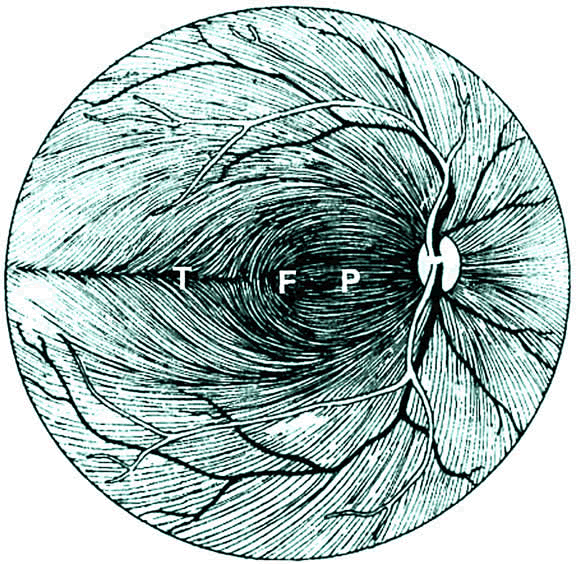
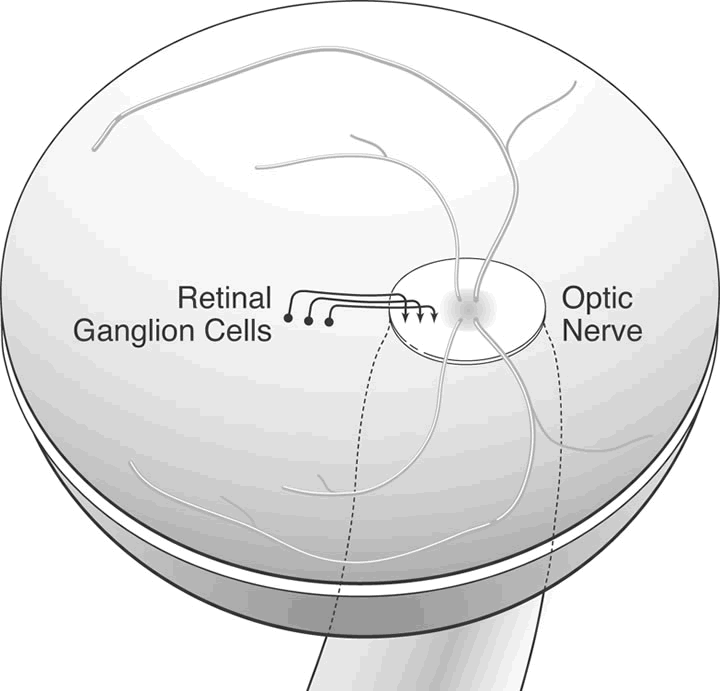
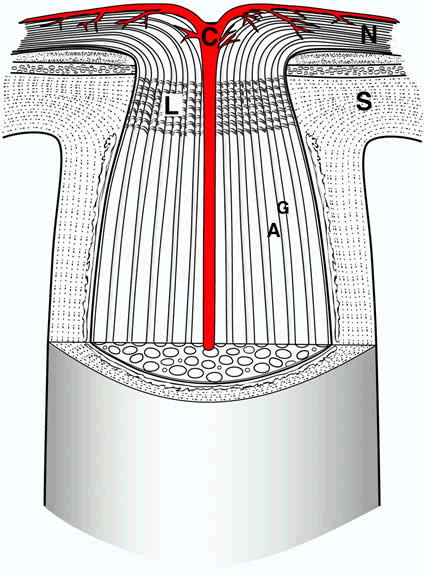
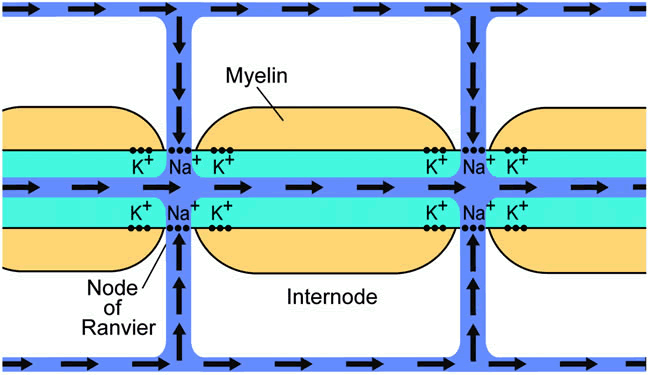
1. Wolff E: The Anatomy of the Eye and Orbit. Philadelphia: Blakiston, 1948:263 2. Fitzgibbon T, Taylor SF: Retinotopy of the human retinal nerve fibre layer and optic nerve head. J Comp Neurol 375:238, 1996 3. Reese BE, Ho KY: Axon diameter distributions across the monkey's optic nerve. Neuroscience 27:205, 1988 4. Jeffery G: Distribution and trajectory of uncrossed axons in the optic nerves of pigmented
and albino rats. J Comp Neurol 289:462, 1989 5. Chan SO, Guillery RW: Changes in fiber order in the optic nerve and tract of rat embryos. J Comp Neurol 344:20, 1994 6. Schmid R, Wilhelm B, Wilhelm H: Naso-temporal asymmetry and contraction anisocoria in the pupillomotor
system. Graefes Arch Clin Exp Ophthalmol 238:123, 2000 7. Balazsi AG, Rootman J, Drance SM et al: The effect of age on the nerve fiber population of the human optic nerve. Am J Ophthalmol 97:760, 1984 8. Mikelberg FS, Drance SM, Schulzer M et al: The normal human optic nerve: Axon count and axon diameter distribution. Ophthalmology 96:1325, 1989 9. Friedrich VL, Mugnaini E: Myelin sheath thickness in the CNS is regulated near the axon. Brain Res 274:329, 1983 10. Bussow H: Schwann cell myelin ensheathing CNS axons in the nerve fibre layer of the
cat retina. J Neurocytol 7:207, 1978 11. Jung HJ, Raine CS, Suzuki K: Schwann cells and peripheral nervous system myelin in the rat retina. Acta Neuropathol Berl 44:245, 1978 12. Anderson DR, Hoyt WF: Ultrastructure of intraorbital portion of human and monkey optic nerve. Arch Ophthalmol 82:506, 1969 13. Turner DL, Cepko CL: A common progenitor for neurons and glia persists in rat retina late in
development. Nature 328:131, 1987 14. Schnitzer J: Astrocytes in the guinea pig, horse, and monkey retina: Their occurrence
coincides with the presence of blood vessels. Glia 1:74, 1988 15. Bussow H: The astrocytes in the retina and optic nerve head of mammals: A special
glia for the ganglion cell axons. Cell Tissue Res 206:367, 1980 16. Hickey WF, Kimura H: Perivascular microglial cells of the CNS are bone marrow-derived and present
antigen in vivo. Science 239:290, 1988 17. Stoll G, Trapp BD, Griffin JW: Macrophage function during wallerian degeneration of rat optic nerve: Clearance
of degenerating myelin and Ia expression. J Neurosci 9:2327, 1989 18. Jeffery G, Evans A, Albon J et al: The human optic nerve: Fascicular organisation and connective tissue types
along the extra-fascicular matrix. Anat Embryol 191:491, 1995 19. Hayreh SS: The sheath of the optic nerve. Ophthalmologica 189:54, 1984 20. Levin LA, Albert DM, Johnson D: Mast cells in human optic nerve. Invest Ophthalmol Vis Sci 34:3147, 1993 21. Nowak JZ, Nawrocki J, Maslinski C: Distribution and localization of histamine in bovine and rabbit eye. Agents Actions 14:335, 1984 22. Bowling DB, Michael CR: Projection patterns of single physiologically characterized optic tract
fibres in cat. Nature 286:899, 1980 23. Kondo Y, Takada M, Kayahara T et al: Single retinal ganglion cells sending axon collaterals to the bilateral
superior colliculi: A fluorescent retrograde double-labeling study in
the Japanese monkey (Macaca fuscata). Brain Res 597:155, 1992 24. Hendry SH, Reid RC: The koniocellular pathway in primate vision. Annu Rev Neurosci 23:127, 2000 25. Itoh K, Takada M, Yasui Y et al: A pretectofacial projection in the cat: A possible link in the visually-triggered
blink reflex pathways. Brain Res 275:332, 1983 26. Baleydier C, Magnin M, Cooper HM: Macaque accessory optic system: II: Connections with the pretectum. J Comp Neurol 302:405, 1990 27. Gamlin PD, Reiner A, Erichsen JT et al: The neural substrate for the pupillary light reflex in the pigeon (Columba
livia). J Comp Neurol 226:523, 1984 28. Young MJ, Lund RD: The retinal ganglion cells that drive the pupilloconstrictor response in
rats. Brain Res 787:191, 1998 29. Lagreze WA, Kardon RH: Correlation of relative afferent pupillary defect and estimated retinal
ganglion cell loss. Graefes Arch Clin Exp Ophthalmol 236:401, 1998 30. Wurtz RH: Vision for the control of movement. The Friedenwald Lecture. Invest Ophthalmol Vis Sci 37:2130, 1996 31. Sadun AA, Schaechter JD, Smith LE: A retinohypothalamic pathway in man: Light mediation of circadian rhythms. Brain Res 302:371, 1984 32. Schaecter JD, Sadun AA: A second hypothalamic nucleus receiving retinal input in man: The paraventricular
nucleus. Brain Res 340:243, 1985 33. Johnson RF, Morin LP, Moore RY: Retinohypothalamic projections in the hamster and rat demonstrated using
cholera toxin. Brain Res 462:301, 1988 34. Bieda MC, Copenhagen DR: Sodium action potentials are not required for light-evoked release of GABA
or glycine from retinal amacrine cells. J Neurophysiol 81:3092, 1999 35. Waxman SG, Black JA, Kocsis JD et al: Low density of sodium channels supports action potential conduction in
axons of neonatal rat optic nerve. Proc Natl Acad Sci U S A 86:1406, 1989 36. Morin PJ, Liu NG, Johnson RJ et al: Isolation and characterization of rapid transport vesicle subtypes from
rabbit optic nerve. J Neurochem 56:415, 1991 37. Lorenz T, Willard M: Subcellular fractionation of intra-axonally transport polypeptides in the
rabbit visual system. Proc Natl Acad Sci U S A 75:505, 1978 38. Aschner M: Changes in axonally transported proteins in the mature and developing rat
nervous system during early stages of methyl mercury exposure. Pharmacol Toxicol 60:81, 1987 39. Crossland WJ: Fast axonal transport in the visual pathway of the chick and rat. Brain Res 340:373, 1985 40. Matthews MA, Cornell WJ, Alchediak T: Inhibition of axoplasmic transport in the developing visual system of the
rat: I: Structural changes in the retina and optic nerve with graded
doses of intraocular colchicine. Neuroscience 7:365, 1982 41. Black MM, Lasek RJ: Axonal transport of actin: Slow component b is the principal source of
actin for the axon. Brain Res 171:401, 1979 42. Willard M, Wiseman M, Levine J et al: Axonal transport of actin in rabbit retinal ganglion cells. J Cell Biol 81:581, 1979 43. Giorgi PP, DuBois H: Labelling by axonal transport of myelin-associated proteins in the rabbit
visual pathway. Biochem J 196:537, 1981 44. Frizell M, McLean WG, Sjostrand J: Slow axonal transport of proteins: Blockade by interruption of contact
between cell body and axon. Brain Res 86:67, 1975 45. Levy NS: The effect of interruption of the short posterior ciliary arteries on slow
axoplasmic transport and histology within the optic nerve of the rhesus
monkey. Invest Ophthalmol 15:495, 1976 46. Radius RL: Pressure-induced fast axonal transport abnormalities and the anatomy at
the lamina cribrosa in primate eyes. Invest Ophthalmol Vis Sci 24:343, 1983 47. Anderson DR, Hendrickson A: Effect of intraocular pressure on rapid axoplasmic transport in monkey
optic nerve. Invest Ophthalmol 13:771, 1974 48. Minckler DS, Tso MO: A light microscopic, autoradiographic study of axoplasmic transport in
the normal rhesus optic nerve head. Am J Ophthalmol 82:1, 1976 49. Minckler DS, Tso MO, Zimmerman LE: A light microscopic, autoradiographic study of axoplasmic transport in
the optic nerve head during ocular hypotony, increased intraocular pressure, and
papilledema. Am J Ophthalmol 82:741, 1976 50. Tso MO, Hayreh SS: Optic disc edema in raised intracranial pressure. IV. Axoplasmic transport
in experimental papilledema. Arch Ophthalmol 95:1458, 1977 51. Guy J, Ellis EA, Tark EF et al: Axonal transport reductions in acute experimental allergic encephalomyelitis: Qualitative
analysis of the optic nerve. Curr Eye Res 8:261, 1989 52. Olver JM, Spalton DJ, McCartney AC: Quantitative morphology of human retrolaminar optic nerve vasculature. Invest Ophthalmol Vis Sci 35:3858, 1994 53. Orgul S, Cioffi GA: Embryology, anatomy, and histology of the optic nerve vasculature. J Glaucoma 5:285, 1996 54. Lieberman MF, Maumenee AE, Green WR: Histologic studies of the vasculature of the anterior optic nerve. Am J Ophthalmol 82:405, 1976 55. Olver JM, Spalton DJ, McCartney AC: Microvascular study of the retrolaminar optic nerve in man: The possible
significance in anterior ischaemic optic neuropathy. Eye 4:7, 1990 56. Olver JM: Functional anatomy of the choroidal circulation: Methyl methacrylate casting
of human choroid. Eye 4:262, 1990 57. Guy J, McGorray S, Fitzsimmons J, Beck B et al: Disruption of the blood-brain barrier in experimental optic neuritis: Immunocytochemical
co-localization of H2O2 and extravasated serum albumin. Invest Ophthalmol Vis Sci 35:1114, 1994 58. Guy J, Fitzsimmons J, Ellis EA et al: Intraorbital optic nerve and experimental optic neuritis. Correlation of
fat suppression magnetic resonance imaging and electron microscopy. Ophthalmology 99:720, 1992 59. Geijer C, Bill A: Effects of raised intraocular pressure on retinal, prelaminar, laminar, and
retrolaminar optic nerve blood flow in monkeys. Invest Ophthalmol Vis Sci 18:1030, 1979 60. Weinstein JM, Duckrow RB, Beard D et al: Regional optic nerve blood flow and its autoregulation. Invest Ophthalmol Vis Sci 24:1559, 1983 61. Riva CE, Grunwald JE, Petrig BL: Autoregulation of human retinal blood flow. An investigation with laser
Doppler velocimetry. Invest Ophthalmol Vis Sci 27:1706, 1986 62. Francois J, Fryczkowski A: The blood supply of the optic nerve. Adv Ophthalmol 36:164, 1978 63. Murray M, Grafstein B: Changes in the morphology and amino acid incorporation of regenerating
goldfish optic neurons. Exp Neurol 23:544, 1969 64. Humphrey MF, Beazley LD: Retinal ganglion cell death during optic nerve regeneration in the frog
Hyla moorei. J Comp Neurol 263:382, 1985 65. Humphrey MF: A morphometric study of the retinal ganglion cell response to optic nerve
severance in the frog Rana pipiens. J Neurocytol 17:293, 1988 66. Radius RL, Anderson DR: Retinal ganglion cell degeneration in experimental optic atrophy. Am J Ophthalmol 86:673, 1978 67. Quigley HA, Anderson DR: Descending optic nerve degeneration in primates. Invest Ophthalmol Vis Sci 16:841, 1977 68. Kupfer C: Retinal ganglion cell degeneration following chiasmal lesions in man. Arch Ophthalmol 70:256, 1963 69. Kerr JF, Wyllie AH, Currie AR: Apoptosis: A basic biological phenomenon with wide-ranging implications
in tissue kinetics. Br J Cancer 26:239, 1972 70. Gavrieli Y, Sherman Y, Ben-Sasson SA: Identification of programmed cell death in situ via specific labeling of
nuclear DNA fragmentation. J Cell Biol 119:493, 1992 71. Berkelaar M, Clarke DB, Wang YC et al: Axotomy results in delayed death and apoptosis of retinal ganglion cells
in adult rats. J Neurosci 14:4368, 1994 72. Garcia-Valenzuela E, Gorczyca W, Darzynkiewicz Z et al: Apoptosis in adult retinal ganglion cells after axotomy. J Neurobiol 25:431, 1994 73. Rehen SK, Linden R: Apoptosis in the developing retina: Paradoxical effects of protein synthesis
inhibition. Braz J Med Biol Res 27:1647, 1994 74. Quigley HA, Nickells RW, Kerrigan LA et al: Retinal ganglion cell death in experimental glaucoma and after axotomy
occurs by apoptosis. Invest Ophthalmol Vis Sci 36:774, 1995 75. Levin LA, Louhab A: Apoptosis of retinal ganglion cells in anterior ischemic optic neuropathy. Arch Ophthalmol 114:488, 1996 76. Cragg BG: What is the signal for chromatolysis? Brain Res 23:1, 1970 77. Thanos S, Pavlidis C, Mey J et al: Specific transcellular staining of microglia in the adult rat after traumatic
degeneration of carbocyanine-filled retinal ganglion cells. Exp Eye Res 55:101, 1992 78. Watson WE: Cellular responses to axotomy and to related procedures. Br Med Bull 30:112, 1974 79. Grafstein B, Ingoglia NA: Intracranial transection of the optic nerve in adult mice: Preliminary
observations. Exp Neurol 76:318, 1982 80. Schnitzer J, Scherer J: Microglial cell responses in the rabbit retina following transection of
the optic nerve. J Comp Neurol 302:779, 1990 81. Mansour-Robaey S, Clarke DB, Wang YC et al: Effects of ocular injury and administration of brain-derived neurotrophic
factor on survival and regrowth of axotomized retinal ganglion cells. Proc Natl Acad Sci U S A 91:1632, 1994 82. Cui Q, Harvey AR: At least two mechanisms are involved in the death of retinal ganglion cells
following target ablation in neonatal rats. J Neurosci 15:8143, 1995 83. Yoles E, Muller S, Schwartz M: NMDA-receptor antagonist protects neurons from secondary degeneration after
partial optic nerve crush. J Neurotrauma 14:665, 1997 84. Aguayo AJ, Clarke DB, Jelsma TN et al: Effects of neurotrophins on the survival and regrowth of injured retinal
neurons. Ciba Foundation Symp 196:135, 1996 85. Mey J, Thanos S: Intravitreal injections of neurotrophic factors support the survival of
axotomized retinal ganglion cells in adult rats in vivo. Brain Res 602:304, 1993 86. Maffei L, Carmignoto G, Perry VH et al: Schwann cells promote the survival of rat retinal ganglion cells after
optic nerve section. Proc Natl Acad Sci U S A 87:1855, 1990 87. Castillo BJ, del CM, Breakefield XO et al: Retinal ganglion cell survival is promoted by genetically modified astrocytes
designed to secrete brain-derived neurotrophic factor (BDNF). Brain Res 647:30, 1994 88. Weibel D, Kreutzberg GW, Schwab ME: Brain-derived neurotrophic factor (BDNF) prevents lesion-induced axonal
die-back in young rat optic nerve. Brain Res 679:249, 1995 89. Peinado-Ramon P, Salvador M, Villegas-Perez MP et al: Effects of axotomy and intraocular administration of NT-4, NT-3, and brain-derived
neurotrophic factor on the survival of adult rat retinal ganglion
cells. A quantitative in vivo study. Invest Ophthalmol Vis Sci 37:489, 1996 90. Lessell S: Nonarteritic anterior ischemic optic neuropathy: Enigma variations. Arch Ophthalmol 117:386, 1999 91. Johnson MW, Kincaid MC, Trobe JD: Bilateral retrobulbar optic nerve infarctions after blood loss and hypotension. A
clinicopathologic case study. Ophthalmology 94:1577, 1987 92. Connolly SE, Gordon KB, Horton JC: Salvage of vision after hypotension-induced ischemic optic neuropathy. Am J Ophthalmol 117:235, 1994 93. Rizzo JFd, Lessell S: Risk of developing multiple sclerosis after uncomplicated
optic neuritis: A long-term prospective study. Neurology 38:185, 1988 94. The 5-year risk of MS after optic neuritis. Experience of the optic neuritis
treatment trial. Optic Neuritis Study Group [see comments]. Neurology 49:1404, 1997 95. Trapp BD, Peterson J, Ransohoff RM et al: Axonal transection in the lesions of multiple sclerosis. N Engl J Med 338:278, 1998 96. Perry VH, Anthony DC: Axon damage and repair in multiple sclerosis. Philos Trans R Soc Lond B Biol Sci 354:1641, 1999 97. Evangelou N, Esiri MM, Smith S et al: Quantitative pathological evidence for axonal loss in normal appearing
white matter in multiple sclerosis. Ann Neurol 47:391, 2000 98. MacFadyen DJ, Drance SM, Douglas GR et al: The retinal nerve fiber layer, neuroretinal rim area, and visual evoked
potentials in MS. Neurology 38:1353, 1988 99. Hayreh SS, Massanari RM, Yamada T et al: Experimental allergic encephalomyelitis. I. Optic nerve and central nervous
system manifestations. Invest Ophthalmol Vis Sci 21:256, 1981 100. Sergott RC, Brown MJ, Silberberg DH et al: Antigalactocerebroside serum demyelinates optic nerve in vivo. J Neurol Sci 64:297, 1984 101. Zhu B, Moore GR, Zwimpfer TJ et al: Axonal cytoskeleton changes in experimental optic neuritis. Brain Res 824:204, 1999 102. Guy J, Ellis EA, Tark EFd et al: Axonal transport reductions in acute experimental allergic encephalomyelitis: Qualitative
analysis of the optic nerve. Curr Eye Res 8:261, 1989 103. Guy J, Ellis EA, Kelley K et al: Quantitative analysis of labelled inner retinal proteins in experimental
optic neuritis. Curr Eye Res 8:253, 1989 104. Rao NA, Guy J, Sheffield PS: Effects of chronic demyelination on axonal transport in experimental allergic
optic neuritis. Invest Ophthalmol Vis Sci 21:606, 1981 105. Fontana L, Bhandari A, Fitzke FW et al: In vivo morphometry of the lamina cribrosa and its relation to visual field
loss in glaucoma. Curr Eye Res 17:363, 1998 106. Clifford-Jones RE, McDonald WI, Landon DN: Chronic optic nerve compression. An experimental study. Brain 108:241, 1985 107. Clifford-Jones RE, Landon DN, McDonald WI: Remyelination during optic nerve compression. J Neurol Sci 46:239, 1980 108. Guyer DR, Miller NR, Long DM et al: Visual function following optic canal decompression via craniotomy. J Neurosurg 62:631, 1985 109. Giles CL, Soble AR: Intracranial hypertension and tetracycline therapy. Am J Ophthalmol 72:981, 1971 110. Minckler DS: Histology of optic nerve damage in ocular hypertension and early glaucoma. Surv Ophthalmol 33(Suppl):401, 1989 111. Quigley HA: Ganglion cell death in glaucoma: Pathology recapitulates ontogeny. Aust N Z J Ophthalmol 23:85, 1995 112. Kerrigan LA, Zack DJ, Quigley HA et al: TUNEL-positive ganglion cells in human primary open-angle glaucoma. Arch Ophthalmol 115:1031, 1997 113. Okisaka S, Murakami A, Mizukawa A et al: Apoptosis in retinal ganglion cell decrease in human glaucomatous eyes. Jpn J Ophthalmol 41:84, 1997 114. Dkhissi O, Chanut E, Wasowicz M et al: Retinal TUNEL-positive cells and high glutamate levels in vitreous humor
of mutant quail with a glaucoma-like disorder. Invest Ophthalmol Vis Sci 40:990, 1999 115. Quigley HA, Dunkelberger GR, Green WR: Retinal ganglion cell atrophy correlated with automated perimetry in human
eyes with glaucoma. Am J Ophthalmol 107:453, 1989 116. Hoyt WF, Frisen L, Newman NM: Funduscopy of nerve fiber layer defects in glaucoma. Invest Ophthalmol 12:814, 1973 117. Quigley HA, Miller NR, George T: Clinical evaluation of nerve fiber layer atrophy as an indicator of glaucomatous
optic nerve damage. Arch Ophthalmol 98:1564, 1980 118. Airaksinen PJ, Drance SM, Douglas GR et al: Diffuse and localized nerve fiber loss in glaucoma. Am J Ophthalmol 98:566, 1984 119. Iwata K, Kurosawa A, Sawaguchi S: Wedge-shaped retinal nerve fiber layer defects in experimental glaucoma
preliminary report. Graefes Arch Clin Exp Ophthalmol 223:184, 1985 120. Drance SM: The early structural and functional disturbances of chronic open-angle
glaucoma. Robert N. Shaffer lecture. Ophthalmology 92:853, 1985 121. Stroman GA, Stewart WC, Golnik KC et al: Magnetic resonance imaging in patients with low-tension glaucoma. Arch Ophthalmol 113:168, 1995 122. Iwata F, Patronas NJ, Caruso RC et al: Association of visual field, cup-disc ratio, and magnetic resonance imaging
of optic chiasm. Arch Ophthalmol 115:729, 1997 123. Weber AJ, Chen H, Hubbard WC et al: Experimental glaucoma and cell size, density, and number in the primate
lateral geniculate nucleus. Invest Ophthalmol Vis Sci 41:1370, 2000 124. Yucel YH, Zhang Q, Gupta N et al: Loss of neurons in magnocellular and parvocellular layers of the lateral
geniculate nucleus in glaucoma. Arch Ophthalmol 118:378, 2000 125. Vickers JC, Hof PR, Schumer RA et al: Magnocellular and parvocellular visual pathways are both affected in a
macaque monkey model of glaucoma. Aust N Z J Ophthalmol 25:239, 1997 126. Chaturvedi N, Hedley-Whyte ET, Dreyer EB: Lateral geniculate nucleus in glaucoma. Am J Ophthalmol 116:182, 1993 127. Hernandez MR, Pena JD: The optic nerve head in glaucomatous optic neuropathy. Arch Ophthalmol 115:389, 1997 128. Pena JD, Netland PA, Vidal I et al: Elastosis of the lamina cribrosa in glaucomatous optic neuropathy. Exp Eye Res 67:517, 1998 129. Thale A, Tillmann B, Rochels R: SEM studies of the collagen architecture of the human lamina cribrosa: Normal
and pathological findings. Ophthalmologica 210:142, 1996 130. Hollander H, Makarov F, Stefani FH et al: Evidence of constriction of optic nerve axons at the lamina cribrosa in
the normotensive eye in humans and other mammals. Ophthalmic Res 27:296, 1995 131. Quigley H, Pease ME, Thibault D: Change in the appearance of elastin in the lamina cribrosa of glaucomatous
optic nerve heads. Graefes Arch Clin Exp Ophthalmol 232:257, 1994 132. Fukuchi T, Sawaguchi S, Yue BY et al: Sulfated proteoglycans in the lamina cribrosa of normal monkey eyes and
monkey eyes with laser-induced glaucoma. Exp Eye Res 58:231, 1994 133. Hernandez MR: Ultrastructural immunocytochemical analysis of elastin in the human lamina
cribrosa. Changes in elastic fibers in primary open-angle glaucoma. Invest Ophthalmol Vis Sci 33:2891, 1992 134. Fukuchi T, Sawaguchi S, Hara H et al: Extracellular matrix changes of the optic nerve lamina cribrosa in monkey
eyes with experimentally chronic glaucoma. Graefes Arch Clin Exp Ophthalmol 230:421, 1992 135. Spileers W, Goethals M: Structural changes of the lamina cribrosa and of the trabeculum in primary
open angle glaucoma (POAG). Bull Soc Belge Ophthalmol 244:27, 1992 136. Quigley HA, Hohman RM, Addicks EM et al: Morphologic changes in the lamina cribrosa correlated with neural loss
in open-angle glaucoma. Am J Ophthalmol 95:673, 1983 137. Susanna R Jr: The lamina cribrosa and visual field defects in open-angle glaucoma. Can J Ophthalmol 18:124, 1983 138. Quigley HA, Addicks EM: Regional differences in the structure of the lamina cribrosa and their
relation to glaucomatous optic nerve damage. Arch Ophthalmol 99:137, 1981 139. Emery JM, Landis D, Paton D et al: The lamina cribrosa in normal and glaucomatous
human eyes. Trans Am Acad Ophthalmol Otolaryngol 78:OP290, 1974 140. Shihab ZM, Lee PF, Hay P: The significance of disc hemorrhage in open-angle glaucoma. Ophthalmology 89:211, 1982 141. Airaksinen PJ, Mustonen E, Alanko HI: Optic disc haemorrhages precede retinal nerve fibre layer defects in ocular
hypertension. Acta Ophthalmol Copenh 59:627, 1981 142. Drance SM, Fairclough M, Butler DM et al: The importance of disc hemorrhage in the prognosis of chronic open angle
glaucoma. Arch Ophthalmol 95:226, 1977 143. Anderson DR: What happens to the optic disc and retina in glaucoma? Ophthalmology 90:766, 1983 144. Quigley HA, Addicks EM: Chronic experimental glaucoma in primates. II. Effect of extended intraocular
pressure elevation on optic nerve head and axonal transport. Invest Ophthalmol Vis Sci 19:137, 1980 145. Quigley HA, Addicks EM, Green WR et al: Optic nerve damage in human glaucoma. II. The site of injury and susceptibility
to damage. Arch Ophthalmol 99:635, 1981 146. Pease ME, McKinnon SJ, Quigley HA et al: Obstructed axonal transport of BDNF and its receptor TrkB in experimental
glaucoma. Invest Ophthalmol Vis Sci 41:764, 2000 147. Allcutt D, Berry M, Sievers J: A qualitative comparison of the reactions of retinal ganglion cell axons
to optic nerve crush in neonatal and adult mice. Brain Res 318:231, 1984 148. Allcutt D, Berry M, Sievers J: A quantitative comparison of the reactions of retinal ganglion cells to
optic nerve crush in neonatal and adult mice. Brain Res 318:219, 1984 149. Barron KD, Dentinger MP, Krohel G et al: Qualitative and quantitative ultrastructural observations on retinal ganglion
cell layer of rat after intraorbital optic nerve crush. J Neurocytol 15:345, 1986 150. Cioffi GA, Sullivan P: The effect of chronic ischemia on the primate optic nerve. Eur J Ophthalmol 9(Suppl 1):S34, 1999 151. Neufeld AH, Hernandez MR, Gonzalez M: Nitric oxide synthase in the human glaucomatous optic nerve head. Arch Ophthalmol 115:497, 1997 152. Neufeld AH: Nitric oxide: A potential mediator of retinal ganglion cell damage in glaucoma. Surv Ophthalmol 43(Suppl 1):S129, 1999 |