1. Manschot WA: Primary congenital aphakia. Arch Ophthalmol 69:571, 1963 2. Lowe RF: Aetiology of the anatomical basis of primary angle-closure glaucoma: Biomedical
comparisons between normal eyes and eyes with primary angle-closure
glaucoma. Br J Ophthalmol 54:161, 1970 3. Durham DG: Cutis hyperelastica (Ehlers-Danlos syndrome) with blue scleras, microcornea, and
glaucoma. Arch Ophthalmol 49:220, 1953 4. McKusick VA: Megalocornea. In McKusick VA (ed): Mendelian Inheritance in
Man, p 639. Baltimore, Johns Hopkins University Press, 1975 5. Skuta GL, Sugar J, Ericson ES: Corneal endothelial cell measurements in megalocornea. Arch Ophthalmol 101:51, 1983 6. Francois J: La gonioscopie. III. Colobome de l'iris. Adv Ophthalmol 4:44, 1955 7. Malbran E, Dodds R: Megalocornea and its relation to congenital glaucoma. Am J Ophthalmol 49:908, 1960 8. Vail DT Jr: Adult hereditary anterior megalophthalmos sine glaucoma: A definite disease
entity, with special reference to extraction of the cataract. Arch Ophthalmol 6:39, 1931 9. Duke-EIder S: System of Ophthalmology, Vol III, Normal and Abnormal Development, Part 2, Congenital Deformities, p 501. London, Kimpton, 1963 10. Calamandrei D: Megalocornea in due pazienti con syndrome craniosinostocia. Q Ital Ofthalmol 3:278, 1950 11. Libert J, Vanhoof F, Farreaux JP et al: Ocular findings in I cell disease (mucolipidosis type II). Am J Ophthalmol 83:617, 1977 12. Desvignes P, Pouliquen Y, Legras M et al: Aspect iconographique d'une cornea plana dans une maladie de Lobstein. Arch Ophtalmol (Paris) 72:585, 1967 13. Rubel E: Kongenitale familiare flachheit der kornea (cornea plana). Klin Monatsbl Augenheilkd 50:427, 1912 14. Duke-EIder S: System of Ophthalmology, Vol III, Normal and Abnormal Development, Part 2, Abnormalities in the Curvature of the Cornea. Cornea
Plana, p 505. London, Kimpton, 1963 15. Kolkott W: Cornea plana und Mikrocornea periplana. Klin Monatsbl Augenheilkd 98:372, 1937 16. Reese AB, Ellsworth RM: The anterior chamber cleavage syndrome. Arch Ophthalmol 75:307, 1966 17. Kenyon, KR: Mesenchymal dysgeneses of the cornea. Metab Ophthalmol 2: 173, 1978 18. Waring GO, Bourne WM, Edelhauser HF et al: The corneal endothelium: Normal and pathologic structure and function. Ophthalmology 89:531, 1982 19. Townsend WM, Font RL, Zimmerman LE: Congenital corneal leukomas. II. Histopathologic findings in 19 eyes with
central defect in Descemet's membrane. Am J Ophthalmol 77:192, 1974 20. Townsend WM, Font RL, Zimmerman LE: Congenital corneal leukomas. III Histopathologic findings in 13 eyes with
noncentral defect in Descemet's membrane. Am J Ophthalmol 77:400, 1974 21. Bahn CF, Falls HF, Varley GA: Classification of corneal endothelial disorders based on neural crest origin. Ophthalmology 91:558, 1984 22. Kenyon KR: Mesenchymal dysgenesis in Peters' anomaly, sclerocornea and congenital
endothelial dystrophy. Exp Eye Res 21:125, 1975 23. Waring GO III, Rodrigues MM, Laibson PR: Anterior chamber cleavage syndrome: A stepladder classification. Surv Ophthalmol 20:3, 1975 24. Axenfeld T: Embryotoxon corneae posterius. Dtsch Ophthalmol Gesamte 42:301, 1920 25. Sugar HS: Juvenile glaucoma with Axenfeld's syndrome: A histologic report. Am J Ophthalmol 59:1012, 1965 26. Dark AJ, Kirkham TH: Congenital corneal opacities in a patient with Rieger's anomaly and
Down's syndrome. Br J Ophthalmol 52:631, 1968 27. Henkind P, Siegel IM, Carr RE: Mesodermal dysgenesis of the anterior segment: Rieger's anomaly. Arch Ophthalmol 73:810, 1965 28. Tabbara KF, Khouri FP, Kaloustian VM der: Rieger's syndrome with chromosomal anomaly (report of a case). Can J Ophthalmol 8:488, 1973 29. Collier M: Le keratocone posterieur. Arch Ophtalmol (Paris) 22:376, 1962 30. Jacobs HB: Posterior conical cornea. Br J Ophthalmol 41:31, 1957 31. Krachmer JH, Rodrigues MM: Posterior keratoconus. Arch Ophthalmol 96: 1867, 1978 32. Wolter JR, Haney WP: Histopathology of keratoconus posticus circumscriptus. Arch Ophthalmol 69:357, 1963 33. Ingram HU: Keratoconus posticus. Trans Ophthalmol Soc UK 56:563, 1963 34. Alkemade PPH: Dysgenesis Mesodermalis of the Iris and the Cornea. Springfield, IL, Charles
C Thomas, 1969 35. Peters' A: Uber angeborene Defektbildung der Descemetschen Membran. Klin Monatsbl Augenheilkd 44:27, 1906 36. Townsend WM: Congenital anomalies of the cornea. In Kaufman HE, Barron
BA, McDonald MB et al (eds): Cornea, p 333. New York, Churchill Livingstone, 1989 37. Ide CH, Matta C, Holt JE et al: Dysgenesis mesordermalis of the cornea (Peters' anomaly) associated with
cleft lip and palate. Ann Ophthalmol 7:841, 1975 38. Nakanishi I, Brown SI, The histopathology and ultrastructure of congenital
central corneal opacity
(Peters' anomaly). Am J Ophthalmol 72:801, 1971 39. Stone DL, Kenyon KR, Green WR et al: Congenital central corneal leukoma (Peters' anomaly). Am J Ophthalmol 81:173, 1976 40. Fogle JA, Green WR, Kenyon KR et al: Peripheral Peters' anomaly: A histopathologic case report. J Pediatr Ophthalmol 15:71, 1978 41. Townsend WM: Congenital corneal leukomas. I. Central defect in Descemet's membrane. Am J Ophthalmol 77:80, 1974 42. Waring GO, Laibson PR, Rodrigues M: Clinical and pathological alterations of Descemet's membrane: With
emphasis on endothelial metaplasia. Surv Ophthalmol 18:325, 1974 43. Waring GO: Ultrastructural classification of abnormal collagen tissue on the posterior
cornea (posterior collagen layer) (abstr). Invest Ophthalmol Vis Sci (suppl) 18:124, 1979 44. Howard RO, Abrahams IW: Sclerocornea. Am J Ophthalmol 71:1254, 1971 45. Goldstein JE, Cogan DG, Kaufman HE: Sclerocornea and associated congenital anomalies. Arch Ophthalmol 67:761, 1962 46. Kanai A, Wood TC, Polack FM et al: The fine structure of sclerocornea. Invest Ophthalmol 10:687, 1971 47. Rodrigues MM, Calhoun J, Weinreb S: Sclerocornea with unbalanced translocation (17p, 10q). Am J Ophthalmol 78:49, 1974 48. Weinzenblatt S: Congenital malformations of cornea associated with embryonic arrest of
ectodermal and mesodermal structures. Arch Ophthalmol 52:415, 1954 49. Fogle JA, Kenyon KR, Stark WJ et al: Defective epithelial adhesion in anterior corneal dystrophies. Am J Ophthalmol 79:925, 1975 50. Bron AJ, Tripathi RC: Cystic disorders of the corneal epithelium. I. Clinical aspects. Br J Ophthalmol 57:361, 1973 51. Guerry D III: Fingerprint-like lines in the cornea. Am J Ophthalmol 33:724, 1950 52. Guerry D III: Observations on Cogan's microcystic dystrophy of the corneal epithelium. Trans Am Ophthalmol Soc 63:320, 1965 53. DeVoe AG: Certain abnormalities of Bowman's membrane with particular reference
to fingerprint lines in the cornea. Trans Am Ophthalmol Soc 60: 195, 1962 54. Cogan DG, Donaldson DD, Kuwabara T et al: Microcystic dystrophy of the corneal epithelium. Trans Am Ophthalmol Soc 62:213, 1964 55. Cogan DG, Kuwabara T, Donaldson DD et al: Microcystic dystrophy of the cornea: A partial explanation for its pathogenesis. Arch Ophthalmol 92:470, 1974 56. Laibson PR: Microcystic corneal dystrophy. Trans Am Ophthalmol Soc 74:488, 1977 57. Franceschetti A: Hereditare rezidivierende Erosion der Hornhaut. Z Augenheilkd 66:309, 1928 58. Chandler PA: Recurrent erosion of the cornea. Am J Ophthalmol 28:355, 1945 59. Bron AJ, Brown NA: Some superficial corneal disorders. Trans Ophthalmol Soc UK 91:13, 1971 60. Tripathi RC, Bron AJ: Ultrastructural study of non-traumatic recurrent corneal erosion. Br J Ophthalmol 56:73, 1972 61. Trobe JD, Laibson PR: Dystrophic changes in the anterior cornea. Arch Ophthalmol 87:378, 1972 62. Tripathi RC, Bron AJ: Cystic disorders of the corneal epithelium. It. Pathogenesis. Br J Ophthalmol 57:376, 1973 63. Brown N, Bron A: Recurrent erosion of the cornea. Br J Ophthalmol 60:84, 1976 64. Brodrick JD, Dark AJ, Peace GW: Fingerprint dystrophy of the cornea: A histologic study. Arch Ophthalmol 92:483, 1974 65. Rodrigues MM, Fine BS, Laibson PR et al: Disorders of the corneal epithelium: A clinocopathologic study of dot, geographic, and
fingerprint patterns. Arch Ophthalmol 92:475, 1974 66. Brown NA, Bron A J: Superficial lines and associated disorders of the cornea. Am J Ophthalmol 81:34, 1976 67. Laibson PR, Krachmer JH: Familial occurrence of dot (microcystic), map, fingerprint dystrophy of
the cornea. Invest Ophthalmol 14:397, 1975 68. Meesmann A: Uber eine bisher nicht beschriebene dominant vererbte Dystrophia epithelialis
corneae. Ber Dtsch Opthalmol Ges 52: 154, 1938 69. Meesmann A, Wilke F: Klinische und anatomische Untersuchungen uber eine bisher unbekannte, dominant
vererbte Epitheldystrophie der Hornhaut. Klin Monatsbl Augenheilkd 103:361, 1939 70. Kuwabara T, Ciccarelli EC: Meesmann's corneal dystrophy: A pathological study. Arch Ophthalmol 71:676, 1964 71. Behnke H, Thiel HJ: Uber die Hereditare Epitheldystrophie der Hornhaut (typ Meesmann-Wilke) in
Schleswig-Holstein. Klin Monatsbl Augenheilkd 147:662, 1965 72. Thiel HJ, Behnke H: Uber die Variationsbreite der hereditaren Hornhautepitheldystrophie (Typ
Meesmann-Wilke). Ophthalmologica 155:81, 1968 73. Burns RP: Meesmann corneal dystrophy. Trans Am Ophthalmol Soc 66:530, 1968 74. Nakanishi I, Brown SI: Ultrastructure of the epithelial dystrophy of Meesmann. Arch Ophthalmol 93:259, 1975 75. Fine BS, Yannof M, Pitts E et al: Meesmann's epithelial dystrophy of the cornea. Am J Ophthalmol 83:633, 1977 76. Stocker FW, Holt LB: Rare form of hereditary epithelial dystrophy: Genetic, clinical
and pathologic study, Arch Ophthalmol 53:536, 1955 77. Pameijer JK: Ueber eine fremdartige familiare oberflachliche Hornhautveranderung. Klin Monatsbl Augenheilkd 95:516, 1935 78. Grayson M, Wilbrandt H: Dystrophy of the anterior limiting membrane of the cornea (Reis-Bucklers
type). Am J Ophthalmol 61:345, 1966 79. Fogle JA, Green WR, Kenyon KR: Anterior corneal dystrophy. Am J Ophthalmol 77:529, 1974 80. Reis W: Familiare, fleckige Hornhautentartung. Dtsch Med Wochenschr 43:575, 1917 81. Bucklers M: Uber eine weitere familiare Hornhautdystrophie (Reis). Klin Monatsbl Augenheilkd 114:386, 1949 82. Wood TO, Fleming JC, Dotson RS et al: Treatment of Reis-Bucklers corneal dystrophy by removal of subepithelial
fibrous tissue. Am J Ophthalmol 85:360, 1978 83. Caldwell DR: Postoperative recurrence of Reis-Bucklers dystrophy. Am J Ophthalmol 35:567, 1978 84. Olson RJ, Kaufman H: Recurrence of Reis-Bucklers corneal dystrophy in a graft. Am J Ophthalmol 85:349, 1978 85. Yamaguchi T, Polack FM, Valenti J: Electron microscopic study of recurrent Reis-Bucklers corneal dystrophy. Am J Ophthalmol 90:95, 1980 86. Lohse E, Stock EL, Jones L et al: Reis-Bucklers dystrophy: Immunofluorescent and electron microscopic studies. Cornea 8:200, 1989 87. Griffith DG, Fine BS: Light and electron microscopic observations in a superficial corneal dystrophy: Probable
early Reis-Bucklers type. Am J Ophthalmol 63:1659, 1967 88. Rice NSC, Ashton N, Jay B et al: Reis-Bucklers dystrophy: A clinicopathologic study. Br J Ophthalmol 52:577, 1968 89. Jones ST, Stauffer LK: Reis-Bucklers corneal dystrophy: A clinicopathological study. Trans Am Acad Ophthalmol Otolaryngol 74:417, 1970 90. Akiya S, Brown SI: The ultrastructure of Reis-Bucklers dystrophy. Am J Ophthalmol 72:549, 1971 91. Hogan MJ, Wood I: Reis-Bucklers corneal dystrophy. Trans Ophthalmol Soc UK 91:41, 1971 92. Franceschetti AT: La cornea verticillata (Gruber) et ses relations avec la maladie de Fabry. Ophthalmologica 156:232, 1968 93. Francois J: Glycolipid lipoidosis. In Symposium on Surgical and Medical
Management of Congenital Anomalies of the Eye. Transactions of the New
Orleans Academy of Ophthalmology. St. Louis, CV Mosby, 1968 94. D'Amico DJ, Kenyon KR, Ruskin JN: Amiodarone keratopathy: A drug-induced lipid storage disorder. Arch Ophthalmol 99:257, 1981 95. Tripathi RC, Bron AJ: Secondary anterior crocodile shagreen of Vogt. Br J Ophthalmol 59:5, 1975 96. Pouliquen Y, Dhermy P, Presles D et al: Degenerescence en Chagrin de crocodile
de Vogt ou degenerescence en mosaique de Valerio, Arch Ophthalmol (Paris) 36:395, 1976 97. Charney SM: Idiopathic band keratopathy. Arch Ophthalmol 75:505, 1966 98. Rodrigues MM, Gaster RN, Pratt MV: Unusual superficial confluent form of granular corneal dystrophy. Ophthalmology 90:1507, 1983 99. Jones ST, Zimmerman LE: Histopathologic differentiation of granular, macular and lattice dystrophies
of the cornea. Am J Ophthalmol 51:394, 1961 100. Garner A: Histochemistry of corneal granular dystrophy. Br J Ophthalmol 53:799, 1969 101. Rodrigues MM, Streeten BW, Krachmer JH et al: Micro-fibrillar protein and phospholipid in granular corneal dystrophy. Arch Ophthalmol 101:802, 1983 102. Johnson BL, Brown SI, Zaidman GW: A light and electron microscopic study of recurrent granular dystrophy
of the cornea. Am J Ophthalmol 92:49, 1981 103. Akiya S, Brown SI: Granular dystrophy of the cornea: Characteristic electron microscopic lesion. Arch Ophthalmol 84: 179, 1970 104. Waardenburg PJ, Jonkers GH: A specific type of dominant progressive dystrophy of the cornea developing
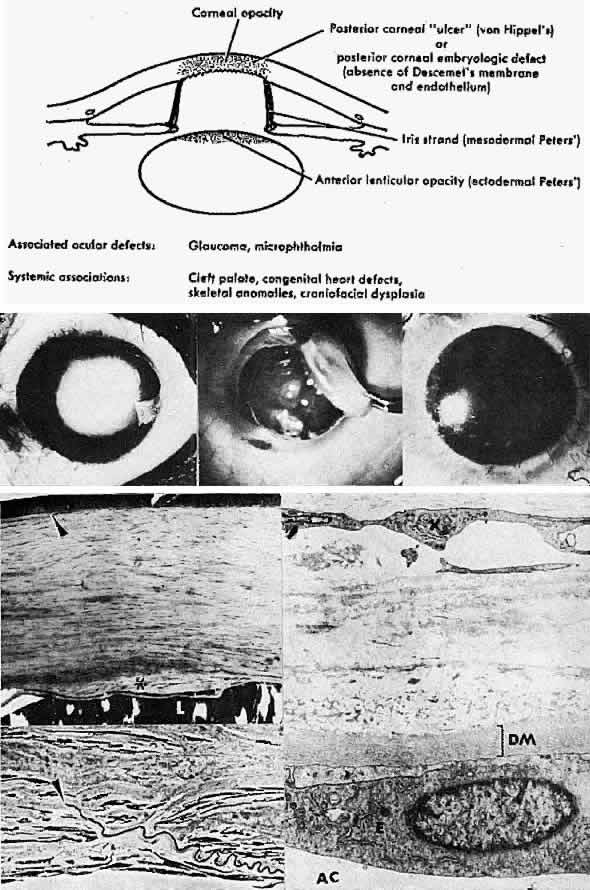
after birth. Acta Ophthalmol (Copenh) 39:919, 1961 105. Haddad R, Font RL, Fine BS: Unusual superficial variant of granular dystrophy of the cornea. Am J Ophthalmol 83:213, 1977 106. Folberg R, Alfonso E, Croxatto O et al: Clinically atypical granular corneal dystrophy with pathologic features
of lattice-like amyloid deposits. Ophthalmology 95:47, 1988 107. Tripathi RC, Garner A: Corneal granular dystrophy: A light and electron microscopical study of
its recurrence in a graft. Br J Ophthalmol 54:361, 1970 108. Brownstein S, Fine BS, Sherman ME et al: Granular dystrophy of the cornea: Light and electron microscopic confirmation
of recurrence in a graft. Am J Ophthalmol 77:701, 1974 109. Rodrigues MM, McGavic JS: Recurrent corneal granular dystrophy: A clinicopathologic study. Trans Am Ophthalmol Soc 73:306, 1975 110. Stuart JC, Mund ML, Iwamoto T et at: Recurrent granular corneal dystrophy. Am J Ophthalmol 79: 18, 1975 111. Klintworth Gk: Lattice corneal dystrophy: An inherited variety of amyloidosis restricted
to the cornea. Am J Pathol 50:371, 1967 112. Bowen RA, Hassard DTR, Wong VG et al: Lattice dystrophy of the cornea as a variety of amyloidosis. Am J Ophthalmol 70:822, 1970 113. Mondino BJ, Raj CVS, Skinner M et al: Protein AA and lattice corneal dystrophy. Am J Ophthalmol 89:337, 1980 114. Wheeler GE, Eiferman RA: Immunohistochemical identifications of the AA protein in lattice dystrophy. Exp Eye Research 36: 181, 1983 115. McMullan FD, DeLellis RA, Albert D et al: Corneal amyloidosis: An immunohistochemical
analysis. ARVO abstract. Invest Ophthalmol Vis Sci (suppl)25:6, 1984 116. Gorevic PD, Rodrigues MM, Krachmer JH et al: Lack of evidence for AA reactivity in amyloid deposits of lattice corneal
dystrophy and corneal amyloid degeneration. Am J Ophthalmol 98:216, 1984 117. Goldberg M: Genetic and Metabolic Eye Disease, pp 283–285. Boston, Little
Brown & Co. 1974 118. Frayer WC, Blodi FC: The lattice type of familial corneal degeneration. Arch Ophthalmol 61:712, 1959 119. Lorenzetti DWC, Kaufman HE: Macular and lattice dystrophies and their recurrences after keratoplasty. Trans Am Acad Ophthalmol Otolaryngol 71:112, 1967 120. Lanier JD, Fine M, Togni B: Lattice corneal dystrophy. Arch Ophthalmol 94:921, 1976 121. Meisler DM, Fine M: Recurrence of the clinical signs of lattice corneal dystrophy (type I) in
corneal transplants. Am J Ophthalmol 97:210, 1984 122. Meretoja J: Genetic aspects of familial amyloidosis with corneal lattice
dystrophy and cranial neuropathy, Clin Genet 4:173, 1973 123. Purcell JJ, Rodrigues M, Chisthi MI et al: Lattice corneal dystrophy associated with familial systemic amyloidosis (Meretoja's
syndrome). Ophthalmology 90:1512, 1983 124. Meretoja J: Comparative histopathological and clinical findings in eyes with lattice
corneal dystropy of the two different types. Ophthalmological 165: 15, 1972 125. Maury CPJ, Teppo AM, Kariniemi AL et at: Amyloid fibril protein in familial amyloidosis with cranial neuropathy
and corneal lattice dystrophy (FAP type IV) is related to transthyretin. Am J Pathol 89:359, 1988 126. Hida T, Proia AD, Kigasawa K et al: Histopathologic and immunochemical features of lattice corneal dystrophy
type III. Am J Ophthalmol 104:249, 1987 127. Hida T, Tsubota K, Kigasawa K et al: Clinical features of a newly recognized type of lattice corneal dystrophy. Am J Ophthalmol 104:241, 1987 128. Rabb MF, Blodi F, Boniuk M: Unilateral lattice dystrophy of the cornea. Trans
Am Acad Ophthalmol Otolaryngol 78:OP440, 1974 129. Donnenfeld ED, Cohen EJ, Ingraham HJ et al: Corneal thinning in macular corneal dystrophy. Am J Ophthalmol 101:112, 1986 130. Garner A: Histochemistry of macular dystrophy. Invest Ophthalmol 8:475, 1969 131. Klintworth GK, Smith CF: Macular corneal dystrophy: Studies of sulfated glycosaminoglycans in corneal
explant and confluent stromal cell cultures. Am J Pathol 89:167, 1977 132. Francois J, Victoria-Troncoso V, Maudgal PC et al: Study of the lysosomes by vital strains in normal keratocytes and in keratocytes
from macular dystrophy of the cornea. Invest Ophthalmol 15:559, 1976 133. Klintworth GK, Vogel FS: Macular corneal dystrophy: An inherited acid mucopolysaccharide storage
disease of the corneal fibroblast. Am J Pathol 45:565, 1964 134. Snip RC, Kenyon KR, Green WR: Macular corneal dystrophy: Ultrastructural pathology of corneal endothelium
and Descemet's membrane. Invest Ophthalmol 12:88, 1973 135. Deepak E, Thonar EJ-MA, Srinivasan M et al: Macular corneal dystrophy of the cornea: A systemic disorder of keratan
sulfate metabolism. Ophthalmology 97:1194, 1990 136. Robin AL, Green WR, Lapsa TP et al: Recurrence of macular corneal dystrophy after lamellar keratoplasty. Am J Ophthalmol 84:457, 1977 137. Thomsitt J, Bron AJ: Polymorphic stromal dystrophy. Br J Ophthalmol 59: 125, 1975 138. Pillar A: Zur Frage der familiaren Hornhautentartung: Uber eine eigenartige tiefe
schollige und periphere gitterformige familare Horhautdystrophie. Klin Monatsbl Augenheilkd 104:571, 1940 139. Krachmer JH, Dubord PJ, Rodrigues MM et al: Corneal posterior crocodile shagreen and polymorphic amyloid degeneration. Arch Ophthalmol 101:54, 1983 140. Mannis MJ, Krachmer JH, Rodrigues MM et al: Polymorphic amyloid degeneration of the cornea: A clinical and histopathologic
study. Arch Ophthalmol 99:1217, 1981 141. Kirk JG, Rabb M, Hattenhauer J et al: Primary familial amyloidosis of the cornea. Trans Am Acad Ophthalmol Otolaryngol 77:411, 1973 142. Stock EL, Kielar RA: Primary familial amyloidosis of the cornea. Am J Ophthalmol 82:266, 1976 143. Weber FL, Babel J: Gelatinous drop-like dystrophy: A form of primary corneal amyloidosis. Arch Ophthalmol 98:144, 1980 144. Mondino BJ, Rabb MJ, Sugar J et al: Primary familial amyloidosis of the cornea. Am J Ophthalmol 92:732, 1981 145. Schnyder WF: Ein bisher nicht bekannter Typus von familiarer, praseniler, subkapsularer
Schalenkatarakt und gleichzeitiges familiares Vorkommen von Glaskorper-veranderungen. Schwewiz Med Wochenschr 57:403, 1927 146. Schnyder WF: Scheibenformige Kristalleinlagerungen in der Hornhautmitte als Erbleiden. Klin Monatsbl Augenheilkd 103:494, 1939 147. Paufique L, Ravault MP, Bonnet M et al: Dystrophie crystalline de Schnyder. Bull Soc Ophtalmol Fr 64: 104, 1964 148. Luxemburg M: Hereditary crystalline dystrophy of the cornea. Am J Ophthalmol 63:507, 1967 149. Garner A, Tripathi RC: Hereditary crystalline stromal dystrophy of Schnyder. II. Histopathology
and ultrastructure. Br J Ophthalmol 56:400, 1972 150. Bron A, Williams HP, Carruthers ME: Hereditary crystalline stromal dystrophy of Schnyder. I. Clinical features
of a family with hyperlipoproteinaemia. Br J Ophthalmol 56:383, 1972 151. Bietti G: Ueber familares Vorkommen von “Retinitis punctata albescens” (verbunden
mit “Dystrophia marginalis cristallinea cornea”), Glitzern
des Glaskorpers und anderen degenerativen Augenveranderungen. Klin Monatsbl Augenheilkd 99:737, 1937 152. Welch RB: Bietti's tapetoretinal degeneration with marginal corneal dystrophy: Crystalline
retinopathy. Trans Am Ophthalmol Soc 75:176, 1977 153. Strachan IM: Cloudy central corneal dystrophy of Francois: Five cases in the same family. Br J Ophthalmol 53:192, 1969 154. Bramsen T, Ehlers N, Baggesen LH: Central cloudy corneal dystrophy of Francois. Acta Ophthalmol (Copenh) 54:221, 1976 155. Carpel EF, Sigelman RJ, Doughman DJ: Posterior amorphous corneal dystrophy. Am J Ophthalmol 83:629, 1977 156. Dunn SP, Krachmer JH, Ching SST: New findings in posterior amorphous corneal dystrophy. Arch Ophthalmol 102:236, 1984 157. Johnson AT, Folberg R, Vrabec MP et al: The pathology of posterior amorphous corneal dystrophy. Ophthalmology 97:104, 1990 158. Witschel H, Fine BS, Grutzner P et al: Congenital hereditary stromal dystrophy of the cornea. Arch Ophthalmol 96:1043, 1978 159. Goodside V: Posterior crocodile shagreen: A corneal dystrophy. Am J Ophthalmol 46:748, 1958 160. Krachmer JH, Dubord PJ, Rodrigues MM et al: Corneal posterior crocodile shagreen and polymorphic amyloid degeneration: A
histopathologic study. Arch Ophthalmol 101:54, 1983 161. Francois J, Neetens A: L'heredo-dystrophie mouchetee du parenchyme corneen. Acta Genet Med Gemellol (Roma) 6:387, 1957 162. Streeten BW, Falls HF: Hereditary fleck dystrophy of the cornea. Am J Ophthalmol 51:275, 1961 163. Aracena T: Hereditary fleck dystrophy of the cornea: Report of a family. J Pediatr Ophthalmol 12:223, 1975 164. Patten JT, Hyndiuk RA, Donaldson DD et al: Fleck (mouchettee) dystrophy
of the cornea, Ann Ophthalmol 8:25, 1976 165. Nicholson DH, Green WR, Cross HE et al: A clinical and histopathological study of Francois-Neetens speckled corneal
dystrophy. Am J Ophthalmol 83:554, 1977 166. Purcell JJ Jr, Krachmer JH, Weingeist TA: Fleck corneal dystrophy. Arch Ophthalmol 95:440, 1977 167. Pippow G: Zur Erbbedingheit der Cornea farinata. (Mehlstaubartige Hornhautdegeneration). Albrecht von Graefes Arch Ophtalmol 144:276, 1941 168. Paufique L, Etienne R: La “cornea farinata.” Bull Soc Ophtalmol Fr 50:522, 1950 169. Franceschetti A, Maeder G: Dystrophie profonde de la cornee dans un cas d'itchtyose congenitale. Bull Soc Ophtalmol Fr 67: 146, 1954 170. Grayson M, Wilbrandt H: Pre-Descemet dystrophy. Am J Ophthalmol 64:276, 1967 171. Curran RE, Kenyon KR, Green WR: Pre-Descemet's membrane corneal dystrophy. Am J Ophthalmol 77:711, 1974 172. Maeder G, Danis P: Sur une nouvelle forme de dystrophic corneenne (dystrophia filiformis profunda
corneae) associee a un keratocone. Ophthalmologica 114:246, 1947 173. Maumenee AE: Congenital hereditary corneal dystrophy. Am J Ophthalmol 50:1114, 1960 174. Judisch GF, Maumenee IH: Clinical differentiation of recessive congenital hereditary endothelial
dystrophy and dominant hereditary endothelial dystrophy. Am J Ophthalmol 85:606, 1978 175. Kenyon KR, Maumenee AE: The histological and ultrastructural pathology of congenital hereditary
corneal dystrophy: A case report. Invest Ophthalmol 7:475, 1968 176. Pearce WG, Tripathi RC, Morgan G: Congenital endothelial lial corneal dystrophy: Clinical, pathological and
genetic study. Br J Ophthalmol 53:577, 1969 177. Kanai A, Kaufman HE: Further electron microscopic study of the hereditary corneal edema. Invest Ophthalmol 10:545, 1971 178. Kanai A, Waltman S, Polack FM et al: Electron microscopic study of hereditary corneal edema. Invest Ophthalmol 10:89, 1971 179. Antine B: Histology of congenital hereditary corneal dystrophy. Am J Ophthalmol 69:964, 1970 180. Kenyon KR, Maumenee AE: Further studies of congenital hereditary endothelial dystrophy of the cornea. Am J Ophthalmol 76:419, 1973 181. Rodrigues MM, Waring GO, Laibson PR et al: Endothelial alterations in congenital corneal dystrophies. Am J Ophthalmol 80:678, 1975 182. Wolter JR: Secondary cornea guttata in interstitial keratopathy. Ophthalmologica 148:289, 1964 183. Laing RA, Sandstrom MM, Leibowitz HM: In vivo photomicrography of the corneal endothelium. Arch Ophthalmol 93: 143, 1975 184. Bourne WM, Kaufman HE: Specular microscopy of human corneal endothelium in vivo. Am J Ophthalmol 81:319, 1976 185. Cross HE, Maumenee AE, Cantolino SJ: Inheritance of Fuchs' endothelial dystrophy. Arch Ophthalmol 85:268, 1971 186. Krachmer JH, Purcell JJ Jr, Young CW et al: Corneal endothelial dystrophy: A study of 64 families. Arch Ophthalmol 96:2036, 1978 187. Polack FM: The posterior corneal surface in Fuchs' dystrophy: Scanning electron microscope
study. Invest Ophthalmol 13:913, 1974 188. Kenyon KR: The synthesis of basement membrane by the corneal epithelium in bullous
keratopathy. Invest Ophthalmol 8:156, 1969 189. Hogan MJ, Wood I, Fine M: Fuchs' endothelial dystrophy of the cornea. Am J Ophthalmol 78:363, 1974 190. Rosenblum P, Stark WJ, Maumenee IH et al: Hereditary Fuchs' dystrophy. Am J Ophthalmol 90:455, 1980 191. Bourne WM, Johnson DH, Campbell RJ: The ultrastructure of Descemet's membrane. III. Fuchs' dystrophy. Arch Ophthalmol 100: 1952, 1982 192. Cibis GW, Krachmer JA, Phelps CD et al: The clinical spectrum of posterior polymorphous dystrophy. Arch Ophthalmol 95: 1529, 1977 193. Snell AC Jr, Irwin ES: Hereditary deep dystrophy of the cornea. Am J Ophthalmol 45:636, 1958 194. Morgan G, Patterson A: Pathology of posterior polymorphous degeneration of the cornea. Br J Ophthalmol 51:433, 1967 195. Rubenstein RA, Sliverman J J: Hereditary deep dystrophy of the cornea associated with glaucoma and ruptures
in Descemet's membrane. Arch Ophthalmol 79:123, 1968 196. Hogan MJ, Bietti G: Hereditary deep dystrophy of the cornea (polymorphous). Am J Ophthalmol 68:777, 1969 197. Boruchoff SA, Kuwabara T: Electron microscopy of posterior polymorphous degeneration. Am J Ophthalmol 72:879, 1971 198. Hanselmayer H: Zur Histopathologie der hinteren polymorphen Hornhautdystrophie
nach Schlichting. 1. Licht mikroskopische Befunde in Beziehung
zum Klinischen Bild, Graefes Arch Klin Ophthalmol 184:345, 1972 199. Hanselmayer H: Zur Histopathologie der hinteren polymorphen Hornhautdystrophie nach Schlichting: II. Ultrastrukturelle Befunde pathogenetische und pathophysiologische
Bemerkungen. Graefes Arch Klin Ophthalmol 185:53, 1972 200. Grayson M: The nature of hereditary deep polymorphous dystrophy of the cornea: Its
association with iris and anterior chamber dysgenesis. Trans Am Ophthalmol Soc 72:516, 1974 201. Tripathi RC, Casey TA, Wise G: Hereditary posterior polymorphous dystrophy: An ultrastructural and clinical
report. Trans Ophthalmol Soc UK 94:211, 1974 202. Cibis GW, Krachmer JH, Phelps CD, Weingeist TA: Iridocorneal adhesions
in posterior polymorphous dystrophy. Trans Am Acad Ophthalmol Otolaryngol 81:OP770, 1976 203. Johnson BL, Brown SI: Posterior polymorphous dystrophy: A light and electron microscopic study. Br J Ophthalmol 62:89, 1978 204. Cibis GW, Tripathi RC: The differential diagnosis of Descemet's tear (Haab's striae) and
posterior polymorphous dystrophy bands: A clinicopathologic study. Ophthalmology 89:614, 1982 205. Brooks AMV, Grant G, Gillies WE: Differentiation of posterior polymorphous dystrophy from other posterior
corneal opacities by specular microscopy. Ophthalmology 96:1639, 1989 206. Hirst L, Waring GO: Clinical specular microscopy of posterior polymorphous endothelial dystrophy. Am J Ophthalmol 95:143, 1983 207. Boruchoff SA, Weiner MJ, Albert DM: Recurrence of posterior polymorphous corneal dystrophy after penetrating
keratoplasty. Am J 0phthalmol 109:323, 1990 208. Rodrigues MM, Phelps CD, Krachmer JH et al: Glaucoma due to endothelialization of the anterior chamber angle: A comparison
of posterior polymorphous dystrophy of the cornea and Chandler's
syndrome. Arch Ophthalmol 98:688, 1980 209. Campbell DG, Shields MB, Smith TR: The corneal endothelium and the spectrum of essential iris atrophy. Am J Ophthalmol 86:317, 1978 210. Rodrigues MM, Streeten BW, Spaeth GL: Chandler's syndrome as a variant of essential iris atrophy. Arch Ophthalmol 96:643, 1978 211. Shields MB, Campbell DG, Simmons RJ: The essential iris atrophies. Am J Ophthalmol 85:749, 1978 212. Shields MB: Progressive essential iris atrophy, Chandler's syndrome, and the iris
nevus (Cogan-Reese) syndrome: A spectrum of disease. Surv Ophthalmol 24:3, 1979 213. Yanoff M: Iridocorneal endothelial syndrome: Unification of a disease spectrum. Surv Ophthalmol 24: 1, 1979 214. Shields MB, Hirst LW, Quigley HA et al: Endothelial specular microscopy of iridocorneal endothelial syndrome (abstr). Invest Ophthalmol Visual Sci (suppl) 18:40, 1979 215. Patel A, Kenyon KR, Hirst LW et al: Clinicopathologic features Of Chandler's syndrome. Surv Ophthalmol 27:327, 1983 216. Kenyon KR, Van Horn DL, Edelhauser HF: Endothelial degeneration and posterior collagenous proliferation in aphakic
bullous keratopathy. Am J Ophthalmol 85:329, 1978 217. Perry HD, Buxton JN, Fine BS: Round and oval cones in keratoconus. Ophthalmology 87:905, 1980 218. Chi HH, Katzin HM, Teng CC: Histopathology of keratoconus. Am J Ophthalmol 42:847, 1956 219. Teng CC: Electron microscope study of the pathology of keratoconus. Am J Ophthalmol 55: 18, 1963 220. Pataa C, Joyon L, Roucher F: Ultrastructure du keratocone. Arch Ophthalmol (Paris) 30:403, 1970 221. Pouliquen Y, Graf B, De Kozak Y et al: Etude morphologique du keratocone. Arch Ophthalmol (Paris) 30:497, 1970 222. Stone DL, Kenyon KR, Stark W J: Ultrastructure of keratoconus with healed hydrops. Am J Ophthalmol 82:450, 1976 223. Spencer WH, Fisher JJ: The association of keratoconus with atopic dermatitis. Am J Ophthalmol 47:332, 1959 224. Copeman PW: Eczema and keratoconus. Br Med J 5468:977, 1965 225. Streiff EB: Keratocone et retinite pigmentaire, Bull Mem Soc Fr Ophtalmol 65:323, 1952 226. Cameron JA, Al-Rajhi AA, Badr IA: Corneal ectasias in vernal keratoconjunctivitis. Ophthalmology 96: 1915, 1989 227. Macsai MS, Varley GA, Krachmer JH: Development of keratoconus after contact lens wear. Arch Ophthalmol 108:534, 1990 228. McDonald MB, Kaufman HE, Durrie DS et al: Epikeratophakia for keratoconus: The nationwide study. Arch Ophthalmol 104: 1294, 1986 229. Fogle JA, Kenyon KR, Stark WJ: Damage to epithelial basement membrane by thermokeratoplasty. Am J Ophthalmol 83:392, 1977 230. Gasset AR, Kaufman HE: Thermokeratoplasty in the treatment of keratoconus. Am J Ophthalmol 79:226, 1975 231. Varley GA, Macsai MS, Krachmer JH: The results of penetrating keratoplasty for pellucid marginal degeneration. Am J Ophthalmol 110:149, 1990 232. Krachmer JH: Pellucid marginal corneal degeneration. Arch Ophthalmol 96: 1217, 1978 233. Cavara V: Keratoglobus and keratoconus. Br J Ophthalmol 34:621, 1950 234. Grayson M: Acute keratoglobus. Am J Ophthalmol 56:300, 1963 235. Cogan DG, Kuwabara T: Arcus senilis: Its pathology and histochemistry. Arch Ophthalmol 61:553, 1959 236. Andrews JS: The lipids of arcus senilis. Arch Ophthalmol 68:264, 1962 237. Walton KW: Studies on the pathogenesis of corneal arcus formation. I. The human corneal
arcus and its relation to atherosclerosis as studied by immunofluorescence. J Pathol 111:263, 1973 238. Sugar HS, Kobernick S: The white limbus girdle of Vogt. Am J Ophthalmol 50:101, 1960 239. Macfaul PA, Bedford MA: Ocular complications after therapeutic irradiation. Br J Ophthalmol 54:237, 1970 240. Suveges I, Levai G, Alberth B: Pathology of Terrien's disease: Histochemical and electron microscopic
study. Am J Ophthalmol 74:1191, 1972 241. Austin P, Brown SI: Inflammatory Terrien's marginal corneal disease. Am J Ophthalmol 92: 189, 1981 242. Edwards WC, Reed RE: Mooren's ulcer: A pathologic case report. Arch Ophthalmol 89:361, 1968 243. Kietzman B: Mooren's ulcer in Nigeria. Am J Ophthalmol 65:679, 1968 244. Schaap OL, Feltkamp TEW, Breebaart AC: Circulating antibodies to corneal tissue in a patient suffering from Mooren's
ulcer (ulcus rodens corneae). Clin Exp Immunol 5:365, 1969 245. Wood TO, Kaufman HE: Mooren's ulcer. Am J Ophthalmol 71:417, 1971 246. Foster CS, Kenyon KR, Greiner J et al: The immunopathology of Mooren's ulcer. Am J Ophthalmol 88:149, 1979 247. Feder RS, Krachmer JH: Conjunctival resection for the treatment of the rheumatoid corneal ulceration. Ophthalmology 91:11, 1984 248. Kenyon KR: Decision-making in the therapy of external eye disease. Ophthalmology 89:44, 1982 249. Foster CS: Systemic immunosuppressive therapy for progressive bilateral Mooren's
ulcer. Ophthalmology 92:1436, 1985 250. Foster CS: Immunosuppressive therapy for external ocular inflammatory disease. Ophthalmology 87: 140, 1980 251. Tauber J, Sainz de la Maza M, Hoang-Xuan T et al: An analysis of therapeutic decision making regarding immunosuppressive
chemotherapy for peripheral ulcerative keratitis. Cornea 9:66, 1990 252. Gass JD: the iron lines of the superficial cornea. Arch Ophthalmol 71:348, 1964 253. Barraquer-Somers E, Chart CC, Green WR: Corneal epithelial iron deposition. Ophthalmology 90:729, 1983 254. Ferry AP: A “new” line of the superficial cornea: Occurrence in patients
with filtering blebs. Arch Ophthalmol 79:142, 1968 255. Coats G: Small superficial opaque white rings in the cornea. Trans Ophthalmol Soc UK 32:53, 1912 256. Nevins RC Jr, Davis WH Jr, Elliot JH: Coat's white ring of the cornea: Unsettled metal fettle (correspondence). Arch Ophthalmol 80: 145, 1968 257. Shapiro LA, Frakas TG: Lipid keratopathy following corneal hydrops. Arch Ophthalmol 95:456, 1977 258. Fine BS, Townsend WM, Zimmerman LE et al: Primary lipoidal degeneration of the cornea. Am J Ophthalmol 78:12, 1974 259. Friedlaender MH, Cavanagh HD, Sullivan WR et al: Bilateral central lipid infiltrates of the cornea. Am J Ophthalmol 84:781, 1977 260. Stafford WR, Fine BS: Amyloidosis of the cornea: Report of a case without conjunctival involvement. Arch Ophthalmol 75:53, 1966 261. McPherson SD Jr, Kiffney GT Jr, Freed CC: Corneal amyloidosis. Am J Ophthalmol 62: 1025, 1966 262. Garner A: Amyloidosis of the cornea. Br J Ophthalmol 53:73, 1969 263. Ramsey MS, Fine BS, Cohen SW: Localized corneal amyloidosis: Case report
with electron microscopic observations. Am J Ophthalmol 73:560. 1972 264. Rodrigues MM, Zimmerman LE: Secondary amyloidosis in ocular leprosy. Arch Ophthalmol 85:277, 1971 265. Garner A: Keratinoid corneal degeneration. Br J Ophthalmol 54:769, 1970 266. reedman A: Climatic droplet keratopathy. I. Clinical aspects. Arch Ophthalmol 89: 193, 1973 267. Garner A, Morgan G, Tripathi RC: Climatic droplet keratopathy. II. Pathologic findings. Arch Ophthalmol 89: 198, 1973 268. Anderson J, Fuglsang H: Droplet degeneration of the cornea in North Cameroon: Prevalence and clinical
appearances. Br J Ophthalmol 60:256, 1976 269. Ahmad A, Hogan M, Wood I et al: Climatic droplet keratopathy in a 16-year-old boy. Arch Ophthalmol 95:149, 1977 270. Christensen GR: Proteinaceous corneal degeneration: A histochemical study. Arch Ophthalmol 89:30, 1973 271. Freedman A: Labrador keratopathy. Arch Ophthalmol 74:198, 1965 272. D'Alena P, Wood IS: Labrador keratopathy: A microscopic study. Am J Ophthalmol 74:430, 1972 273. Johnson CJ, Ghosh M: Labrador keratopathy: Clinical and pathological findings. Can
J Ophthalmol 10:119, 1975 274. Klintworth GK: Chronic actinic keratopathy: A condition associated with conjunctival elastosis (pingueculae) and
typified by characteristic extracellular concretions. Am J Pathol 67:327, 1972 275. Hanna C, Fraunfelder FT: Spheroidal degeneration of the cornea and conjunctiva. 1I. Pathology, Am
J Ophthalmol 74:829, 1972 276. Fraunfelder FT, Hanna C: Spheroidal degeneration of cornea and conjunctiva. III. Incidences, classification
and etiology. Am J Ophthalmol 76:41, 1973 277. Young YDH, Finlay RD: Primary spheroidal degeneration of the cornea in Labrador and Northern
Newfoundland. Am J Ophthalmol 79:129, 1975 278. Fraunfelder FT, Hanna C, Parker JM: Spheroid degeneration of the cornea and conjunctiva. 1. Clinical course
and characteristics. Am J Ophthalmol 74:821, 1972 279. Rodrigues MM, Laibson PR, Weinreb S: Corneal elastosis: Appearance of band-like keratopathy and spheroidal degeneration. Arch Ophthalmol 93:11 I, 1975 280. Johnson GJ, Overall M: Histology of spheroidal degeneration of the cornea in Labrador. Br J Ophthalmol 62:53, 1978 281. Pouliquen Y, Haye C, Bisson J et al: Ultrastructure de la keratopathie en bandelette. Arch Ophtalmol (Paris) 27:149, 1967 282. O'Conner GR: Calcific band keratopathy. Trans Am Ophthalmol Soc 70:58, 1972 283. Zeiter H J: Calcification and ossification in ocular tissue. Am J Ophthalmol 53:265, 1962 284. Barber CW: Physiological chemistry of the eye. Arch Ophthalmol 87:72, 1972 285. Fishman RS, Sunderman FW: Band keratopathy in gout. Arch Ophthalmol 75:367, 1966 286. Kennedy RE, Roca PD, Landers PH: Atypical band keratopathy in glaucomatous patients. Am J Ophthalmol 72:917, 1971 287. Kennedy RE, Roca PD, Platt DS: Further observations on atypical band keratopathy
in glaucoma patients, Trans Am Ophthalmol Soc 72:107, 1975 288. Letup MA, Ralph RA: Rapid development of band keratopathy in dry eyes. Am J Ophthalmol 83:657, 1977 289. Walsh FB, Howard JE: Conjunctival and corneal lesions in hypercalcemia. J Clin Endocrinol 7:644, 1947 290. Walsh FB, Murray RG: Ocular manifestations of disturbances in calcium metabolism. Am J Ophthalmol 36: 1657, 1953 291. Lessel S, Norton EWD: Band keratopathy and conjunctival calcification in hypophosphatasia. Arch Ophthalmol 71:497, 1964 292. Berkow JW, Fine BS, Zimmerman LE: Unusual ocular calcification in hyperparathyroidism. Am J Ophthalmol 66:812, 1968 293. Schumacher H, Scheler F: Metastatische Kalzifizierungen an Kornea und Konjunktiva bei chronischer
Niereninsuffizienz. KIln Monatsbl Augenheilkd 154:815, 1969 294. Porter R, Crombie AL: Corneal calcification as a presenting and diagnostic sign in hyperparathyroidism. Br J Ophthalmol 57:665, 1973 295. Wood TO, Walker GG: Treatment of band keratopathy. Am J Ophthalmol 80:553, 1975 296. Wood TO: Salzmann's nodular degeneration. Cornea 9:17, 1990 297. Holbach LM, Font RL, Shivitz IA et al: Bilateral keloidlike myofibroblastic proliferations of the cornea in children. Ophthalmology 97:1198, 1990 298. Austin P, Jakobiec FA, Iwamoto T: Elastodysplasia and elastodystrophy as pathologic bases of ocular pterygium
and pinguecula. Ophthalmology 90:96, 1983 299. Kenyon KR, Wagoner MD, Hettinger ME: Conjunctival autograft transplantation for advanced and recurrent pterygium. Ophthalmology 92: 1461, 1985 300. Singh G, Wilson MR, Foster CS: Mitomycin eye drops as treatment for pterygium. Ophthalmology 95:813, 1988 301. Singh G, Wilson MR, Foster CS: Long-term follow-up study of mitomycin eye drops as adjunctive treatment
for pterygia and its comparison with conjunctival autograft transplantation. Cornea 9:331, 1990 |