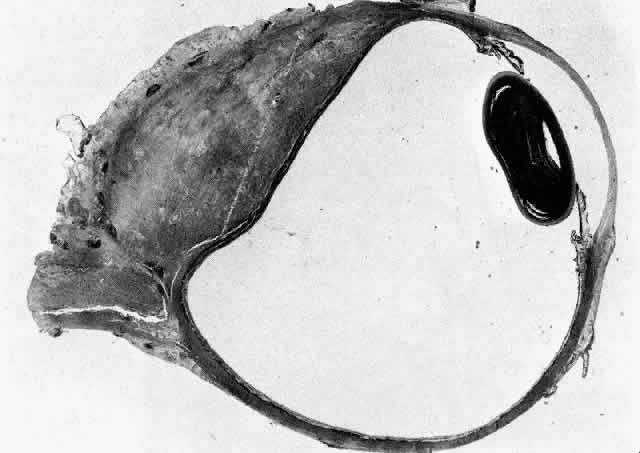
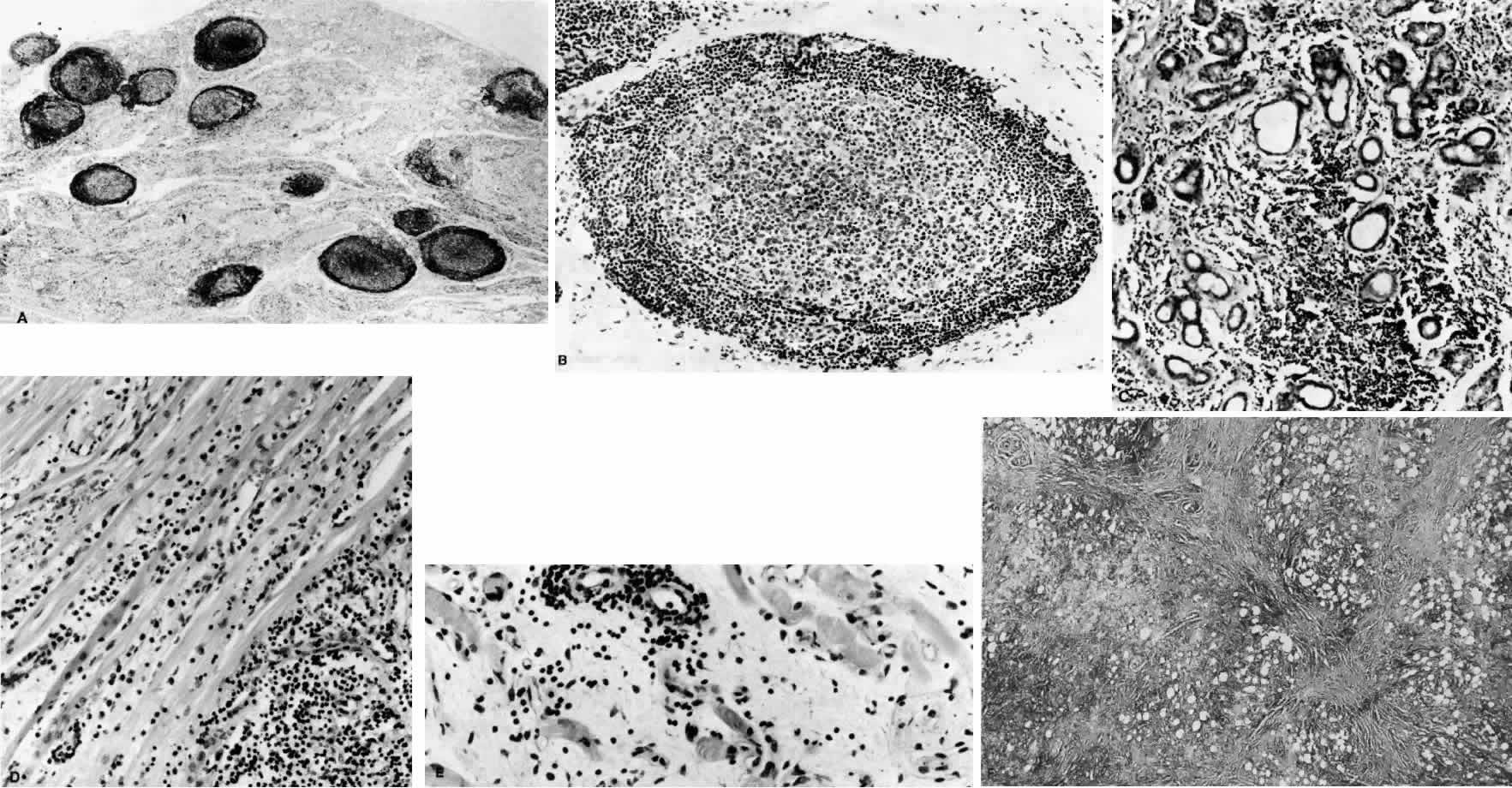
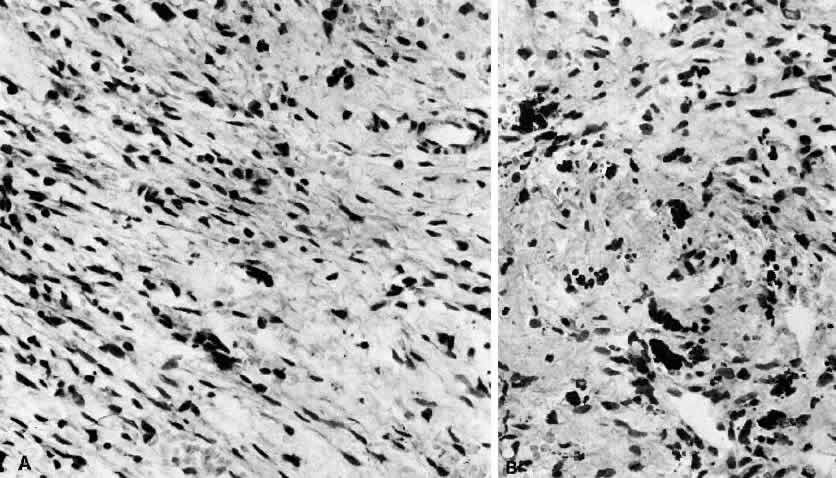
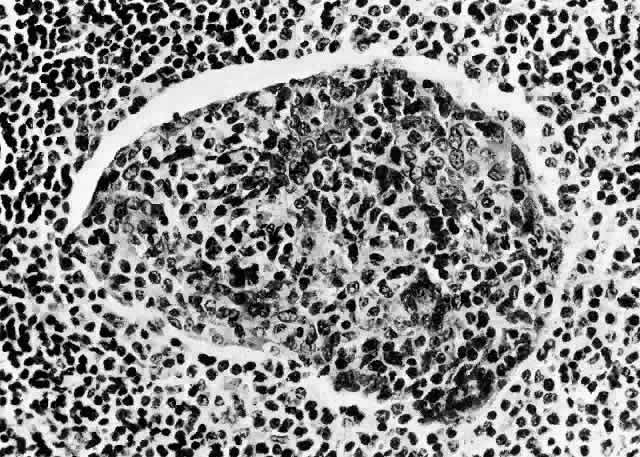
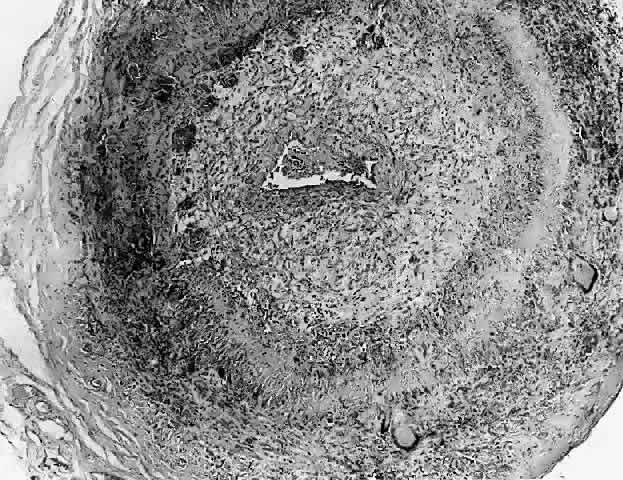
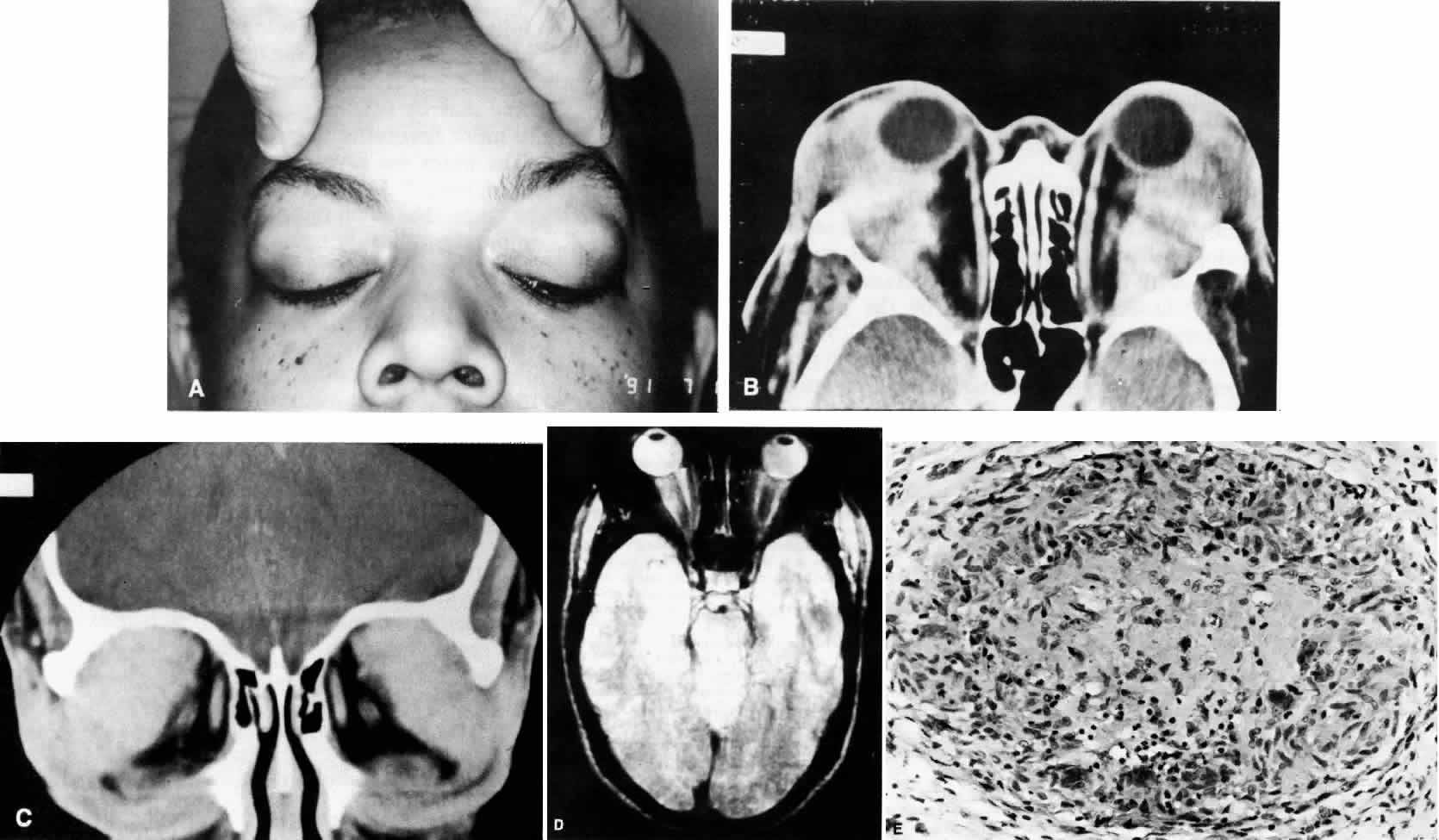
1. Rootman J: Frequency and differential diagnosis of orbital disease. In
Rootman J (ed): Diseases of the Orbit, pp 119–139. Philadelphia, JB
Lippincott, 1988 2. Kennedy R: An evaluation of 820 orbit cases. Trans Am Ophthalmol Soc 82:134, 1984 3. Jakobiec FA, Font RL: Orbit. In Spencer WH (ed): Ophthalmic Pathology, 3rd
ed, vol 3, chap 12. Philadelphia, WB Saunders, 1986 4. Kennerdell JS, Dresner SC: The nonspecific orbital inflammatory syndromes. Surv Ophthalmol 29:93, 1984 5. Nugent RA, Rootman J, Robertson WD et al: Acute orbital pseudotumors: Classification and CT features. AJR 137:957, 1981 6. Rootman J, Robertson W, Lapointe JS: Inflammatory diseases. In Rootman
J (ed): Diseases of the Orbit: A Multidisciplinary Approach, pp 143–204. Philadelphia, JB
Lippincott, 1988 7. Jakobiec F: Orbital inflammations and lymphoid tumors. Trans New Orleans Acad Ophthalmol 30:52, 1982 8. Birch-Hirschfeld A: Die Krankheiten der Orbita. In Graefe A, Saemisch T (eds): Handbuch
der Gesamten Augenheilkunden, vol 9, part 1. Berlin, Julius
Springer, 1930 9. Bullen C, Younge B: Chronic orbital myositis. Arch Ophthalmol 100:1749, 1982 10. Grimson BS, Simons KB: Orbital inflammation, myositis, and systemic lupus erythematosus. Arch Ophthalmol 101:736, 1983 11. Jakobiec FA, Yeo JH, Trokel SL et al: Combined clinical and computed tomographic diagnosis of lacrimal fossa
lesions. Am J Ophthalmol 94:785, 1982 12. Rush J, Kennerdell J, Donin J: Acute periscleritis: A variant of idiopathic orbital inflammation. Orbit 1:221, 1982 13. Rootman J, Nugent R: The classification and management of acute orbital pseudotumors. Ophthalmology 89:1040, 1982 14. Slavin M, Glaser J: Idiopathic orbital myositis. Arch Ophthalmol 100:1261, 1982 15. Weinstein GS, Dresner SC, Slamovits TL, Kennerdell JS: Acute and subacute orbital myositis. Am J Ophthalmol 96:209, 1983 16. Tychsen L, Tse DT, Ossoinig K, Anderson RL: Trochleitis with superior oblique myositis. Ophthalmology 91:1075, 1984 17. Aron-Rosa D, Doyon D, Salamon G, Michotey P: Tolosa-Hunt syndrome. Ann Ophthalmol 10:1161, 1978 18. Hallpike JF: Superior orbital fissure syndrome: Some clinical and radiological observations. J Neurol Neurosurg Psychiatry 36:486, 1973 19. Lakke J: Superior orbital fissure syndrome: Report of a case caused by local pachymeningitis. Arch Neurol 7:289, 1962 20. Edwards M, Zanel D, Gilkor R, Muller J: Invasive orbital pseudotumor: CT demonstration of extension beyond orbit. Neuroradiology 23:215, 1982 21. Eshaghian J, Anderson R: Sinus involvement in inflammatory orbital pseudotumor. Arch Ophthalmol 99:113, 1981 22. Kaye AH, Hahn JF, Craciun A et al: Intracranial extension of inflammatory pseudotumor of the orbit: Case report. J Neurosurg 60:625, 1984 23. Nishi T, Saito Y, Watanabe K, Uozumi T: Intracranial extension of an orbital pseudotumor accompanied by internal
carotid artery occlusion: Case report. Neurol Med Chir 32:758, 1992 24. Noble SC, Chandler WF, Lloyd RV: Intracranial extension of orbital pseudotumor: A case report. Neurosurgery 18:798, 1986 25. Frohman LP, Kupersmith MJ, Lang J et al: Intracranial extension and bone destruction in orbital pseudotumor. Arch Ophthalmol 104:380, 1986 26. Perrone T, De Wolf-Peeters C, Frizzera G: Inflammatory pseudotumor of lymph nodes: A distinctive pattern of nodal
reaction. Am J Surg Pathol 12:351, 1988 27. Randin JP, Merot Y, Anani P, Waridel D: Inflammatory pseudotumor of the lung: A propos of a case. Schweiz Med Wochenschr 111:596, 1981 28. Paineau J, Gaillard F, Visset J: Inflammatory pseudotumor of the liver: Unusual course of an unrecognized
abscess? Apropos of a case. J Chir 120:529, 1983 29. Salzstein S: Extranodal malignant lymphoma and pseudolymphomas. In Sommer
S (ed): Pathology Annual, p 159. New York, Appleton-Century-Crofts, 1969 30. Brooks J, Enterline H: Gastric pseudolymphoma: Its three subtypes and relation to lymphoma. Cancer 51:476, 1983 31. Caro WA, Helwig HB: Cutaneous lymphoid hyperplasia. Cancer 24:487, 1969 32. Jakobiec F, Mottow L: Pediatric orbital pseudotumor. In Jakobiec F (ed): Ocular
and Adnexal Tumors, pp 644–658. Birmingham, AL, Aesculapius, 1978 33. Mottow-Lippa L, Jakobiec F, Smith M: Idiopathic inflammatory orbital pseudotumor in childhood: II. Results of
diagnostic tests and biopsies. Ophthalmology 88:565, 1981 34. Mottow-Lippa L, Jakobiec F: Idiopathic inflammatory orbital pseudotumor in childhood: I. Clinical characteristics. Arch Ophthalmol 96:1410, 1978 35. Grimson BS, Cohen KL, Peiffer RL, Bouldin TW: Isolated, bilateral orbital mass lesions during childhood. J Pediatr Ophthalmol Strabismus 19:42, 1982 36. Keane JR: Alternating proptosis: A case report of acute orbital myositis defined
by the computerized tomographic scan. Arch Neurol 34:642, 1977 37. Hatsuda TA, Tanaka J: Bilateral necrotizing scleritis. Am J Ophthalmol 86:710, 1978 38. Spoor TC, Hartel WC: Orbital myositis. J Clin Neuroophthalmol 3:67, 1983 39. Green WR: Uveal tract. In Spencer W (ed): Ophthalmic Pathology, vol 3, pp 1776–1791. Philadelphia, WB Saunders, 1986 40. Schepens C, Brockhurst R: Uveal effusion: I. Clinical picture. Arch Ophthalmol 70:189, 1963 41. Haynes BF, Fishman ML, Fauci AS, Wolff SM: The ocular manifestations of Wegener's granulomatosis: Fifteen years
experience and review of the literature. Am J Med 63:131, 1977 42. Bullen CL, Liesegang TJ, McDonald TJ, DeRemee RA: Ocular complications of Wegener's granulomatosis. Ophthalmology 90:279, 1983 43. Fowler T, Earl C, McAllister V, McDonald W: Tolosa-Hunt syndrome. Br J Ophthalmol 59:149, 1975 44. Hunt W: Tolosa-Hunt syndrome: One cause of painful ophthalmoplegia. J Neurosurg 44:544, 1976 45. Kline L: Tolosa-Hunt syndrome. Surv Ophthalmol 27:79, 1982 46. Lenzi GL, Fieschi C: Superior orbital fissure syndrome: Review of 130 cases. Eur Neurol 16:23, 1977 47. Dornan TL, Espir ML, Gale EA et al: Remittent painful ophthalmoplegia: The Tolosa-Hunt syndrome? A report of
seven cases and review of the literature. J Neurol Neurosurg Psychiatry 42:270, 1979 48. Campbell RJ, Okazaki H: Painful ophthalmoplegia (Tolosa-Hunt variant): Autopsy findings in a patient
with necrotizing intracavernous carotid vasculitis and inflammatory
disease of the orbit. Mayo Clin Proc 62:520, 1987 49. Goto Y, Hosokawa S, Goto I et al: Abnormality in the cavernous sinus in three patients with Tolosa-Hunt syndrome: MRI
and CT findings. J Neurol Neurosurg Psychiatry 53:231, 1990 50. Rosenbaum DH, Davis MJ, Song IS: The syndrome of painful ophthalmoplegia: A case with intraorbital mass
and hypervascularity. Arch Neurol 36:41, 1979 51. Andersson BI: Unusual course of painful ophthalmoplegia: Report of a case. Acta Ophthalmol 58:841, 1980 52. Spirn FH, Wolintz AH, Tenner MS, Gombos GM: Tolosa-Hunt syndrome. Ann Ophthalmol 7:1087, 1975 53. Hannerz J, Ericson K, Bergstrand G: A new etiology for visual impairment and chronic headache: The Tolosa-Hunt
syndrome may be only one manifestation of venous vasculitis. Cephalalgia 6:59, 1986 54. Muhletaler CA, Gerlock A Jr: Orbital venography in painful ophthalmoplegia (Tolosa-Hunt syndrome). AJR 133:31, 1979 55. Hannerz J: Orbital phlebography and signs of inflammation in episodic and chronic
cluster headache. Headache 31:540, 1991 56. Rowed DW, Kassel EE, Lewis AJ: Transorbital intracavernous needle biopsy in painful ophthalmoplegia: Case
report. J Neurosurg 62:776, 1985 57. Sondheimer FK, Knapp J: Angiographic findings in the Tolosa-Hunt syndrome: Painful ophthalmoplegia. Radiology 106:105, 1973 58. Wilner HI, Gupta KL, Kelly JK: Orbital pseudotumor: Association of orbital vein deformities and myositis. Am J Neuroradiol 1:305, 1980 59. Ross WH: Myositic pseudotumour of the orbit. Can J Ophthalmol 18:199, 1983 60. Hankey GJ, Silbert PL, Edis RH, Nicoll AM: Orbital myositis: A study of six cases. Aust NZ J Med 17:585, 1987 61. Goldberg L, Tao A, Romano P: Severe exophthalmos secondary to orbital myopathy not due to Graves' disease. Br J Ophthalmol 66:392, 1982 62. Wesley RE, Cheij G, Bond JB, Davis WG: Orbital pseudotumor of the levator muscle. Ophthalmic Plast Reconstr Surg 2:139, 1986 63. Klein BR, Hedges TR III, Dayal Y, Adelman LS: Orbital myositis and giant cell myocarditis. Neurology 39:988, 1989 64. Ludwig I, Tomsak RL: Acute recurrent orbital myositis. J Clin Neuroophthalmol 3:41, 1983 65. Trokel S, Hilal S: Recognition and differential diagnosis of enlarged extraocular muscles
in computed tomography. Am J Ophthalmol 87:503, 1979 66. Trokel S, Jakobiec F: Correlation of CT scanning and pathologic features of ophthalmic Graves' disease. Ophthalmology 88:553, 1981 67. Dresner SC, Rothfus WE, Slamovits TL et al: Computed tomography of orbital myositis. AJR 143:671, 1984 68. Rothfus WE, Curtin HD: Extraocular muscle enlargement: A CT review. Radiology 151:677, 1984 69. Purcell J Jr, Taulbee WA: Orbital myositis after upper respiratory tract infection. Arch Ophthalmol 99:437, 1981 70. Casteels I et al: Orbital myositis following an upper respiratory tract infection: Contribution
of high resolution CT and MRI. J Belge Radiol 74:45, 1991 71. Harcourt R: Orbital granulomata associated with widespread angiitis. Br J Ophthalmol 48:673, 1964 72. Hannerz J, Greitz D, Hansson P, Ericson K: SUNCT may be another manifestation of orbital venous vasculitis. Headache 32:384, 1992 73. Zimmerman L, Rodgers J: Idiopathic thrombophlebitis of orbital veins simulating primary tumor of
orbit. Trans Am Acad Ophthalmol Otolaryngol 6:609, 1957 74. Hannerz J, Blomback M: Coagulation factors in orbital venous vasculitis. Cephalalgia 10:83, 1990 75. Hannerz J: Systemic symptoms associated with orbital venous vasculitis. Cephalalgia 8:255, 1988 76. Margo CE, Naugle T Jr, Karcioglu ZA: Ectopic lacrimal gland tissue of the orbit and sclerosing dacryoadenitis. Ophthalmic Surg 16:178, 1985 77. Harr D, Quencer R, Abrams G: Computed tomography and ultrasound in the evaluation of orbital infection
and pseudotumor. Radiology 142:395, 1982 78. Mauriello J Jr, Flanagan JC: Management of orbital inflammatory disease: A protocol. Surv Ophthalmol 29:104, 1984 79. Curtin HD: Pseudotumor. Radiol Clin North Am 25:583, 1987 80. Hurwitz BS, Citrin CM: Use of computerized axial tomography (CAT scan) in evaluating therapy of
orbital pseudotumor. Ann Ophthalmol 11:217, 1979 81. Kenney AH, Hafner JN: Ultrasonic evidence of inflammatory thickening and fluid collection within
the retrobulbar fascia: The T sign. Ann Ophthalmol 9:1557, 1977 82. Byrne SF, Green RL: Ultrasound of the Eye and Orbit, pp 283–287. St. Louis, Mosby-Year
Book, 1992 83. Kalina PH, Lie JT, Campbell RJ, Garrity JA: Diagnostic value and limitations of orbital biopsy in Wegener's granulomatosis. Ophthalmology 99:120, 1992 84. Dua HS, Smith FW, Singh AK, Forrester JV: Diagnosis of orbital myositis by nuclear magnetic resonance imaging. Br J Ophthalmol 71:54, 1987 85. Jaikishen P, Bateman JL, Shreeve WW: Orbital pseudotumor imaged with Ga-67 citrate. Clin Nucl Med 14:838, 1989 86. Wesley RE, Cooper J, Litchford DW: Orbital inflammatory pseudotumor associated with multifocal systemic neoplastic
immunoincompetence. Ann Ophthalmol 20:150, 1988 87. Chavis R, Garner A, Wright J: Inflammatory orbital pseudotumor: A clinicopathologic study. Arch Ophthalmol 96:1817, 1978 88. Garner A: Pathology of “pseudotumours” of the orbit: A review. J Clin Pathol 26:639, 1973 89. Satorre J et al: Orbital lesions with granulomatous inflammation. Can J Ophthalmol 26:174, 1991 90. Kennerdell JS: The management of sclerosing nonspecific orbital inflammation. Ophthalmic Surg 22:512, 1991 91. Abramovitz J, Kasdon D, Satala F et al: Sclerosing orbital pseudotumor. Neurosurgery 12:463, 1983 92. Garner A, Rahi AH, Wright JE: Lymphoproliferative disorders of the orbit: An immunological approach to
diagnosis and pathogenesis. Br J Ophthalmol 67:561, 1983 93. Jakobiec FA, Iwamoto T, Knowles DM II: Ocular adnexal lymphoid tumors: Correlative ultrastructural and immunologic
marker studies. Arch Ophthalmol 100:84, 1982 94. Noble AG, Tripathi RC, Levine RA: Indomethacin for the treatment of idiopathic orbital myositis. Am J Ophthalmol 108:336, 1989 95. Leone CR Jr, Lloyd WC III: Treatment protocol for orbital inflammatory disease. Ophthalmology 92:1325, 1977 96. Bielory L, Frohman LP: Low-dose cyclosporine therapy of granulomatous optic neuropathy and orbitopathy. Ophthalmology 98:173, 1991 97. Osher RH, Schatz NJ, Duane TD: Acquired orbital retraction syndrome. Arch Ophthalmol 98:1798, 1980 98. Donaldson SS, McDougall IR, Egbert PR et al: Treatment of orbital pseudotumor (idiopathic inflammation) by radiation
therapy. Int J Radiat Oncol Biol Phys 6:79, 1980 99. Kennerdell JS, Johnson BL, Deutsch M: Radiation treatment of orbital lymphoid hyperplasia. Ophthalmology 86:942, 1979 100. Orcutt JC, Garner A, Henk JM, Wright JE: Treatment of idiopathic inflammatory orbital pseudotumours by radiotherapy. Br J Ophthalmol 67:570, 1983 101. Clay C, Bilaniuk L, Vignaud J: Preliminary report on a new type of therapy. Neuro-Ophthalmology 1:101, 1980 102. Font R, Zimmerman L: Nodular fasciitis of the eye and adnexa: A report of 10 cases. Arch Ophthalmol 75:475, 1966 103. Levitt J, deVeer J, Oguzhan M: Orbital nodular fasciitis. Arch Ophthalmol 81:235, 1969 104. Meacham C: Pseudosarcomatous fasciitis. Am J Ophthalmol 77:747, 1974 105. Perry RH, Ramani PS, McAllister V et al: Nodular fasciitis causing unilateral proptosis. Br J Ophthalmol 59:404, 1975 106. Tolls R, Mohr S, Spencer W: Benign nodular fasciitis originating in Tenon's capsule. Arch Ophthalmol 75:482, 1966 107. Font R, Yanoff M, Zimmerman L: Benign lymphoepithelial lesion of the lacrimal gland and its relationship
to Sjögren's syndrome. Am J Clin Pathol 48:365, 1967 108. Saku T, Okabe HL: Immune histochemical and ultrastructural demonstration of keratin in epimyoepithelial
bloods of autoimmune sialadenitis in man. Arch Oral Biol 9:687, 1984 109. Knowles DM II, Jakobiec FA, Rosen M, Howard G: Amyloidosis of the orbit and adnexae. Surv Ophthalmol 19:367, 1975 110. Cohen MM, Lessell S: Amyloid tumor of the orbit. Neuroradiology 18:157, 1979 111. Simpson GT II, Strong MS, Skinner M, Cohen AS: Localized amyloidosis of the head and neck and upper aerodigestive and
lower respiratory tracts. Ann Otol Rhinol Laryngol 93:374, 1984 112. Raab EL: Intraorbital amyloid. Br J Ophthalmol 54:445, 1970 113. Howard GM. Amyloid tumours of the orbit. Br J Ophthalmol 50:421. 1966 114. Gean-Marton AD, Kirsch CF, Vezina LG, Weber AL: Focal amyloidosis of the head and neck: Evaluation with CT and MR imaging. Radiology 181:521, 1991 115. Nehen JH: Primary localized orbital amyloidosis. Acta Ophthalmol 57:287, 1979 116. Kaiser-Kupfer MI, McAdam KP, Kuwabara T: Localized amyloidosis of the orbit and upper respiratory tract. Am J Ophthalmol 84:721, 1977 117. Liesegang T: Amyloid infiltration of the levator palpebrae superioris muscle: Case report. Ann Ophthalmol 15:610, 1983 118. Savino PJ, Schatz NJ, Rodrigues MM: Orbital amyloidosis. Can J Ophthalmol 11:252, 1976 119. Erie JC, Garrity JA, Norman ME: Orbital amyloidosis involving the extraocular muscles. Arch Ophthalmol 107:1428, 1989 120. Levine MR, Buckman G: Primary localized orbital amyloidosis. Ann Ophthalmol 18:165, 1986 121. Finlay KR, Rootman J, Dimmick J: Optic neuropathy in primary orbital amyloidosis. Can J Ophthalmol 15:189, 1980 122. Holmstrom GE, Nyman KG: Primary orbital amyloidosis localised to an extraocular muscle. Br J Ophthalmol 71:32, 1987 123. Kyle A, Grepp P: Amyloidosis (AL): Clinical and laboratory features in 229 cases. Mayo Clin Proc 58:665, 1983 124. Lucas D, Knox F, Davies S: Apparent monoclonal origin of lymphocytes and plasma cells infiltrating
ocular adnexal amyloid deposits: Report of 2 cases. Br J Ophthalmol 66:600, 1982 125. Glenner G: Amyloid deposits and amyloidosis. The B-fibrilloses (in two
parts). N Engl J Med 302:1283, 1333, 1980 126. Maury C, Wegelius O: Pathogenesis of AA amyloidosis (editorial). Acta Med Scand 215:289, 1984 127. Sood GC, Malik SR, Gupta DK, Kakar PK: Reparative granuloma of the orbit causing unilateral proptosis. Am J Ophthalmol 63:524, 1967 128. Sebag J, Chapman P, Truman J, Riemersma RR: Giant cell granuloma of the orbit with intracranial extension. Neurosurgery 16:75, 1985 129. Hoopes PC, Anderson RL, Blodi FC: Giant cell (reparative) granuloma of the orbit. Ophthalmology 88:1361, 1981 130. Naiman J, Green WR, d'Heurle D et al: Brown tumor of the orbit associated with primary hyperparathyroidism. Am J Ophthalmol 90:565, 1980 131. Parrish CM, O'Day DM: Brown tumor of the orbit: Case report and review of the literature. Arch Ophthalmol 104:1199, 1986 132. Guccion JG, Enzinger FM: Malignant giant cell tumor of soft parts: An analysis of 32 cases. Cancer 29:1518, 1972 133. O'Sullivan RM, Nugent RA, Satorre J, Rootman J: Granulomatous orbital lesions: Computed tomographic features. Can Assoc Radiol J 43:349, 1992 134. Sen DK: Orbital granuloma projecting through the upper fornix with a retained foreign
body (record of an unusual case). Eye Ear Nose Throat Mon 51:467, 1972 135. Reifler DM, Leder D, Rexford T: Orbital hemorrhage and eyelid ecchymosis in acute orbital myositis. Am J Ophthalmol 107:111, 1989 136. Wolter JR, Fralick FB, Tanton JH: Late results of orbital hemorrhage simulating orbital neoplasm: A false
aneurysm and a blood cyst with foreign body granuloma of the orbit. Am J Ophthalmol 62:528, 1966 137. Parke DW II, Font RL, Boniuk M, McCrary JA III: “Cholesteatoma” of the orbit. Arch Ophthalmol 100:612, 1982 138. Raflo GT, Hurwitz JJ: Lipid granuloma of the frontal bone presenting as a space-occupying lesion
of the orbit. Can J Ophthalmol 16:153, 1981 139. Bergin DJ, McCord CD, Dutton JJ, Garrett SN: Chronic hematic cyst of the orbit. Ophthalmic Plast Reconstr Surg 4:31, 1988 140. Goldberg RA et al: Orbital inflammation and optic neuropathies associated with chronic sinusitis
of intranasal cocaine abuse: Possible role of contiguous inflammation. Arch Ophthalmol 107:831, 1989 141. Weber A, Mikulis D: Inflammatory disorders of the paraorbital sinuses and their complications. Radiol Clin North Am 25:615, 1987 142. Chawla HS, Goodwin JA, Ticho BH, Feist RM: Orbital and sinus inflammation with secondary optic neuropathy. Ann Ophthalmol 23:231, 1991 143. Kronschnabel EF: Orbital apex syndrome due to sinus infection. Laryngoscope 84:353, 1974 144. Straatsma B: Ocular manifestations of Wegener's granulomatosis. Am J Ophthalmol 44:789, 1957 145. Soukiasian S, Foster C, Niles J, Raizman M: Diagnostic value of antineutrophil cytoplasmic antibodies in scleritis
associated with Wegener's granulomatosis. Ophthalmology 99:125, 1983 146. Spalton D, Graham E, Page N, Sanders M: Ocular changes in limited forms of Wegener's granulomatosis. Br J Ophthalmol 65:553, 1981 147. Fauci A, Haynes B, Costa J et al: Lymphomatoid granulomatosis: Prospective clinical and therapeutic experience
over 10 years. N Engl J Med 306:68, 1982 148. Cupps T, Fauci A: Wegener's granulomatosis. In Smith L (ed): Major
Problems in Internal Medicine, vol XXI, pp 72–87. Philadelphia, WB
Saunders 1981 149. Kalina PH, Garrity JA, Herman DC et al: Role of testing for anticytoplasmic autoantibodies in the differential
diagnosis of scleritis and orbital pseudotumor. Mayo Clin Proc 65:1110, 1990 150. Allen J, France T: Pseudotumor as the presenting sign of Wegener's granulomatosis in
a child. J Pediatr Ophthalmol 14:158, 1977 151. Liliequist B, Link H: Wegener's granulomatosis: Report of a case. Angiology 19:215, 1968 152. Fauci A, Wolff S: Wegener's granulomatosis: Studies in eighteen patients and a review
of the literature. Medicine 52:535, 1973 153. Cassan SM, Divertie MB, Hollenhorst RW, Harrison E Jr: Pseudotumor of the orbit and limited Wegener's granulomatosis. Ann Intern Med 72:687, 1970 154. Kay C, McCrary J: Multiple cranial nerve palsies in late metastasis of midline malignant
reticulosis. Am J Ophthalmol 88:1087, 1979 155. Hardwig P, Bartley G, Garrity J: Surgical management of nasolacrimal duct obstruction in patients with Wegener's
granulomatosis. Ophthalmology 99:133, 1992 156. Cogan D: Corneoslceral lesions in periarteritis nodosa and Wegener's granulomatosis. Trans Am Ophthalmol Soc 53:321, 1965 157. Brubaker R, Font R, Shepherd E: Granulomatous sclerouveitis: Regression of ocular lesions with cyclophosphamide
and prednisone. Arch Ophthalmol 86:517, 1971 158. Specks U, Wheatley C, McDonald T et al: Anticytoplasmic autoantibodies in the diagnosis and follow-up of Wegener's
granulomatosis. Mayo Clin Proc 64:28, 1989 159. Seidman J: Case records of the Massachusetts General Hspital: Case 2-1990 (letter; comment). N Engl J Med 324:1371, 1991 160. Leavitt R, Fauci A: Wegener's granulomatosis and other systemic granulomatous conditions. Curr Opin Rheumatol 2:55, 1990 161. Liebow AA, Carrington CR, Friedman PJ: Lymphomatoid granulomatosis. Hum Pathol 3:457, 1972 162. DeRemee R, Weiland L, McDonald T: Respiratory vasculitis. Mayo Clin Proc 55:492, 1980 163. Cupps T, Fauci A: Lymphomatoid granulomatosis. In Smith L (ed): Major Problems
in Internal Medicine, vol XXI, pp 88–98. Philadelphia, WB
Saunders, 1981 164. Myers J: Lymphomatoid granulomatosis: Past, present, future? Mayo Clin Proc 65:274, 1990 165. Pearson A, Craft A, Howe J: Choroidal involvement in lymphomatoid granulomatosis. Br J Ophthalmol 75:688, 1991 166. Pisani RJ, DeRemee RA: Clinical implications of the histopathologic diagnosis of. Mayo Clin Proc 65:151, 1990 167. Katzenstein A, Carrington C, Liebow A: Lymphomatoid granulomatosis: A clinicopathologic study of 152 cases. Cancer 43:360, 1979 168. Colby T: Central nervous system lymphomatoid granulomatosis in AIDS? Hum Pathol 20:361, 1989 169. Anders K, Latta H, Chang BS et al: Lymphomatoid granulomatosis and malignant lymphoma of the central nervous
system in the acquired immunodeficiency syndrome. Hum Pathol 20:326, 1989 170. Mittal K, Neri A, Feiner H et al: Lymphomatoid granulomatosis in the acquired immunodeficiency syndrome. Cancer 65:1343, 1990 171. Najjar T, Gadol C, Khan M: Immune deficiency with polymorphic reticulosis. Oral Surg Oral Med Oral Pathol 67:322, 1989 172. Fauci A, Haynes B, Katz P, Wolf S: Wegener's granulomatosis: Prospective clinical and therapeutic experience
with 85 patients for 21 years. Ann Intern Med 98:76, 1983 173. Carlson K, Gibson L: Cutaneous signs of lymphomatoid granulomatosis. Arch Dermatol 127:1693, 1991 174. Eichel B, Harrison E, Devine K et al: Primary lymphoma of the nose including a relationship to lethal midline
granuloma. Am J Surg 112:597, 1966 175. McDonald TJ, DeRemee RA, Harrison E Jr et al: The protean clinical features of polymorphic reticulosis (lethal midline
granuloma). Laryngoscope 86:936, 1976 176. Gaulard P, Henni T, Marolleau JP et al: Lethal midline granuloma (polymorphic reticulosis) and lymphomatoid granulomatosis: Evidence
for a monoclonal T-cell lymphoproliferative disorder. Cancer 62:705, 1988 177. Kornblut AD: Case records of the Massachusetts General Hospital: Weekly clinicopathological
exercises: Case 2-1990: Current concepts of the lethal midline
granuloma syndrome (letter; comment). N Engl J Med 322:116, 1990 178. Robin JB, Schanzlin DJ, Meisler DM et al: Ocular involvement in the respiratory vasculitides. Surv Ophthalmol 30:127, 1985 179. McKay D, Ell J, Williams R, Taylor F: Lymphomatoid granulomatosis presenting as sudden blindness. Aust NZ J Ophthalmol 182:215, 1990 180. Whittaker S, Foroni L, Luzzatto L et al: Lymphomatoid granulomatosis: Evidence of a clonal T-cell origin and an
association with lethal midline granuloma. Q J Med 68:645, 1988 181. Font RL, Rosenbaum PS, Smith J Jr: Lymphomatoid granulomatosis of eyelid and brow with progression to lymphoma. J Am Acad Dermatol 23:334, 1990 182. Lin-Greenberg A, Villacin A, Moussa G: Lymphomatoid granulomatosis presenting as ultrodestructive gastrointestinal
tract lesions in patients with human immunodeficiency virus infection. Arch Intern Med 150:2581, 1990 183. Gold J, Castella A: Polymorphic reticulosis: T- or B-cell malignancy. Laryngoscope 99:989, 1989 184. Ratech H, Burke J, Blayney D et al: A clinicopathologic study of malignant lymphomas of the nose, paranasal
sinus, hard palate, including cases of lethal midline granuloma. Cancer 64:2525, 1989 185. Harabuchi Y, Yamanaka N, Kataura A et al: Epstein-Barr virus in nasal T-cell lymphomas in patients with lethal midline
granuloma. Lancet 335:128, 1990 186. Stewart J: Progressive lethal granulomatosis ulceration of the nose. J Laryngol Otol 48:675, 1933 187. Spear G, Walker WG Jr: Lethal midline granuloma (granuloma gangrenescens) at autopsy. Bull Johns Hopkins Hosp 99:313, 1956 188. Daggett RB, Haghighi P, Terkeltaub RA: Nasal cocaine abuse causing an aggressive midline intranasal and pharyngeal
destructive process mimicking midline reticulosis and limited Wegener's
granulomatosis. J Rheumatol 17:838, 1990 189. Pickens JP, Modica L: Current concepts of the lethal midline granuloma syndrome (see comments). Otolaryngol Head Neck Surg 100:623, 1989 190. Kornblut A: Current concepts of the lethal midline granuloma syndrome. Otolaryngol Head Neck Surg 102:201, 1990 191. von Rokitansky C: Ueger einige der wichtigsten Krankheitin der Arterien. Denkscher Akad Wissensch 4:49, 1852 192. Kussmaul A, Maier K: Uber eine bisher nicht beschriebene eigenthumliche Arterienerkrankung (periarteritis
nodosa), die mit Morbus Brightii un rapid fortschreitender
allgemeiner Muskellahmung einhergeht. Dtsch Arch Klin Med 1:484, 1866 193. Cupps T, Fauci A: Systemic necrotizing vasculitis of the polyarteritis
nodosa group. In Smith L (ed): Major Problems in Internal Medicine, vol
XXI, pp 26–49. Philadelphia, WB Saunders, 1981 194. Frohnert P, Sheps S: Long-term follow-up study of periarteritis nodosa. Am J Med 43:8, 1967 195. Moreland LW, Ball GV: Cutaneous polyarteritis nodosa. Am J Med 88:426, 1990 196. Lightfoot R Jr, Michel BA, Bloch DA et al: The American College of Rheumatology 1990 criteria for the classification
of polyarteritis nodosa. Arthritis Rheum 33:1088, 1990 197. Goldsmith J: Periarteritis nodosa with involvement of the choroidal and retinal arteries. Am J Ophthalmol 29:435, 1946 198. Moore JG, Sevel D: Corneoscleral ulceration in periarteritis nodosa. Br J Ophthalmol 50:651, 1966 199. Harbert F, McPherson S: Scleral necrosis in polyarteritis nodosa. Am J Ophthalmol 30:727, 1947 200. Purcell J Jr, Birkenkamp R, Tsai CC: Conjunctival lesions in periarteritis nodosa: A clinical and immunopathologic
study. Arch Ophthalmol 102:736, 1984 201. Koike R, Yamada M, Matsunaga T et al: Polyarteritis nodosa (PN) complicated with unilateral exophthalmos. Intern Med 32:232, 1993 202. Wien SV, Merz E: Exophthalmos secondary to periarteritis nodosa. Am J Ophthalmol 56:204, 1963 203. Ishida K, Yokota T, Wada Y et al: Unilateral facial swelling and exophthalmos in a patient with polyarteritis
nodosa. Intern Med 31:500, 1992 204. Hope-Robertson W: Pseudo-tumor of the orbit as a presenting sign in periarteritis nodosa. Trans Ophthalmol Soc NZ 8:56, 1956 205. Walton E: Pseudotumor of the orbit and polyarteritis nodosa. J Clin Pathol 12:419, 1959 206. Albert DA, Rimon D, Silverstein MD: The diagnosis of polyarteritis nodosa: I. A literature-based decision analysis
approach. Arthritis Rheum 31:1117, 1988 207. Albert DA, Silverstein MD, Paunicka K et al: The diagnosis of polyarteritis nodosa: II. Empirical verification of a
decision analysis model. Arthritis Rheum 31:1128, 1988 208. Guillevin L, Jarrousse B, Lok C et al: Long-term follow-up after treatment of polyarteritis nodosa and Churg-Strauss
angiitis with comparison of steroids, plasma exchange and cyclophosphamide
to steroids and plasma exchange: A prospective randomized
trial of 71 patients. The Cooperative Study Group for Polyarteritis Nodosa (see
comments). J Rheumatol 18:567, 1991 209. Alloway JA, Cupps TR: High dose methylprednisolone for retroorbital Wegener's granulomatosis. J Rheumatol 20:752, 1993 210. Bagegni A, Lyness RW, Johnston PB, Douglas JF: Visual recovery in orbital vasculitis. Br J Ophthalmol 72:737, 1988 211. Churg J, Strauss L: Allergic granulomatosis, allergic angiitis, and periarteritis nodosa. Am J Pathol 27:277, 1951 212. Cury D, Breakey A, Payne F: Allergic granulomatous angiitis associated with uveoscleritis and papilledema. Arch Ophthalmol 55:261, 1966 213. Chumbley LC, Harrison E Jr, DeRemee RA: Allergic granulomatosis and angiitis (Churg-Strauss syndrome): Report and
analysis of 30 cases. Mayo Clin Proc 52:477, 1977 214. Ashton N, Cook C: Allergic granulomatous nodules of the eyelid and conjunctiva: The XXXV
Edward Jackson Memorial Lecture. Am J Ophthalmol 87:1, 1979 215. Meisler DM, Stock EL, Wertz RD et al: Conjunctival inflammation and amyloidosis in allergic granulomatosis and
angiitis (Churg-Strauss syndrome). Am J Ophthalmol 91:216, 1981 216. Nissim F, Von der Valde J, Czernobilsky B: A limited form of Churg-Strauss syndrome: Ocular and cutaneous manifestations. Arch Pathol Lab Med 106:305, 1982 217. Rose G, Spencer H: Polyarteritis nodosa. Q J Med 26:43, 1957 218. Cupps T, Fauci A: Giant cell arteritides. In Smith L (ed): Major Problems
in Internal Medicine, vol XXI, pp 99–115. Philadelphia, WB Saunders, 1981 219. Hauser WA, Ferguson RH, Holley KE, Kurland LT: Temporal arteritis in Rochester, Minnesota, 1951 to 1967. Mayo Clin Proc 46:597, 1971 220. Glaser J: Topical diagnosis: Prechiasmal visual pathways. In Tasman W, Jaeger
E (eds): Duane's Clinical Ophthalmology, vol 2, chap 5. Philadelphia, JB
Lippincott, 1989 221. Miller N: Walsh and Hoyt's Clinical Neuro-Ophthalmology, pp 2601–2627. Baltimore, Williams & Wilkins, 1991 222. Barricks ME, Traviesa DB, Glaser JS, Levy IS: Ophthalmoplegia in cranial arteritis. Brain 100:209, 1977 223. Klein RG, Campbell RJ, Hunder GG, Carney JA: Skip lesions in temporal arteritis. Mayo Clin Proc 51:504, 1976 224. Talal N. Sjögren's syndrome: Historical overview and clinical spectrum
of disease. Rheum Dis Clin North Am 18:507, 1992 225. Baum J: Clinical manifestations of dry eye states. Trans Ophthalmol Soc UK 104:415, 1985 226. Maddison PJ: Dry eyes: Autoimmunity and relationship to other systemic disease. Trans Ophthalmol Soc UK 104:458, 1985 227. Fox RI, Chan EK, Kang HI: Laboratory evaluation of patients with Sjögren's syndrome. Clin Biochem 25:213, 1992 228. Friedlaender MH: Ocular manifestations of Sjögren's syndrome: Keratoconjunctivitis
sicca. Rheum Dis Clin North Am 18:591, 1992 229. Van Bijsterveld OP: Diagnosis and differential diagnosis of keratoconjunctivitis sicca associated
with tear gland degeneration. Clin Exp Rheumatol 5:3, 1990 230. Sreebny LM, Valdini A: Xerostomia: A neglected symptom. Arch Intern Med 147:1333, 1987 231. Schiodt M, Thorn J: Criteria for the salivary component of Sjögren's syndrome: A
review. Clin Exp Rheumatol 7:119, 1989 232. Scully C: Oral parameters in the diagnosis of Sjögren's syndrome. Clin Exp Rheumatol 7:113, 1989 233. Daniels TE, Fox PC: Salivary and oral components of Sjögren's syndrome. Rheum Dis Clin North Am 18:571, 1992 234. Kelly CA, Griffiths ID: Major upper airways obstruction associated with Sjögren's syndrome: A
case report and literature review. Br J Rheumatol 28:543, 1989 235. Wiedemann HP, Matthay RA: Pulmonary manifestations of the collagen vascular diseases. Clin Chest Med 10:677, 1989 236. Constantopoulos SH, Tsianos EV, Moutsopoulos HM: Pulmonary and gastrointestinal manifestations of Sjögren's syndrome. Rheum Dis Clin North Am 18:617, 1992 237. Provost TT, Watson R: Cutaneous manifestations of Sjögren's syndrome. Rheum Dis Clin North Am 18:609, 1992 238. Moutsopoulos HM, Youinou P: New developments in Sjögren's syndrome. Curr Opin Rheumatol 3:815, 1991 239. Kater L, de Wilde PC: New developments in Sjögren's syndrome. Curr
Opin Rheumatol 1992; 4:657-65. 240. Rosenthal DS, Harris NL, Mueller PR et al: Generalized lymphadenopathy with a palatal lesion associated with Sjögren's
syndrome. N Engl J Med 305:153, 1981 241. Alexander E, Provost TT: Sjögren's syndrome: Association of cutaneous vasculitis with
central nervous system disease. Arch Dermatol 123:801, 1987 242. Khan MA, Akhtar M, Taher SM: Membranoproliferative glomerulonephritis in a patient with primary Sjögren's
syndrome: Report of a case with review of the literature. Am J Nephrol 8:235, 1988 243. Alexander E: Central nervous system disease in Sjögren's syndrome: New insights
into immunopathogenesis. Rheum Dis Clin North Am 18:637, 1992 244. Sigal LH: The neurologic presentation of vasculitic and rheumatologic syndromes: A
review. Medicine 66:157, 1987 245. Cleary KR, Batsakis JG: Biopsy of the lip and Sjögren's syndrome. Ann Otol Rhinol Laryngol 99:323, 1990 246. Daniels TE: Salivary histopathology in diagnosis of Sjögren's syndrome. Scand J Rheumatol Suppl 61:36, 1986 247. Carsons S: Newer laboratory parameters for the diagnosis of rheumatic disease. Am J Med 85:34, 1988 248. Chan EK, Andrade LE: Antinuclear antibodies in Sjögren's syndrome. Rheum Dis Clin North Am 18:551, 1992 249. Nakamura RM, Tan EM: Recent advances in laboratory tests and the significance of autoantibodies
to nuclear antigens in systemic rheumatic diseases. Clin Lab Med 6:41, 1986 250. Arnett FC, Goldstein R, Duvic M, Reveille JD: Major histocompatibility complex genes in systemic lupus erythematosus, Sjögren's
syndrome, and polymyositis. Am J Med 85:38, 1988 251. Reveille JD, Arnett FC: The immunogenetics of Sjögren's syndrome. Rheum Dis Clin North Am 18:539, 1992 252. Fox RI, Kang HI: Pathogenesis of Sjögren's syndrome. Rheum Dis Clin North Am 18:517, 1992 253. Gardiner P: Primary Sjögren's syndrome. Baillieres Clin Rheumatol 7:59, 1993 254. Provost TT, Vasily D, Alexander E: Sjögren's syndrome. Cutaneous, immunologic, and nervous system
manifestations. Neurol Clin 5:405, 1987 255. Lemp MA: Recent developments in dry eye management. Ophthalmology 94:1299, 1987 256. Hidayat AA, Cameron JD, Font RL, Zimmerman LE: Angiolymphoid hyperplasia with eosinophilia (Kimura's disease) of
the orbit and ocular adnexa. Am J Ophthalmol 96:176, 1983 257. Sheren SB, Custer PL, Smith ME: Angiolymphoid hyperplasia with eosinophilia of the orbit associated with
obstructive airway disease. Am J Ophthalmol 108:167, 1989 258. Smith DL, Kincaid MC, Nicolitz E: Angiolymphoid hyperplasia with eosinophilia (Kimura's disease) of
the orbit. Arch Ophthalmol 106:793, 1988 259. Fu KK: Orbital Kimura's disease. J R Soc Med 86:234, 1993 260. Francis IC, Kappagoda MB, Smith J, Kneale K: Kimura's disease of the orbit. Ophthalmic Plast Reconstr Surg 4:235, 1988 261. Archer KF, Hurwitz JJ, Heathcote G: Orbital angiolymphoid hyperplasia with eosinophilia: Presentation as chalazion. Ophthalmic Plast Reconstr Surg 7:208, 1991 262. Feinfield RE, Hesse RJ, Rosenberg SA: Orbital inflammatory disease associated with systemic lupus erythematosus. South Med J 84:98, 1991 263. Brenner EH, Shock JP: Proptosis secondary to systemic lupus erythematosus. Arch Ophthalmol 91:81, 1974 264. Magee KL, Hymes SR, Rapini RP et al: Lupus erythematosus profundus with periorbital swelling and proptosis. J Am Acad Dermatol 24:288, 1991 265. Kennedy AC, McGavin DD: Rheumatoid scleritis producing exophthalmos. Br J Clin Pract 29:73, 1975 266. Jordan DR, McDonald H, Olberg B et al: Orbital panniculitis as the initial manifestation of systemic lupus erythematosus. Ophthalmic Plast Reconstr Surg 9:71, 1993 267. Nowinski T, Bernardino V, Naidoff M, Parrish R: Ocular involvement in lupus erythematosus profundus (panniculitis). Ophthalmology 89:1149, 1982 268. Winkelmann RK: Panniculitis in connective tissue disease. Arch Dermatol 119:336, 1983 269. Alegre VA, Winkelmann RK: Histiocytic cytophagic panniculitis (see comments). J Am Acad Dermatol 20:177, 1989 270. Bankhurst AD, Carlow TJ, Reidy RW: Exophthalmos in systemic lupus erythematosus. Ann Ophthalmol 16:669, 1984 271. Haynes BF, Pikus A, Kaiser-Kupfer M, Fauci AS: Successful treatment of sudden hearing loss in Cogan's syndrome with
corticosteroids. Arthritis Rheum 24:501, 1981 272. Vollertsen RS, McDonald TJ, Younge BR et al: Cogan's syndrome: 18 cases and a review of the literature. Mayo Clin Proc 61:344, 1986 273. Haynes BF, Kaiser-Kupfer MI, Mason P, Fauci AS: Cogan syndrome: Studies in thirteen patients, long-term follow-up, and
a review of the literature. Medicine 59:426, 1980 274. Allen NB, Cox CC, Cobo M et al: Use of immunosuppressive agents in the treatment of severe ocular and vascular
manifestations of Cogan's syndrome. Am J Med 88:296, 1990 275. Gray RE, Jenkins EA, Hall MA et al: Recurrent acute proptosis in atypical systemic lupus erythematosus. Clin Rheumatol 8:528, 1989 276. Kattah JC, Zimmerman LE, Kolsky MP et al: Bilateral orbital involvement in fatal giant cell polymyositis. Ophthalmology 97:520, 1990 277. Cheson BD, Bluming AZ, Alroy J: Cogan's syndrome: A systemic vasculitis. Am J Med 60:549, 1976 278. Cochrane AD, Tatoulis J: Cogan's syndrome with aortitis, aortic regurgitation, and aortic arch
vessel stenoses. Ann Thorac Surg 52:1166, 1991 279. McDonald TJ, Vollertsen RS, Younge BR: Cogan's syndrome: Audiovestibular involvement and prognosis in 18 patients. Laryngoscope 95:650, 1985 280. Vollertsen RS: Vasculitis and Cogan's syndrome. Rheum Dis Clin North Am 16:433, 1990 281. Bicknell JM, Holland JV: Neurologic manifestations of Cogan syndrome. Neurology 28:278, 1978 282. LaRaja RD: Cogan syndrome associated with mesenteric vascular insufficiency. Arch Surg 111:1028, 1976 283. Gilbert WS, Talbot FJ: Cogan's syndrome: Signs of periarteritis nodosa and cerebral venous
sinus thrombosis. Arch Ophthalmol 82:633, 1969 284. Ishida T, Nakamura H, Hori S et al: Acalculous cholecystitis (panniculitis) associated with Weber-Christian
disease. Clin Imaging 17:56, 1993 285. Allen-Mersh TG: Weber-Christian panniculitis and auto-immune disease: A case report. J Clin Pathol 29:144, 1976 286. Panush RS, Yonker RA, Dlesk A et al: Weber-Christian disease: Analysis of 15 cases and review of the literature. Medicine 64:181, 1986 287. Frayer WC, Wise RT, Tsaltas TT: Ocular and adnexal changes associated with relapsing febrile non-suppurative
panniculitis (Weber-Christian disease). Trans Am Ophthalmol Soc 66:233, 1968 288. Bresnitz E, Strom B: Epidemiology of sarcoidosis. Epidemiol Rev 5:124, 1983 289. Siltzbach LE, James DG, Neville E et al: Course and prognosis of sarcoidosis around the world. Am J Med 57:847, 1974 290. Thomas P, Hunninghake G: Current concepts of the pathogenesis of sarcoidosis. Am Rev Respir Dis 135:747, 1987 291. Hoover DL, Khan JA, Giangiacomo J: Pediatric ocular sarcoidosis. Surv Ophthalmol 30:215, 1986 292. Headings VE, Weston D, Young R Jr, Hackney R Jr: Familial sarcoidosis with multiple occurrences in eleven families: A possible
mechanism of inheritance. Ann NY Acad Sci 278:377, 1976 293. Sharma OP, Neville E, Walker AN, James DG: Familial sarcoidosis: A possible genetic influence. Ann NY Acad Sci 278:386, 1976 294. Kavanaugh AF, Andrew SL, Cooper B et al: Familial associations in sarcoidosis: A report to the Research Committee
of the British Thoracic and Tuberculosis Association: Cyclosporine therapy
of central nervous system sarcoidosis (letter). Tubercle 54:87, 1973 295. Jabs DA, Johns CJ: Ocular involvement in chronic sarcoidosis. Am J Ophthalmol 102:297, 1986 296. Obenauf CD, Shaw HE, Sydnor CF, Klintworth GK: Sarcoidosis and its ophthalmic manifestations. Am J Ophthalmol 86:648, 1978 297. Brewerton DA, Cockburn C, James DC et al: HLA antigens in sarcoidosis. Clin Exp Immunol 27:227, 1977 298. Olenchock SA, Heise ER, Marx JJ Jr et al: HLA-B8 in sarcoidosis. Ann Allergy 47:151, 1981 299. Foster C: Ocular manifestations of sarcoidosis preceding systemic manifestations. In
Grassi C, Rizzato G, Pozzi E (eds): Sarcoidosis and Other
Granulomatous Diseases, pp 177–181. New York, Elsevier Science
Publishers, 1988 300. Spalton DJ, Sanders MD: Fundus changes in histologically confirmed sarcoidosis. Br J Ophthalmol 65:348, 1981 301. Klintworth G: Sarcoidosis. In Gold D, Weingeist T (eds): The Eye in Systemic
Disease, pp 289–293. Phildelphia, JB Lippincott, 1990 302. James DG, Neville E, Langley DA: Ocular sarcoidosis. Trans Ophthalmol Soc UK 96:133, 1976 303. Smith R, Nozik R: Sarcoidosis. In Smith R, Nozik R (eds): Uveitis: A Clinical
Approach to Diagnosis and Management, pp 161–163. Baltimore: Williams & Wilkins, 1983 304. O'Connor G: Endogenous uveitis: Sarcoidosis. In Kraus-Mackiw E, O'Connor
G (eds): Uveitis: Pathophysiology and Therapy, pp 93–97. New York, Thieme
Medical, 1986 305. Nussenblatt R, Palestine A: Sarcoidosis. In Nussenblatt R, Palestine A (eds): Uveitis: Fundamentals and Clinical Practice, pp 198–211. Chicago, Year
Book Medical Publishers, 1989 306. Campo RV, Aaberg TM: Choroidal granuloma in sarcoidosis. Am J Ophthalmol 97:419, 1984 307. Gragoudas ES, Regan CD: Peripapillary subretinal neovascularization in presumed sarcoidosis. Arch Ophthalmol 99:1194, 1981 308. Beardsley TL, Brown SV, Sydnor CF et al: Eleven cases of sarcoidosis of the optic nerve. Am J Ophthalmol 97:62, 1984 309. Gass JD, Olson CL: Sarcoidosis with optic nerve and retinal involvement. Arch Ophthalmol 94:945, 1976 310. Lamkin JC, Raizman MB: Update on laboratory tests for diagnosis of orbital disease. Int Ophthalmol Clin 32:27, 1992 311. Collison JM, Miller NR, Green WR: Involvement of orbital tissues by sarcoid. Am J Ophthalmol 102:302, 1986 312. Spillberg I: Sarcoidosis. In Schumacher H, Klippel J, Robinson D (eds): Primer
on the Rheumatic Diseases, pp 190–192. Atlanta, Arthritis
Foundation, 1988 313. Weinreb RN, Tessler H: Laboratory diagnosis of ophthalmic sarcoidosis. Surv Ophthalmol 28:653, 1984 314. Hunninghake GW, Bedell GN, Zavala DC et al: Role of interleukin-2 release by lung T-cells in active pulmonary sarcoidosis. Am Rev Respir Dis 128:634, 1983 315. Chan CC, Wetzig RP, Palestine AG et al: Immunohistopathology of ocular sarcoidosis: Report of a case and discussion
of immunopathogenesis. Arch Ophthalmol 105:1398, 1987 316. Barth C, Judge M, Mattman L, Hessburg P: Isolation of an acid-fast organism from the aqueous humor in a case of
sarcoidosis. Henry Ford Hosp Med J 27:127, 1979 317. Nichols CW, Eagle R Jr, Yanoff M, Menocal NG: Conjunctival biopsy as an aid in the evaluation of the patient with suspected
sarcoidosis. Ophthalmology 87:287, 1980 318. Merritt JC, Lipper SL, Peiffer RL, Hale LM: Conjunctival biopsy in sarcoidosis. J Natl Med Assoc 72:347, 1980 319. Merritt JC, Ross G, Avery A: Conjunctival biopsy in sarcoidosis: 4 year NCMH experience. NC Med J 44:636, 1983 320. Weinreb RN: Diagnosing sarcoidosis by transconjunctival biopsy of the lacrimal gland. Am J Ophthalmol 97:573, 1984 321. Karcioglu ZA, Brear R: Conjunctival biopsy in sarcoidosis. Am J Ophthalmol 99:68, 1985 322. Baughman RP, Ploysongsang Y, Roberts RD, Srivastava L: Effects of sarcoid and steroids on angiotensin-converting enzyme. Am Rev Respir Dis 128:631, 1983 323. Kavanaugh AF, Andrew SL, Cooper B et al: Cyclosporine therapy of central nervous system sarcoidosis (letter). Am J Med 82:387, 1987 324. Alper MG, Zimmerman LE, Piana FG: Orbital manifestations of Erdheim-Chester disease. Trans Am Ophthalmol Soc 81:64, 1983 325. Shields JA, Karcioglu ZA, Shields CL et al: Orbital and eyelid involvement with Erdheim-Chester disease: A report of
two cases. Arch Ophthalmol 109:850, 1991 326. Murdoch IA, Dos Anjos R, Parsons JM, Calver DM: Spontaneous hyphaema in childhood. Eur J Pediatr 150:717, 1991 327. MacLeod PM: Juvenile xanthogranuloma of the iris managed with superficial radiotherapy. Clin Radiol 37:295, 1986 328. Casteels I, Olver J, Malone M, Taylor D: Early treatment of juvenile xanthogranuloma of the iris with subconjunctival
steroids. Br J Ophthalmol 77:57, 1993 329. Harley RD, Romayananda N, Chan GH: Juvenile xanthogranuloma. J Pediatr Ophthalmol Strabismus 19:33, 1982 330. Lewis JR, Drummond GT, Mielke BW et al: Juvenile xanthogranuloma of the corneoscleral limbus. Can J Ophthalmol 25:351, 1990 331. Kaufmann JG, Driebe W Jr, Hamed LM, Margo CE: Juvenile xanthogranuloma of the corneoscleral limbus. Ophthalmic Surg 24:428, 1993 332. Wertz FD, Zimmerman LE, McKeown CA et al: Juvenile xanthogranuloma of the optic nerve, disc, retina, and choroid. Ophthalmology 89:1331, 1982 333. Rouhiainen H, Nerdrum K, Puustjarvi T, Kosma VM: Xanthogranuloma juvenile: A rare cause of orbital swelling in adulthood. Ophthalmologica 204:162, 1992 334. Shields CL, Shields JA, Buchanon HW: Solitary orbital involvement with juvenile xanthogranuloma. Arch Ophthalmol 108:1587, 1990 335. Gaynes PM, Cohen GS: Juvenile xanthogranuloma of the orbit. Am J Ophthalmol 63:755, 1967 336. Sanders TE: Infantile xanthogranuloma of the orbit: A report of three cases. Am J Ophthalmol 61:1299, 1966 337. Cadera W, Silver MM, Burt L: Juvenile xanthogranuloma. Can J Ophthalmol 18:169, 1983 338. Schwartz TL, Carter KD, Judisch GF et al: Congenital macronodular juvenile xanthogranuloma of the eyelid. Ophthalmology 98:1230, 1991 339. Fischbach J, Sause W: Radiation therapy in the management of ocular juvenile xanthogranuloma. Int J Radiat Oncol Biol Phys 8:959, 1982 340. Ross MJ, Cohen KL, Peiffer R Jr, Grimson BS: Episcleral and orbital pseudorheumatoid nodules. Arch Ophthalmol 101:418, 1983 341. Lawton AW, Karesh JW: Periocular granuloma annulare. Surv Ophthalmol 31:285, 1987 342. Choi KH, Wilbur AC, Duvall J et al: Orbital pseudorheumatoid nodule. AJNR 6:828, 1985 343. Cousin GC: Granuloma annulare of the supra-orbital region: A case report. Br J Oral Maxillofac Surg 29:347, 1991 344. Ferry AP: Subcutaneous granuloma annulare (“pseudorheumatoid nodule”) of
the eyebrow. J Pediatr Ophthalmol 14:154, 1977 345. Floyd BB, Brown B, Isaacs H, Minckler DS: Pseudorheumatoid nodule involving the orbit. Arch Ophthalmol 100:1478, 1982 346. Rose GE, Patel BC, Garner A, Wright JE: Orbital xanthogranuloma in adults. Br J Ophthalmol 75:680, 1991 347. Finan MC, Winkelmann RK: Necrobiotic xanthogranuloma with paraproteinemia: A review of 22 cases. Medicine 65:376, 1986 348. Bullock JD, Bartley GB, Campbell RJ et al: Necrobiotic xanthogranuloma with paraproteinemia: Case report and a pathogenetic
theory. Ophthalmology 93:1233, 1986 349. Cornblath WT, Dotan SA, Trobe JD, Headington JT: Varied clinical spectrum of necrobiotic xanthogranuloma. Ophthalmology 99:103, 1992 350. Luck J, Layton A, Noble BA: Necrobiotic xanthogranuloma with orbital involvement. J R Soc Med 85:357, 1992 351. Bullock JD, Bartley GB, Campbell RJ et al: Necrobiotic xanthogranuloma with paraproteinemia: Case report and a pathogenetic
theory. Trans Am Ophthalmol Soc 84:342, 1986 352. Plotnick H, Taniguchi Y, Hashimoto K et al: Periorbital necrobiotic xanthogranuloma and stage I multiple myeloma: Ultrastructure
and response to pulsed dexamethasone documented by magnetic
resonance imaging. J Am Acad Dermatol 25:373, 1991 353. Char DH, LeBoit PE, Ljung BM, Wara W: Radiation therapy for ocular necrobiotic xanthogranuloma (letter). Arch Ophthalmol 105:174, 1987 354. Schonder AA, Clift RC, Brophy JW, Dane LW: Bilateral recurrent orbital inflammation associated with retroperitoneal
fibrosclerosis. Br J Ophthalmol 69:783, 1985 355. Fruh D, Jaeger W, Kafer O: Orbital involvement in retroperitoneal fibrosis (morbus Ormond). Mod Probl Ophthalmol 14:651, 1975 356. Levine MR, Kaye L, Mair S, Bates J: Multifocal fibrosclerosis: Report of a case of bilateral idiopathic sclerosing
pseudotumor and retroperitoneal fibrosis. Arch Ophthalmol 111:841, 1993 357. Berger JR, Snodgrass S, Glaser J et al: Multifocal fibrosclerosis with hypertrophic intracranial pachymeningitis. Neurology 39:1345, 1989 358. Comings DE, Skubi KB, Van Eyes J, Motulsky AG: Familial multifocal fibrosclerosis: Findings suggesting that retroperitoneal
fibrosis, mediastinal fibrosis, sclerosing cholangitis, Riedel's
thyroiditis, and pseudotumor of the orbit may be different manifestations
of a single disease. Ann Intern Med 66:884, 1967 359. Richards AB, Shalka HW, Roberts FJ, Flint A: Pseudotumor of the orbit and retroperitoneal fibrosis: A form of multifocal
fibrosclerosis. Arch Ophthalmol 98:1617, 1980 |