Axenfeld异常的患者并不十分罕见,但遗憾的是,由于日常工作的繁忙。一些医师可能会对可疑的患者漏诊。通过复习该病的一些相关文献,及一些病例观察。认为应当将Axenfeld异常归纳入Axenfeld-rieger症候群。并且,建议
1、认真进行角膜缘和房角检查
2、谨慎看待UBM结果。避免错误地测量房角
3、对可疑的患者行基因检查,预测家族中子代的发病几率。
患者,女,籍贯河北,1942年9月出生。因双眼视力渐降,干涩三年余就诊。患者自1998年底始觉双眼视力下降。查:双视力0.4,j7。BSCVA 双1.5。Pa 双22mmHg。BUT:双5秒、Schirmer试验:双1~2毫米。眼底:双C/D 0.7,颞下盘沿较窄。拟诊:双屈光不正;双干燥性角结膜炎;双开青待排。1999年1月20日,行SLO-RNFL检查,示:右未见明显异常,左视乳头颞下方可见楔形RNFLD。
1999年3月及2001年11月分别行四次眼压检查:结果阴性。其中最高值为22mmHg,最低为19mmHg。
视野检查:
视力 机型 视标 右 左
2001.10.29Od 0.15,Os 0.4 Octopus30度 Ⅲ 右向心缺损 左上、下缺损,鼻侧阶梯
2001年10月31日外院行UBM检查:
前房深度房角(度):颞鼻上下
右2.188 26.45 25.76 18.38 16.75
左2.442 25.76 22.40 25.33 17.92
1999年至今多次眼压检查双眼均在16~20mmHg水平。期间患者间断使用素高捷疗、人工泪液。
2002年4月16日患者来诊。查:双颧面及四肢未见异常。VOD 1.2, OS 0.9(戴镜)。Pa:右20mmHg,左18mmHg。双上睑结膜少许瘢痕。角膜清澈。右颞侧角膜缘后弹力层沿Schwalbe线区域白线样结构。双前房等深,右颞侧周边前房明显浅于左侧。约1/4CT,左约21/2~1/3CT。双虹膜纹清,瞳孔缘区尚平伏,右颞侧周边轻膨隆。小瞳孔下:双晶体密度略高,视乳头色红界清,C/D 双0.7。双颞下盘沿窄,切迹明显。
房角镜检查:散瞳前:右360度Schwalbe线突出,颞侧为著。其上可见残存粘连之虹膜组织。虹膜根部起伏不平,其上可见环形印迹和点状隆起,与隆起的Schwalbe线有松弛的粘连。滤帘可见色素,睫状体带及房角隐窝可见入射角15~20度。左眼Schwalbe线轻度突出。,其上亦可见少许残存粘连之虹膜组织。虹膜根部轻度点状隆起。Schlemm管充血。散瞳后:右眼上方部分房角关闭。Schwalbe线与虹膜根部附着范围扩大。360度房角进一步变窄。诊断:Axenfeld综合症。
讨论:1、患者此前曾多次眼科检查。但由于患者的特殊不典型性:角膜后胚环不明显;房角入射角部分类似高原虹膜形态;虹膜与Schwalbe线粘连疏松;眼压多次测量正常范围。以上特点导致患者长期未得到确诊。应当引起重视。
2、关于先天性房角异常。
先天性房角异常源于细胞分化异常或胚胎期组织退化滞后。其最常见的良性改变为角膜后胚胎环。正常的Schwalbe线为小梁网和角膜后弹力层终点的连接处。角膜后胚胎环表现为Schwalbe线加重、突起和向中央移位,裂隙灯下显示白色、明亮的不规则弧线,距角膜缘0.5~2.0mm。但在正常眼中约3~15%亦可见这种异常。1920年,Axenfeld在报道首例Axenfeld异常时,引入了“后胚环”的概念。先天性房角异常尚可伴随虹膜发育不良或位置异常;角膜后弹力层异常;瞳孔形态异常或无瞳孔等。与Axenfeld异常关系密切的Rieger异常便是如此。在Rieger综合症的描述中,Rieger首先引入了“角膜虹膜中胚层发育不良”的概念,即前房分裂综合症(旧称)。先天性房角异常还可以成为全身性综合症的眼部表现,如Alagille综合症(肝动脉发育不良)、X-联琐鱼鳞癣病、家族性先天无虹膜、Down’s综合症等。
角膜缘后胚胎环

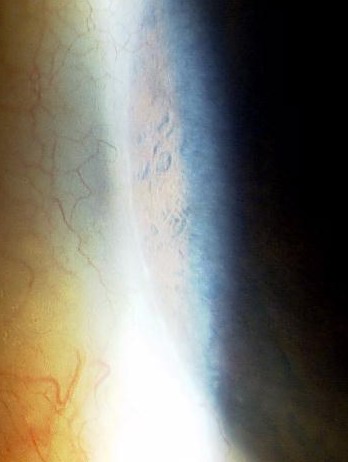
房角检查:增宽前移的schwable氏线和后胚胎环


3、关于Axenfeld先天异常及相关综合症:
Axenfeld异常是一种先天性异常,通常为双眼出现。表现为Schwalbe线向前移位,并与虹膜根部粘连。1920年,Axenfeld报道首例这类罕见的先天性疾患。当同时伴随有小梁网缺陷和青光眼表现时则称Axenfeld综合征。Axenfeld异常可以合并其它先天性前节异常,即胚胎期中胚层发育异常,如Peter’s异常、伊藤黑色素过少症、先天性虹膜外翻、牙齿和骨骼、心脏发育异常,神经性耳聋、白化病等。如当合并Rieger异常,即虹膜、瞳孔异常以及其后发生的青光眼,则称为Axenfeld-Rieger综合症,其中约50%的患者可能在儿童期或成人后发生青光眼。如果Rieger异常合并面部、牙齿和骨骼发育异常时,则称Rieger综合症。由于Axenfeld异常、Axenfeld综合征、Rieger异常、Rieger综合征相互间的表现有很多重叠,又由于该组疾患具有共同的染色体异常表现,所以目前主张将它们归为一组疾患,总称Axenfeld-Rieger组群。该组群通常表现为高外显率的异质性常染色体显性遗传(75%),也可以表现为散发发病。Alkemade通过大样本调查,认为该组群无明显性别差异。另外,半年前有报道,在Ramon综合症中可出现Axenfeld异常,并可能合并视网膜病变和视乳头苍白。
角膜后胚胎环被认为是Axenfeld-Rieger组群早期和轻微的组织学异常。有学者认为,其在组群中发生率不高(为8~15%),且单独发生的角膜后胚胎环并不会增加青光眼发生的危险性,所以单独的角膜后胚胎环不应归入Axenfeld-Rieger组群。
4、关于Axenfeld-Rieger组群的房角胚胎发育:
Rieger在1930年代首先报道了该组群中的Rieger异常、Rieger综合征。由于颌面部、牙齿和骨骼发育与虹膜的发育均来源于相同胚叶,故认为中胚叶,尤其是神经胚细胞(Neural crest cell)发育异常是该病的组织学原因。该理论已经被广泛接受,而对于Neural crest cell异常如何导致组织形态学异常,存在不同的学说。比较流行的学说为“发育停滞”。M.bruce Sheilds等认为Neural crest cell来源的部分前节组织,在其胚胎发育的某个时期,可能是妊娠晚期,存在发育停滞。虹膜原始内皮层和前房角之间的膜性牵拉,以及这些原始细胞在角膜后弹力层的沉积导致了虹膜最终的异常改变;胚胎期发育停滞还导致了房水流出通道的结构异常,从而导致先天性青光眼的发生。另外,通过虹膜荧光造影发可能发现位于虹膜外周细微而纡曲的异常渗漏血管。提示虹膜原发或继发血管异常等也可能是导致Rieger异常的影响因素。
5、关于Axenfeld-Rieger组群的分子遗传生物学研究进展:
近年来,关于Axenfeld-Rieger组群的分子遗传生物学研究成为探索该组疾患的热点。目前公认存在三个染色体位点的基因突变与本组群有关——4q25, 6p25, and 13q。
通过分子遗传生物学研究,目前已经明确了一些致病基因位点和致病机制。同时,研究的结果使我们认识到过去认为应加以明确区分的一些先天性疾患实际上为发病机制类似的一组疾患。例如,虹膜房角发育不良综合症(IGDS)与Axenfeld-Rieger组群(ARS)有部分共同的基因突变。
|















 网友评论:(只显示最新10条。评论内容只代表网友观点,与本站立场无关!)
网友评论:(只显示最新10条。评论内容只代表网友观点,与本站立场无关!)