure glaucoma is more frequent among the Chinese.18 The primary open-angle glaucomas can be further subdivided into two rough groups based on the age at which the disease is manifest. Juvenile open-angle glaucoma has an onset before age 35 and adult open-angle glaucoma (usually called chronic open-angle glaucoma or even simply primary open-angle glaucoma) has an onset after age 35.
JUVENILE PRIMARY OPEN-ANGLE GLAUCOMA
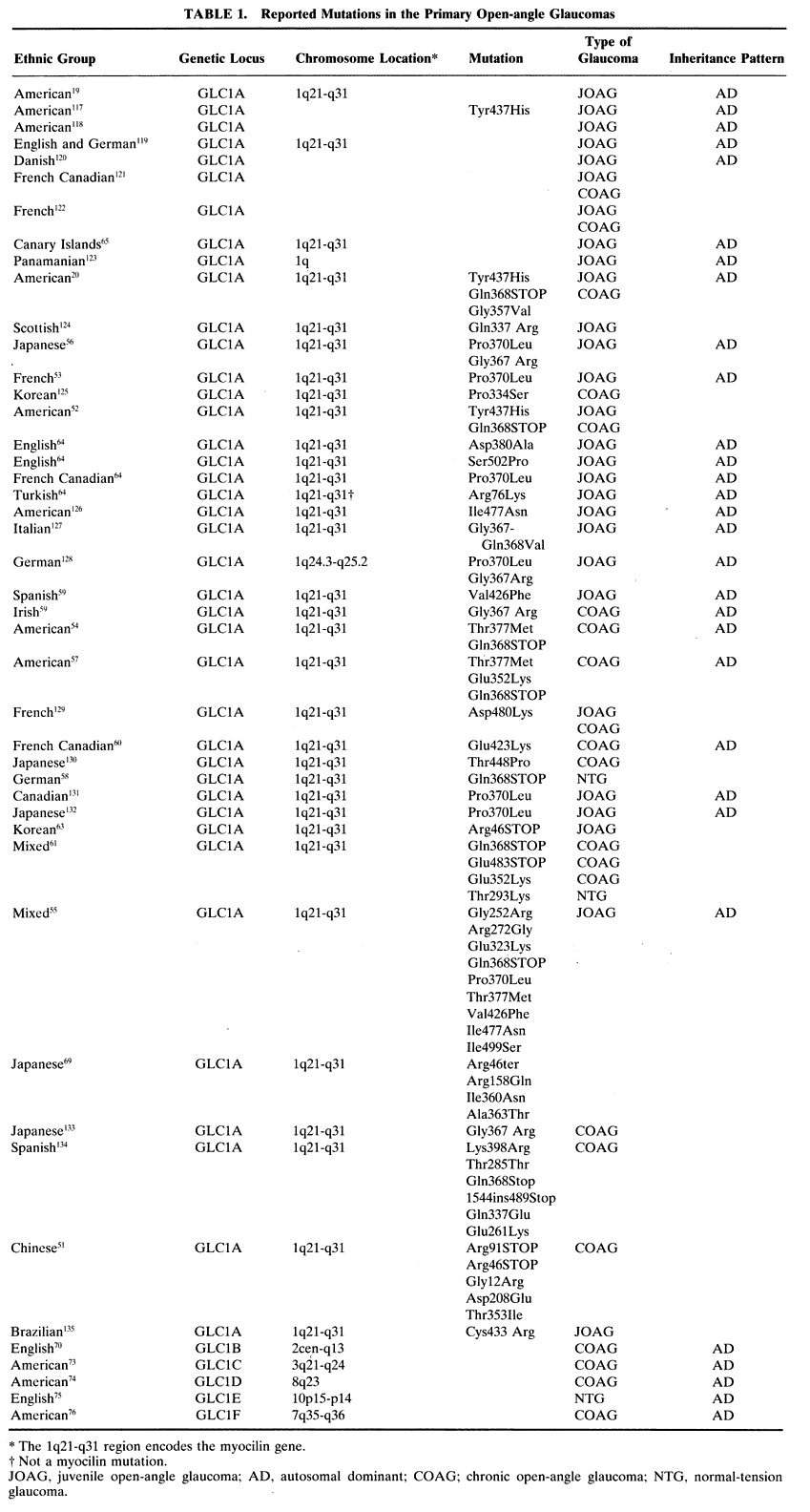
Juvenile open-angle glaucoma (JOAG), although uncommon, has been the most extensively investigated glaucoma. It is a disease that typically affects people between the ages of 3 and 35 in an autosomal dominant fashion and is characterized by elevated IOP with accompanying optic nerve and visual field damage. In 1993 Sheffield and coworkers described an American family with JOAG who appeared to contain an abnormality in the region of the long arm of chromosome 1.19 This locus was given the name GLC1A and several groups subsequently reported pedigrees of JOAG with abnormalities at the GLC1A locus in a variety of ethnic groups (Table 1). In 1997 Stone and coworkers identified the gene associated with the GLC1A locus as the region that codes for the protein myocilin (previously known as the trabecular meshwork-induced glucocorticoid response protein, or TIGR).20 Since then, multiple investigators have reported specific mutations in the myocilin gene and its protein product in families collected from around the world (Table 1).
The TIGR gene (now called the myocilin gene, or MYOC) was originally identified by induction of primary cultured trabecular meshwork cells with glucocorticoids. The protein that was induced was named TIGR. This protein has been found in the trabecular meshwork,21,22 ciliary body,23 and iris, but was not found in other ocular tissues24 until recently. Myocilin has now been identified in almost all ocular tissues,25 including the optic nerve,26 in an intracellular and extracellular manner. Its presence is more variable in the endothelial cells of Schlemm's canal.21,27 Systemically, it is also present in skeletal and heart muscle.24 The MYOC gene encodes a 57-kDa protein that is 504 amino acids in length and consists of three exons separated by two introns. One portion of the protein has a myosinlike domain, and one portion has an olfactomedinlike domain. Olfactomedin is the major secretory protein of the mucus layer surrounding olfactory neurons.28 Additional homologous proteins are present in other human tissues.29 Shortly after its description,30 the TIGR gene and its protein product were found to be identical to the myosinlike protein myocilin and its gene, which had been independently identified.31 Since 1998, the gene has been known as myocilin (MYOC), not TIGR. Of the variety of mutations reported in the myocilin gene, the third exon (the olfactomedinlike domain) contains almost all the known mutations of the MYOC gene.32 This may be related to a translocational pause that occurs in the native myocilin protein at the beginning of this domain.33
Myocilin appears to be part of a group of stress proteins and can be induced by subjecting trabecular cells to various insults.34 This class of proteins may bind important intracellular proteins and prevent them from denaturing under stress.32 Mutations in this protein may alter its ability to function and bind other enzymes.35 Although myocilin is present in greater amounts in the trabecular meshwork of glaucomatous eyes,36 it is also present in healthy eyes,25,26,37 arguing that it has a physiologic role. What that role may be is still unknown, but it is well conserved in other mammalian species,38–43 and analysis of the MYOC gene promoter shows regions that are responsive to glucocorticoids.30 How this finding may relate to glaucoma is still under investigation.44,45 Previous work has found that ultrastructural analysis of the trabecular meshwork in steroid-induced glaucoma and JOAG are similar46–50 and that corticosteroid treatment results in the overproduction of myocilin in some cultured trabecular cells.30 Perhaps overproduction of myocilin is common to both glaucomas. Such evidence supports one hypothesis for why myocilin mutations lead to increased IOP. This hypothesis says that myocilin is secreted by trabecular meshwork cells, which then reduces the amount of aqueous that can drain through the meshwork.51 Mutated myocilin might be oversecreted or might have an abnormal extracellular function once secreted that results in reduced aqueous outflow. A second hypothesis however proposes that alterations in myocilin might alter the cytoskeletal structure, thereby affecting cell shape and thereby reducing the size of the pores through which aqueous humor may exit. This second hypothesis suggests that a mutation in the myocilin protein might prevent it from performing its physiologic intracellular function.51 Whether either of these hypotheses is correc is not known. What is clear is that there is a spectrum of clinical findings associated with different mutations (especially the Gln368STOP mutation)32,52 and even within a single mutation.51 Further research into the myocilin protein and its function will be needed to clarify these issues.
Despite the evidence presented above relating open-angle glaucoma to myocilin, mutations in this gene cannot account for most cases of glaucoma, even those that are juvenile onset. Adam and coworkers found five of eight pedigrees to have a mutation at the GLC1A locus;53 Wiggs and coworkers found only 8% of their JOAG pedigrees had mutations in the myocilin gene;54 and Shimizu and coworkers found 33% of their JOAG pedigrees possessed a GLC1A mutation.55 Additionally, some families with mutations of this protein develop adult onset open-angle glaucoma20,52,54,56,57 or normal-tension glaucoma58 instead of JOAG, and some persons with certain mutations in the myocilin gene do not develop glaucoma at all.59–61 A large number of probably non-disease-causing polymorphisms have been found also.62 Recent work among a group of Chinese patients has found stop mutations in the myocilin gene in persons with and without glaucoma, suggesting that a reduction in the amount of myocilin protein may sometimes be, but not necessarily is, a cause of glaucoma.51 Most interesting is the mutation Arg46STOP. This mutation essentially eliminates production of myocilin altogether in homozygous individuals and reduces its production by 50% in heterozygous persons. A 15-year-old Korean girl with severe JOAG was found to be homozygous for this mutation,63 but another larger survey found this mutation present in 3% of normal Asian persons.51 In fact, one unaffected person in this larger study was a 77-year-old woman who was homozygous for this mutation, and only one heterozygous person had glaucoma. Others have found homozygous persons with a different mutation to be unaffected, whereas heterozygous family members manifest glaucoma.60 In the Korean case, the family members who were heterozygous were also unaffected, but two did have increased IOP. These data suggest that nonpenetrance is occurring, that absenceor alteration of every copy of the myocilin protein may not result in glaucoma, or that the Korean girl had other genetic or environmental factors at work. If reductions in or loss of myocilin is not necessary to cause glaucoma, it seems that other mutations that cause simply an alteration in myocilin structure may not cause glaucoma by inacti-vating a normal myocilin function. Rather, the ab-normal protein probably acquires an abnormalfunction.51
Some families have GLC1A-linked disease without mutations in the MYOC gene,64 and one group has reported cases of JOAG that do not even map to the GLC1A locus.65 The myocilin gene hypothesis may not account for the finding that despite the presence of the mutation at birth, JOAG is not congenital. Taken together, such findings indicate that our current understanding of the genetics of JOAG and the GLC1A locus is incomplete and that other genes, such as cyclooxygenase 2 (which is located near MYOC on chromosome 1q)66 and environmental factors may be involved in this disease.32
ADULT-ONSET PRIMARY OPEN-ANGLE GLAUCOMA
Whereas it appears that some of the JOAG cases studied to date have mutations in the GLC1A locus, the situation in adult-onset primary open-angle glaucoma, or chronic open-angle glaucoma (COAG), is much more heterogeneous. There are cases of COAG with mutations of the myocilin gene at locus GLC1A, but also at multiple other locations.
Some investigators have found pedigrees of COAG with mutations of the GLC1A locus (Table 1),20,51,52,54,57,67 and others have excluded this locus in their pedigrees.55,68 In families with mixed JOAG and COAG, 31% of persons have been found to possess a myocilin mutation.55 Overall, it is estimated that only 3% to 5% of patients with COAG have mutations in the myocilin gene,20,52,56,61,62,69 of which the most common is the Gln368STOP mutation.62 This stop mutation, usually associated with increased IOP in elderly patients,52 was reported to be present in one case of normal-tension glaucoma in a young man, indicating there may be other factors involved.58 There appears to be no difference in the frequency of myocilin mutations in COAG cases among racial groups ranging from blacks to whites to Japanese.62
Five additional loci for COAG, termed GLC1B through GLC1F, have been subsequently identified. All are on chromosomes different from GLC1A (Table 1). There are no data that estimate the percentage of patients with COAG who have one of these mutations. It is possible that these mutations may explain only a small number of COAG cases. Such a situation seems to have occurred with locus GLC1B. This was originally described in a pedigree from the United Kingdom,70 but so far, pedigrees from North America do not possess this mutation.71 A recent genome screen of North American patients of various ethnic backgrounds found possible abnormalities in regions of chromosomes 2, 14, 17, and 19. Genomic regions near loci GLC1A, GLC1B, GLC1D, and GLC1E seemed to be more involved than regions near loci GLC1C and GLC1F in this population.72 To date, no gene has been identified with any of these additional loci of COAG.
Clinically, patients with the GLC1B mutation have normal to moderately increased IOP,70 and patients with the GLC1C mutation have markedly increased IOP.73 Patients with a mutation of the GLC1D locus develop open-angle glaucoma in middle age with an associated moderately increased IOP.74 The fourth locus that has been described, GLC1E, was discovered in a British family with low-tension glaucoma,75 and the fifth locus was discovered in a family with increased IOP and has been termed GLC1F (Table 1).76
Other genetic loci have been associated with glaucoma, but not assigned a GLC name. Two groups have found an association of open-angle glaucoma and certain HLA loci among whites77 and Mexicans,78 but not Spaniards.79 It seems likely that adult-onset open-angle glaucoma is a heterogeneous condition and that not only will many more loci and genes be discovered, but there may well be a complex interplay between genes and other factors that eventually result in glaucoma.
SECONDARY OPEN-ANGLE GLAUCOMAS
The secondary open-angle glaucomas have been so named because the anterior segment, including the trabecular meshwork, does not show peripheral anterior synechiae, yet there is an identifiable likely cause of increased IOP and glaucoma. Even with an open angle, this group of glaucomas often has a clinically apparent abnormality of the trabecular meshwork on gonioscopy. There is also typicallyan increased IOP. Examples of this group of glau-comas include uveitic glaucoma, steroid-inducedglaucoma, angle-recession glaucoma, pigment dis-persion syndrome, and the exfoliation syndrome (pseudoexfoliation).
Pseudoexfoliation syndrome is the most common secondary open-angle glaucoma in the world. First described by Lindberg in 1917,80 it is a multisystem condition.81,82 Clinically, the syndrome features deposits of gray-white amyloidlike material throughout the anterior segment. This is most readily apparent on the lens surface and pupillary margin, but the material can be seen on the zonules. The trabecular meshwork is often more heavily pigmented than normal, and there are pupillary margin transillumination defects and atrophy. The condition is particularly prevalent in Scandinavian countries and in other locations where Scandinavians and their descendants live; it is often not apparent until late in life.
For some time there has been evidence of a genetic basis for this disease. Supporting evidence has come from twin12,83 and family84 studies. Analysis of cases in Iceland and Canada found that the disease shows evidence of maternal transmission and suggests an X-linked inheritance, autosomal inheritance with genetic imprinting, or possibly a mitochondrial disease.6,7 To date, there has been no locus or gene identified for this condition.
Pigment dispersion syndrome is a condition that typically affects young myopic persons. In whites, it may affect 2% to 4% of the population between the ages of 20 and 40 years.85 It appears that a posterior bowing of the iris causes iris-zonule touch with subsequent liberation of pigment.86,87 The pigment then is dispersed throughout the anterior chamber, where it accumulates in the trabecular meshwork and causes increased IOP and glaucoma.
Analysis of families with pigment dispersion syndrome shows a pattern of autosomal dominant inheritance.88 Recent work by Andersen and coworkers localized the gene for this condition to chromosome 7q (Table 2).89

CONGENITAL GLAUCOMA
Congenital glaucoma is defined as glaucoma present at birth. In reality, most cases of this type of glaucoma are probably better-termed infantile glaucoma because the disease is often not manifest until several months of age up to 3 years old. From a clinical standpoint, infantile glaucoma is most often an autosomal recessive disease characterized by increased IOP caused by an angle dysgenesis that leads to the classic symptom triad of corneal clouding attributable to edema, epiphora, and photophobia. If left untreated, buphthalmos and amblyopia often develop. This form of glaucoma must be considered separately from secondary congenital glaucomas in which the glaucoma likely arises as a result of other dysmorphic or multisystem abnormal conditions.
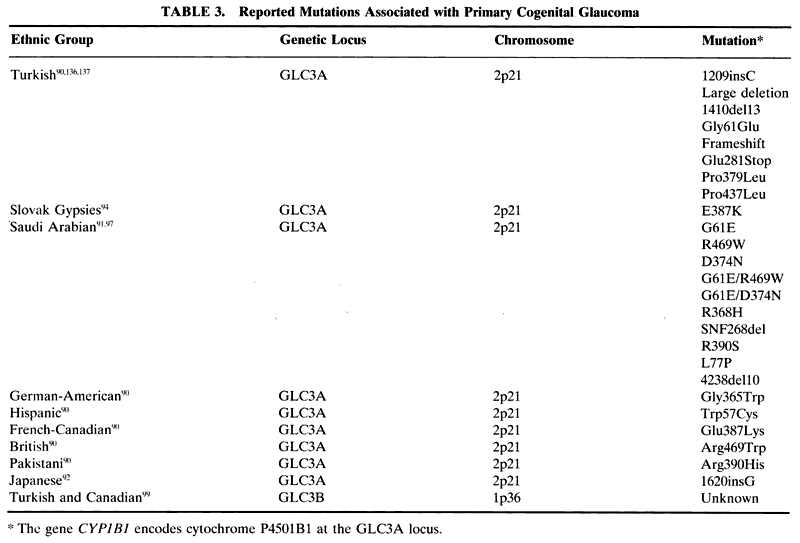
Two loci have been identified that appear to segregate with infantile glaucoma, termed GLC3A and GLC3B. These loci have been localized to chromosome 2 and 1, respectively (Table 3). The gene that is mutated at the GLC3A locus is cytochrome P4501B1. Multiple mutations in this gene havebeen identified in populations around the world, in-cluding German-American, Hispanic, French-Canadian, British, Pakistani, Turkish,90 Saudi Arabian,91 Japanese,92 and Slovak Gypsies.93 In Slovak Gypsies affected with congenital glaucoma, the mutation has been reported to be a homozygous glutamic acid substitution for lysine at position 387.94 This location happens to be in a highly conserved portion of the protein and the various mutations most likely affect the normal protein folding or truncate the protein. Position 387 is absolutely conserved among cytochrome P450 proteins and so is likely a critical portion of the protein. A homozygous mutation of glutamic acid for lysine at this position is always associated with congenital glaucoma.94 This matches well with the original descriptions of congenital glaucoma being markedly worse in the Slovak Gypsy population than in other groups.95,96 This is not the case in other families with different mutations. Such families can have incomplete penetrance and a variable clinical picture depending on the mutation and whether it is heterozygous or homozygous.90,97
Cytochrome P4501B1 is part of a superfamily of cytochrome P450 proteins. These proteins are involved in metabolism in the cell usually by inserting oxygen into a molecule and seem to assist in keeping the cornea clear.98 In patients with the mutation at the GLC3A locus, loss of this function may be responsible for the corneal edema that is seen in congenital glaucoma.
A second locus for congenital glaucoma was recently identified. Termed GLC3B, it has been localized to chromosome 1p.99 To date, there has been no gene identified with this locus.
CHRONIC ANGLE-CLOSURE GLAUCOMA
Angle-closure glaucoma is a term that describes a group of glaucomas associated with functional obstruction of the trabecular meshwork. Such an obstruction can be from iris peripheral anterior synechiae or from any other scar tissue or cellular layer that covers the trabecular meshwork to impede outflow of aqueous from the eye, such as in iridocorneal endothelial syndrome.
Primary angle-closure glaucoma, in which peripheral anterior synechiae acutely or gradually zipper the angle shut, is thought to be the result of pupillary block that pushes the peripheral iris into the angle and results in scarring. Although there is no clear understanding why this disease occurs, it is found most commonly in the Chinese18 and can be halted by a peripheral iridectomy. The secondary angle-closure glaucomas are a diverse group of conditions in which there is a known cause of angle closure. Conditions such as neovascular glaucoma, malignant glaucoma, and iridocorneal endothelial syndrome represent some of the many diseases that can cause a secondary angle-closure glaucoma.
There have been no reports to date describing any loci for primary angle-closure glaucoma. A single report has placed the gene for dominantly inherited nanophthalmos (NNO1) and angle-closure glaucoma on chromosome 11p.100 No other secondary angle-closure glaucoma has been described to have a genetic basis.
OTHER DEVELOPMENTAL GLAUCOMAS
The term anterior segment dysgenesis describes a variety of ocular conditions in which the anterior segment is malformed at birth. Such conditions are typically bilateral and often associated with glaucoma. Of the various syndromes and anomalies that make up this group of ocular diseases, some have been determined to have a genetic basis (Table 2).
Axenfeld-Rieger syndrome describes a spectrum of anterior segment and systemic findings that are inherited in an autosomal dominant fashion with high penetrance. The condition is associated with an onset of glaucoma in childhood or early adulthood in approximately half of patients. Three genetic loci have been identified on chromosomes 4q25,101 6p25,102 and 13q14.103 The DNA at the 4q25 and 6p25 loci has been found to encode the genes PITX2 (paired-like homeodomain transcription factor 2)104 and FKHL7 (forkhead Drosophila homologue-like transcription factor gene).102 No gene has been identified to date at loci 13q14.
The PITX2 gene is a paired homeobox gene that regulates the expression of other genes during development. Mutations in this gene have also been associated with autosomal dominant iris hypoplasia,105,106 iridogoniodysgenesis syndrome,107 and a single case of Peter anomaly.108 FKHL7 is also a gene that regulates the expression of other genes during development. In the mouse, the homologue gene FOXC1, is expressed in mesenchyme from which the drainage structures of the anterior segment are derived. Although mice with this mutation had anterior segment anomalies, they did not have increased IOP. This suggests an interaction of some sort with another gene is responsible for the glaucoma associated with this condition.109 Mutations at the 6p25 locus have been associated with a variety of anterior segment abnormalities, such as Axenfeld anomaly, iris hypoplasia, Rieger anomaly, posterior embryotoxon,102 iridogoniodysgenesis anomaly,110 and familial glaucoma iridogoniodysplasia.111 Not all the patients with mutations in the 6p25 locus have mutations of the FKHL7 gene. Only those with features of Axenfeld-Rieger anomaly had mutations in this gene.112
The important message from the genetics of Axenfeld-Rieger syndrome is that multiple phenotypic variations can be caused by mutations in the same genes. This supports the notion that there is no useful information to be gained from splitting these conditions into separate diseases and the spectrum of phenotypic findings should be lumped under the name Axenfeld-Rieger syndrome.113
The nail-patella syndrome (NPS1) has been localized to chromosome 9q34114 and its gene LMX1B.115 This gene is a transcription factor important in skeletal development.