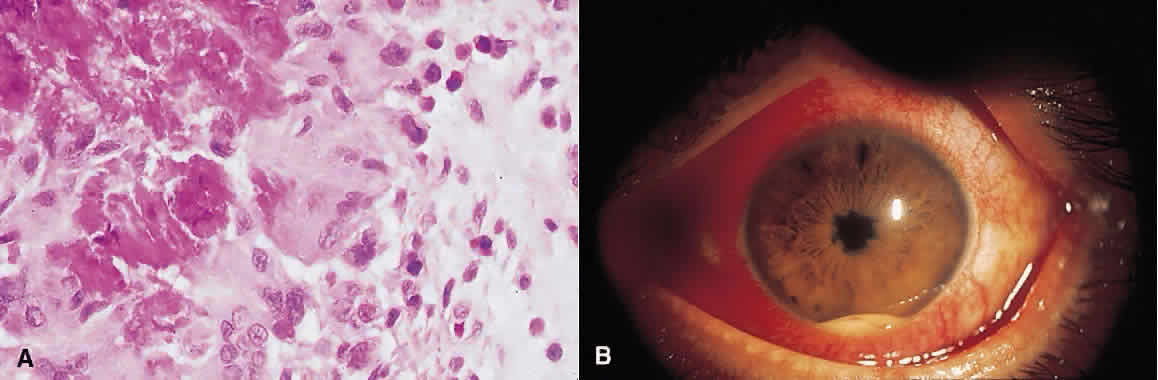
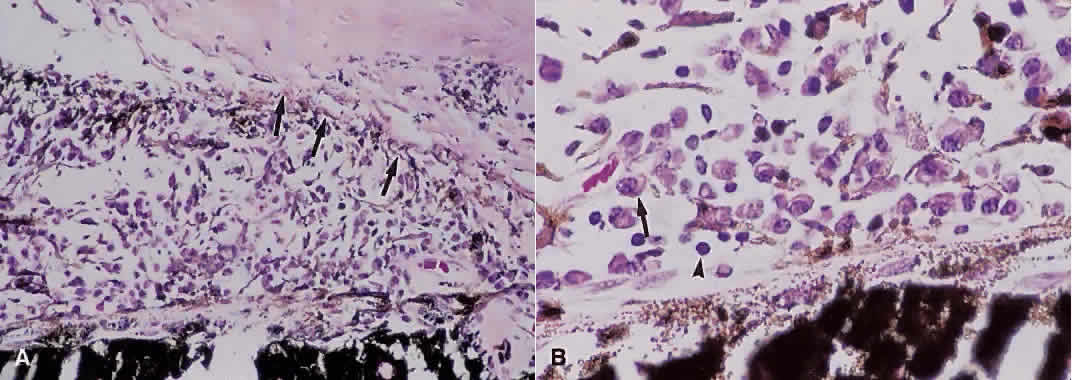
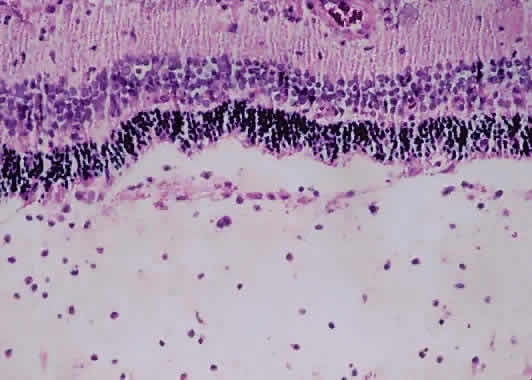
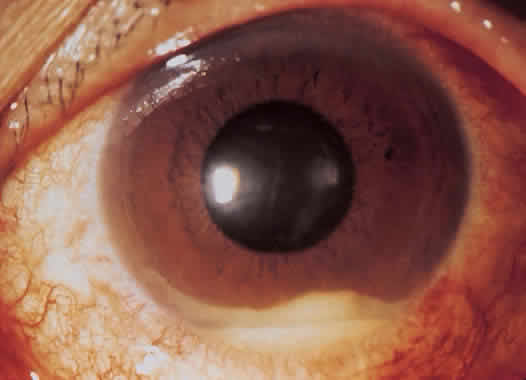
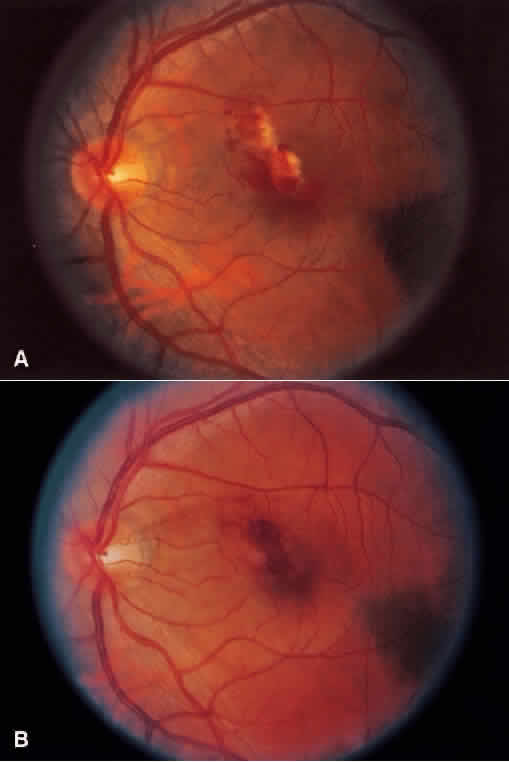
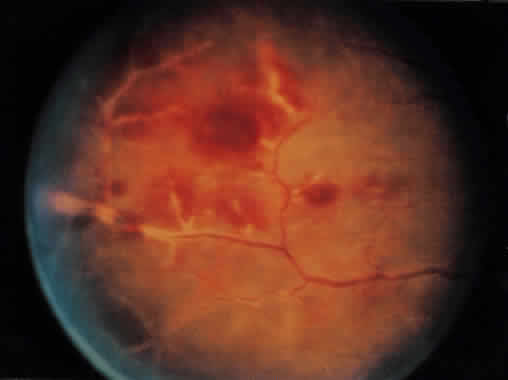
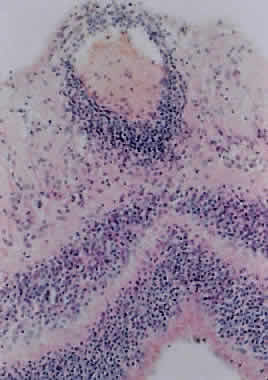
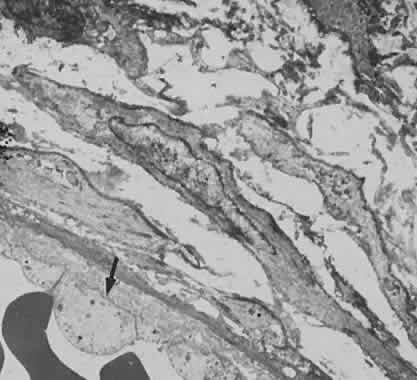
1. Roitt I, Brostoff J, Male D: Immunology. 4th ed. London: Mosby, 1996 2. Morel PA, Oriss TB: Cross-regulation between Th1 and Th2 cells. Crit Rev Immunol 18:275–303, 1998 3. Streilein JW: Immunoregulatory mechanisms of the eye. Progr Retinal Eye Res 18:357–370, 1999 4. Castenholtz A: Studies on the “vascular labeling” phenomenon in the iris and
the ciliary body of the rat after intravenous injection of colloidal
carbon. Graefes Arch Klin Exp Ophthalmol 202:27, 1977 5. Streilein JW: Immune privilege as the result of local tissue barriers and immunopressive
microenvironments. Curr Opin Immunol 5:482–492, 1993 6. Tonjum AM, Pederson OO: The permeability of the human ciliary and iridic epithelium to horseradish
peroxidase: An in vitro study. Acta Ophthalmol 55:781, 1977 7. Raviola G: The structural basis of the blood-ocular barriers. Exp Eye Res 25:27, 1977 8. Cunha-Vaz J: The blood-ocular barriers. Surv Ophthalmol 23:279, 1979 9. Streilein JW, Toews G, Bergstresser P: Corneal allografts fail to express Ia antigens. Nature 282:325–327, 1979 10. Kaiser C, Ksander B, Streilein JW: Inhibition of lymphocyte proliferation by aqueous humor. Reg Immunol 2:42–49, 1989 11. Griffith T, Brunner T, Fletcher S: Fas ligand-induced apoptosis as a mechanism of immune privilege. Science 270:1189–1192, 1995 12. Hogan MJ, Zimmerman LE (eds): Ophthalmic Pathology: An Atlas and Textbook. 2nd
ed. Philadelphia: WB Saunders, 1962 13. Rutzen AR, Ortega-Larrocea G, Dugel PU et al: Retinal and choroidal biopsy in intraocular inflammation: Clinicopathologic
study. Trans Am Ophthalmol Soc 92:431–455, 1994 14. Freeman WR, Wiley CA, Gross JG et al: Endoretinal biopsy in immunosuppressed and healthy patients with retinitis. Indications, utility, and techniques. Ophthalmology 96: 1559–1565, 1989 15. Therese KL, Anand AR, Madhavan HN: Polymerase chain reaction in the diagnosis of bacterial endophthalmitis. Br J Ophthalmol 82:1078–1082, 1998 16. Lohmann CP, Heeb M, Linde HJ et al: Diagnosis of infectious endophthalmitis after cataract surgery by polymerase
chain reaction. J Cataract Refract Surg 24:821–826, 1998 17. Kim MC, Kabeer NH, Tandhasetti MT et al: Immunohistochemical studies on melanin associated antigen (MAA) induced
experimental autoimmune anterior uveitis (EAAU). Curr Eye Res 14:703–710, 1995 18. Broekhuyse RM, Kuhlmann ED, Winkens HJ: Experimental autoimmune anterior uveitis (EAAU). III. Induction by immunization
with purified uveal and skin melanins. Exp Eye Res 56:575–583, 1993 19. Broekhuyse RM, Kuhlmann ED: Experimental autoimmune anterior uveitis. The preparation of uveitogenic
ocular melanin. Invest Ophthalmol Vis Sci 34:698–700, 1993 20. Broekhuyse RM, Kuhlmann ED, Winkens HJ: Experimental autoimmune anterior uveitis (EAAU): Induction by melanin antigen
and suppression by various treatments. Pigment Cell Res 6:1–6, 1993 21. Rosenbaum JT, McDevitt HO, Guss RB et al: Endotoxin-induced uveitis in rats as a model for human disease. Nature 286:611–613, 1980 22. Parnaby-Price A, Stanford MR, Biggerstaff J et al: Leukocyte trafficking in experimental autoimmune uveitis in vivo. J Leukoc Biol 64:434–440, 1998 23. Hamada M, Ogura Y, Miyamoto K et al: Retinal leukocyte behavior in experimental autoimmune uveoretinitis of
rats. Exp Eye Res 65:445–450, 1997 24. Gery I, Wiggert B, Redmond TM et al: Uveoretinitis and pinealitis induced by immunization with interphotoreceptor
retinoid-binding protein. Invest Ophthalmol Vis Sci 27: 1296–1300, 1986 25. Atalla L, Linker-Israeli M, Steinman L et al: Inhibition of autoimmune uveitis by anti-CD4 antibody. Invest Ophthalmol Vis Sci 31:1264–1270, 1990 26. Forrester JV, Liversidge J, Dua HS: Regulation of the local immune response by retinal cells. Curr Eye Res 9:183–191, 1990 27. Sun SH, Silver PB, Caspi RR et al: Identification of genomic regions controlling experimental autoimmune uveoretinitis
in rats. Int Immunol 11:529–534, 1999 28. Ishimoto S, Wu GS, Hayashi S et al: Free radical tissue damages in the anterior segment of the eye in experimental
autoimmune uveitis. Invest Ophthalmol Vis Sci 37:630–636, 1996 29. Wu GS, Zhang J, Rao NA: Peroxynitrite and oxidative damage in experimental autoimmune uveitis. Invest Ophthalmol Vis Sci 38:1333–1339, 1997 30. Goto H, Wu GS, Chen F et al: Lipid peroxidation in experimental uveitis: Sequential studies. Curr Eye Res 11:489–499, 1992 31. Criteria for diagnosis of Behçet's disease: International Study
Group for Behçet's Disease. Lancet 335:1078–1080, 1990 32. Sakane T: New perspective on Behçet's disease. Int Rev Immunol 14:89–96, 1997 33. Mizuki N, Inoko H, Ohno S: Pathogenic gene responsible for the predisposition of Behçet's
disease. Int Rev Immunol 14:33–48, 1997 34. Lehner T, Lavery E, Smith R et al: Association between 65-kilodalton heat shock protein Streptococcus sanguis and the corresponding antibodies in Behçet's syndrome. Infect Immunol 59:1434, 1991 35. Lehner T: The role of heat shock protein, microbial and autoimmune agents in the
aetiology of Behçet's disease. Int Rev Immunol 14:21–32, 1997 36. Yamashita N: Hyperreactivity of neutrophils and abnormal T cell homeostasis: A new insight
for pathogenesis of Behçet's disease. Int Rev Immunol 14:11–19, 1997 37. Stanford MR, Kasp E, Whiston R et al: Heat shock protein peptides reactive in patients with Behçet's
disease are uveitogenic in Lewis rats. Clin Exp Immunol 97:226–231, 1994 38. Chajek-Shaul T, Pisanty S, Knobler H et al: HLA-B51 may serve as an immunogenetic marker for a subgroup of patients
with Behçet's syndrome. Am J Med 83:666–72, 1987 39. Hirohata S, Hashimoto T: Abnormal T cell responses to bacterial superantigens in Behçet's
disease (BD). Clin Exp Immunol 112:317–324, 1998 40. Mishima S, Masuda K, Izawa Y et al: The Eighth Frederick H. Verhoeff Lecture. Presented by Saiichi Mishima, MD, Behçet's
disease in Japan: Ophthalmologic aspects. Trans Am Ophthalmol Soc 77:225–279, 1979 41. Shimada K, Yaoita H, Shikano S: Chemotactic activity for polymorphonuclear leucocytes of the aqueous humor
in patients with Behçet's disease. Nippon Ganka Gakkai Zasshi 75:2100–2105, 1971 42. Nussenblatt RB. Uveitis in Behçet's disease. Int Rev Immunol 14:67–79, 1997 43. George RK, Chan CC, Whitcup SM et al: Ocular immunopathology of Behçet's disease. Surv Ophthalmol 42:157–162, 1997 44. Charteris DG CC, Rosenthal AR et al: Behçet's disease: Activated T-lymphocytes in retinal perivasculitis. Br J Ophthalmol 76:499–501, 1992 45. Charteris DG BK, McCartney ACF et al: CD4+ lymphocyte involvement in ocular Behçet's disease. Autoimmunity 12:201–206, 1992 46. Aracil P, Moyenin P, Ryckewaert M et al: Pars planitis and multiple sclerosis. Bull Soc Ophtalmol Fr 86:485–487, 1986 47. Malinowski SM, Pulido JS, Goeken NE et al: The association of HLA-B8, B51, DR2, and multiple sclerosis in pars planitis. Ophthalmology 100:1199–1205, 1993 48. Palimeris G, Marcomichelakis N, Konstantinidou V et al: Intermediate uveitis: What is the natural course of the disease and its
relationship with other systemic diseases? Eur J Ophthalmol 4:223–227, 1994 49. Tang WM, Pulido JS, Eckels DD et al: The association of HLA-DR15 and intermediate uveitis. Am J Ophthalmol 123:70–75, 1997 50. Biousse V, Trichet C, Bloch-Michel E et al: Multiple sclerosis associated with uveitis in two large clinic-based series. Neurology 52:179–181, 1999 51. Raja SC, Jabs DA, Dunn JP et al: Pars planitis: Clinical features and class II HLA associations. Ophthalmology 106:594–5999, 1999 52. Green WR, Kincaid MC, Michels RG et al: Pars planitis. Trans Ophthalmol Soc UK 101:361–367, 1981 53. Pederson JE, Kenyon KR, Green WR et al: Pathology of pars planitis. Am J Ophthalmol 86:762–774, 1978 54. Yoser SL, Forster DJ, Rao NA: Pathology of intermediate uveitis. Dev Ophthalmol 23:60–70, 1992 55. Eichenbaum JW, Friedman AH, Mamelok AE: A clinical and histopathological review of intermediate uveitis (“pars
planitis”). Bull NY Acad Med 64:164–174, 1988 56. Brockhurst RJ, Schepens CL, Okamura ID: Uveitis. II. Peripheral uveitis: Clinical description, complications and
differential diagnosis. Am J Ophthalmol 49:1257–1266, 1960 57. Kimura SJ, Hogan MJ: Chronic cyclitis. Arch Ophthalmol 71:193–201, 1964 58. Wetzig RP, Chan CC, Nussenblatt RB et al: Clinical and immunopathological studies of pars planitis in a family. Br J Ophthalmol 72:5–10, 1988 59. Gartner J: The fine structure of the vitreous base of the human eye and pathogenesis
of pars planitis. Am J Ophthalmol 71:1317–1327, 1971 60. Gartner J: The vitreous base of the human eye and “pars planitis.”Mod Probl Ophthalmol 10:250–255, 1972 61. Loewenfeld IE, Thompson HS: Fuchs's heterochromic cyclitis: A critical review of the literature. I. Clinical
characteristics of the syndrome. Surv Ophthalmol 17:394–457, 1973 62. Merayo-Lloves J, Power WJ, Rodriguez A et al: Secondary glaucoma in patients with uveitis. Ophthalmologica 213: 300–304, 1999 63. Fearnley IR, Rosenthal AR: Fuchs' heterochromic iridocyclitis revisited. Acta Ophthalmol Scand 73:166–170, 1995 64. La Hey E, de Vries J, Langerhorst CT: Treatment and prognosis of secondary glaucoma in Fuchs' heterochromic iridocyclitis. Am J Ophthalmol 116:327–340, 1993 65. Jones NP: Glaucoma in Fuchs' heterochromic uveitis: Aetiology, management and outcome. Eye 5:662–667, 1991 66. La Hey E, Baarsma GS, De Vries J et al: Clinical analysis of Fuchs' heterochromic cyclitis. Doc Ophthalmol 78:225–235, 1991 67. Henderly DE, Genstler AJ, Smith RE et al: Changing patterns of uveitis. Am J Ophthalmol 103:131–136, 1987 68. Pivetti-Pezzi P, Accorinti M, La Cava M et al: Endogenous uveitis: An analysis of 1,417 cases. Ophthalmologica 210: 234–238, 1996 69. Saari M, Vuorre I, Tiilikainen A et al: Genetic background of Fuchs' heterochromic cyclitis. Can J Ophthalmol 13:240–246, 1978 70. Berger BB, Tessler HH, Kottow MH: Anterior segment ischemia in Fuchs' heterochromic cyclitis. Arch Ophthalmol 98:499–501, 1980 71. Goldberg MF, Erozan YS, Duke JR et al: Cytopathologic and histopathologic aspects of Fuchs' heterochromic iridocyclitis. Arch Ophthalmol 74:604–609, 1965 72. Regenbogen LS, Naveh-Floman N: Glaucoma in Fuchs' heterochromic cyclitis associated with congenital Horner's
syndrome. Br J Ophthalmol 71:844–849, 1987 73. Collier M: Progressive facial hemiatrophy, heterochromic cyclitis, Fuchs type, and
micropapilla. Bull Soc Ophtalmol Fr 67:595–603, 1967 74. Fulmek R: Hemiatrophia progressiva faciei (Rombergsyndrome) associated with heterochromia
complicata (Fuchs-syndrome) (author's translation). Klin Monatsbl Augenheilkd 164:615–628, 1974 75. La Hey E, Baarsma GS: Fuchs' heterochromic cyclitis and retinal vascular abnormalities in progressive
hemifacial atrophy. Eye 7:426–428, 1993 76. Melamed S, Lahav M, Sandbank U et al: Fuch's heterochromic iridocyclitis: An electron microscopic study
of the iris. Invest Ophthalmol Vis Sci 17:1193–1199, 1978 77. Wobmann P: Fuchs's heterochromic cyclitis. Electronmicroscopic study of nine
iris biopsies (author's translation). Albrecht Von Graefes Arch Klin Exp Ophthalmol 199:167–178, 1976 78. La Hey E, Rothova A, Baarsma GS et al: Fuchs' heterochromic iridocyclitis is not associated with ocular toxoplasmosis. Arch Ophthalmol 110:806–811, 1992 79. La Heij E, Rothova A: Fuchs's heterochromic cyclitis in congenital ocular toxoplasmosis. Br J Ophthalmol 75:372–373, 1991 80. La Hey E, de Jong PT, Kijlstra A: Fuchs' heterochromic cyclitis: Review of the literature on the pathogenetic
mechanisms. Br J Ophthalmol 78:307–312, 1994 81. Schwab IR: The epidemiologic association of Fuchs' heterochromic iridocyclitis and
ocular toxoplasmosis. Am J Ophthalmol 111:356–362, 1991 82. Jones NP: Fuchs' heterochromic uveitis: An update. Surv Ophthalmol 37:253–272, 1993 83. van den Born LI, van Schooneveld MJ, de Jong PT et al: Fuchs' heterochromic uveitis associated with retinitis pigmentosa in a
father and son. Br J Ophthalmol 78:504–505, 1994 84. Vuorre I, Saari M, Tiilikainen A et al: Fuchs' heterochromic cyclitis associated with retinitis pigmentosa: A family
study. Can J Ophthalmol 14:10–16, 1979 85. Yalvac IS, Altintas AK, Gokdere A et al: Fuchs' heterochromic uveitis associated with retinitis pigmentosa. Acta Ophthalmol Scand 76:243–244, 1998 86. Chowers I, Zamir E, Banin E et al: Retinitis pigmentosa associated with Fuchs' heterochronic uveitis. Arch Ophthalmol 118:800–802, 2000 87. Fuchs E: Uber Komplicationen der heterochromie. Z Augenheilkd 15:191–212, 1906 88. Dithmar S, Tetz MR, Volcker HE: Fuchs' heterochromic cyclitis. Clinico-histopathologic findings of nodular
iritis.Klin Monatsbl Augenheilkd 209:158–162, 1996 89. Bialasiewicz A, Gierth K, Naumann GO: Heterochromia complicata Fuchs, crystalline iridopathy and increased immunoglobulin
G in aqueous humor: A case report. Klin Monatsbl Augenheilkd 198:205–206, 1991 90. Lam S, Tessler HH, Winchester K et al: Iris crystals in chronic iridocyclitis. Br J Ophthalmol 77:181–182, 1993 91. Zamir E, Margalit E, Chowers I: Iris crystals in Fuchs' heterochromic iridocyclitis. Arch Ophthalmol 116:1394, 1998 92. Callear AB, Reynolds A, Harry J et al: Iris crystals in chronic uveitis. Br J Ophthalmol 83:703–706, 1999 93. Goldstein DA, Edward DP, Tessler HH: Iris crystals in Fuch heterochromic iridocyclitis. Arch Ophthalmol 116: 1692–1693, 1998 94. Bloch-Michel E, Lambin P, Debbia M et al: Local production of IgG and IgG subclasses in the aqueous humor of patients
with Fuchs heterochromic cyclitis, herpetic uveitis and toxoplasmic
chorioretinitis. Int Ophthalmol 21:187–194, 1997 95. Kuo I, Rao NA: Ocular disease in AIDS. Springer Semin Immunopathol 21:161–177, 1999 96. Hogan LH, Weinstock JV, Sandor M: TCR specificity in infection induced granulomas. Immunol Lett 68:115–120, 1999 97. Morinelli EN, Dugel PU, Riffenburgh R et al: Infectious multifocal choroiditis in patients with acquired immune deficiency
syndrome. Ophthalmology 100:1014–1021, 1993 98. Morinelli EN, Dugel PU, Lee M et al: Opportunistic intraocular infections in AIDS. Trans Am Ophthalmol Soc 90:97–108, 1992 99. Perlman DC, El-Helou P, Salomon N: Tuberculosis in patients with human immunodeficiency virus infection. Semin Respir Infect 14:344–352, 1999 100. Pepose JS, Holland GN, Nestor MS et al: Acquired immune deficiency syndrome. Pathogenic mechanisms of ocular disease. Ophthalmology 92:472–484, 1985 101. Read RW, Zhang JA, Ishimoto SI et al: Evaluation of the role of human retinal vascular endothelial cells in the
pathogenesis of CMV retinitis. Ocul Immunol Inflamm 7:139–146, 1999 102. Neuwirth J, Gutman I, Hofeldt AJ et al: Cytomegalovirus retinitis in a young homosexual male with acquired immunodeficiency. Ophthalmology 89:805–808, 1982 103. Yoser SL, Forster DJ, Rao NA: Systemic viral infections and their retinal and choroidal manifestations. Surv Ophthalmol 37:313–352, 1993 104. Palestine AG, Rodrigues MM, Macher AM et al: Ophthalmic involvement in acquired immunodeficiency syndrome. Ophthalmology 91:1092–1099, 1984 105. Anand R, Font RL, Fish RH et al: Pathology of cytomegalovirus retinitis treated with sustained release intravitreal
ganciclovir. Ophthalmology 100:1032–1039, 1993 106. Rodrigues MM, Palestine A, Nussenblatt R et al: Unilateral cytomegalovirus retinochoroiditis and bilateral cytoid bodies
in a bisexual man with the acquired immunodeficiency syndrome. Ophthalmology 90:1577–1582, 1983 107. Faber DW, Crapotta JA, Wiley CA et al: Retinal calcifications in cytomegalovirus retinitis. Retina 13:46–49, 1993 108. Engstrom RE, Jr., Holland GN, Margolis TP et al: The progressive outer retinal necrosis syndrome; A variant of necrotizing
herpetic retinopathy in patients with AIDS. Ophthalmology 101:1488–1502, 1994 109. Rutzen AR, Ortega-Larrocea G, Dugel PU et al: Clinicopathologic study of retinal and choroidal biopsies in intraocular
inflammation. Am J Ophthalmol 119:597–611, 1995 110. Moorthy RS, Smith RE, Rao NA: Progressive ocular toxoplasmosis in patients with acquired immunodeficiency
syndrome. Am J Ophthalmol 115:742–747, 1993 111. Smith RE: Toxoplasmic retinochoroiditis as an emerging problem in AIDS patients [editorial]. Am J Ophthalmol 106:738–739, 1988 112. Holland GN, Engstrom RE Jr, Glasgow BJ et al: Ocular toxoplasmosis in patients with the acquired immunodeficiency syndrome. Am J Ophthalmol 106:653–667, 1988 113. Park KL, Rao NA: Ocular pathology of opportunistic infections and neoplasms associated with
AIDS. Ophthalmol Clin North Am 10:73, 1997 114. Aaberg TM Jr, Flynn HW Jr, Schiffman J et al: Nosocomial acute-onset postoperative endophthalmitis survey: A 10-year
review of incidence and outcomes. Ophthalmology 105:1004–1010, 1998 115. Powe NR, Schein OD, Gieser SC et al: Synthesis of the literature on visual acuity and complications after cataract
extraction with intraocular lens implantation. Arch Ophthalmol 112:239–252, 1994 116. Kattan HM, Flynn HW, Pfugfelder SC et al: Nosocomial endophthalmitis survey: Current incidence of infection after
intraocular surgery. Ophthalmology 98:227–238, 1991 117. Javitt JC, Vitale S, Canner JK et al: National outcomes of cataract extraction: Endophthalmitis after inpatient
surgery. Arch Ophthalmol 109:1085–1089, 1991 118. Norregaard JC, Thoning H, Bernth-Petersen P et al: Risk of endophthalmitis after cataract extraction: Results from the International
Cataract Surgery Outcomes study. Br J Ophthalmol 81:102–106, 1997 119. Han DP, Wisniewski SR, Wilson LA et al: Spectrum and susceptibilities of
microbiologic isolates in the Endophthalmitis Vitrectomy Study. Am J
Ophthalmol 122:1–17, 1996; published erratum appears in Am J Ophthalmol 122:920, 1996 120. Kunimoto DY, Das T, Sharma S et al: Microbiologic spectrum and susceptibility of isolates: Part I. Postoperative
endophthalmitis. Endophthalmitis Research Group. Am J Ophthalmol 128:240–242, 1999 121. Mistlberger A, Ruckhofer J, Raithel E et al: Anterior chamber contamination during cataract surgery with intraocular
lens implantation. J Cataract Refract Surg 23:1064–1069, 1997 122. Egger SF, Huber-Spitzy V, Skorpik C et al: Different techniques of extracapsular cataract extraction: Bacterial contamination
during surgery: Prospective study on 230 consecutive patients. Graefes Arch Clin Exp Ophthalmol 232:308–311, 1994 123. Egger SF, Huber-Spitzy V, Scholda C et al: Bacterial contamination during extracapsular cataract extraction. Prospective
study on 200 consecutive patients. Ophthalmologica 208:77–81, 1994 124. Beigi B, Westlake W, Chang B et al: The effect of intracameral, per-operative antibiotics on microbial contamination
of anterior chamber aspirates during phacoemulsification. Eye 12:390–394, 1998 125. Beigi B, Westlake W, Mangelschots E et al: Peroperative microbial contamination of anterior chamber aspirates during
extracapsular cataract extraction and phacoemulsification. Br J Ophthalmol 81:953–955, 1997 126. Ariyasu RG, Nakamura T, Trousdale MD et al: Intraoperative bacterial contamination of the aqueous humor. Ophthalmic Surg 24:367–373, 1993 127. Maxwell DP, Brent BD, Orillac R et al: A natural history study of experimental Staphylococcus epidermidis endophthalmitis. Curr Eye Res 12:907–912, 1993 128. Pleyer U, Mondino BJ, Adamu SA et al: Immune response to Staphylococcus epidermidis-induced endophthalmitis in a rabbit model. Invest Ophthalmol Vis Sci 33:2650–2663, 1992 129. Davis JL, Winward KR, Lonardo EC et al: Association of Propionibacterium acnes endophthalmitis with HLA-DQw5. Ocular Immunol Inflam 4:25–32, 1996 130. Endophthalmitis Vitrectomy Study Group: Results of the endophthalmitis
vitrectomy study: A randomized trial of immediate vitrectomy and of intravenous
antibiotics for the treatment of postoperative bacterial endophthalmitis. Arch
Ophthalmol 113:1479–1496, 1995 131. Ormerod LD, Puklin JE, Giles CL: Chronic Propionibacterium acnes endophthalmitis as a cause of intermediate
uveitis. Ocul Immunol Inflamm 5:67–68, 1997 132. Doyle A, Beigi B, Early A et al: Adherence of bacteria to intraocular lenses: A prospective study. Br J Ophthalmol 79:347–349, 1995 133. Menikoff JA, Speaker MG, Marmor M et al: A case-control study of risk factors for postoperative endophthalmitis. Ophthalmology 98:1761–1768, 1991 134. Rao NA, Nerenberg AV, Forster DJ: Torulopsis Candida (Candida famata) endophthalmitis simulating Propionibacterium acnes syndrome. Arch Ophthalmol 109:1718–1721, 1991 135. Busin M, Cusumano A, Spitznas M: Intraocular lens removal from eyes with chronic low-grade endophthalmitis. J Cataract Refract Surg 21:679–684, 1995 136. Ciulla TA, Beck AD, Topping TM et al: Blebitis, early endophthalmitis, and late endophthalmitis after glaucoma-filtering
surgery. Ophthalmology 104:986–995, 1997 137. Waheed S, Ritterband DC, Greenfield DS et al: New patterns of infecting organisms in late bleb-related endophthalmitis: A
ten year review. Eye 12:910–915, 1998 138. Kangas TA, Greenfield DS, Flynn HW, Jr. et al: Delayed-onset endophthalmitis associated with conjunctival filtering blebs. Ophthalmology 104:746–52, 1997 139. Kunimoto DY, Das T, Sharma S et al: Microbiologic spectrum and susceptibility of isolates: Part II. Posttraumatic
endophthalmitis. Endophthalmitis Research Group. Am J Ophthalmol 128:242–244, 1999 140. Abu el-Asrar AM, al-Amro SA, al-Mosallam AA et al: Post-traumatic endophthalmitis: Causative organisms and visual outcome. Eur J Ophthalmol 9:21–31, 1999 141. Duch-Samper AM, Chaques-Alepuz V, Menezo JL et al: Endophthalmitis following open-globe injuries. Curr Opin Ophthalmol 9:59–65, 1998 142. Okada AA, Johnson RP, Liles WC et al: Endogenous bacterial endophthalmitis: Report of a ten-year retrospective
study. Ophthalmology 101:832–838, 1994 143. Wong JS, Balakrishnan V: Neisseria meningitidis endogenous endophthalmitis: Case report and literature
review. J Pediatr Ophthalmol Strabismus 36:145–152, 1999 144. Lohmann CP, Gabel VP, Heep M et al: Listeria monocytogenes-induced endogenous endophthalmitis in an otherwise healthy
individual: Rapid PCR-diagnosis as the basis for effective treatment. Eur J Ophthalmol 9:53–57, 1999 145. Marinella MA, Warwar R: Endogenous endophthalmitis due to Serratia marcescens. South Med J 91:388–391, 1998 146. Mendivil A, Cuartero V: Endogenous endophthalmitis caused by Rickettsia conorii. Acta Ophthalmol Scand 76: 121–122, 1998 147. Davitt B, Gehrs K, Bowers T: Endogenous Nocardia endophthalmitis. Retina 18:71–73, 1998 148. Berman AJ, Del Priore LV, Fischer CK: Endogenous Ochrobactrum anthropi endophthalmitis. Am J Ophthalmol 123:560–562, 1997 149. Essman TF, Flynn HW Jr, Smiddy WE et al: Treatment outcomes in a 10-year study of endogenous fungal endophthalmitis. Ophthalmic Surg Lasers 28:185–194, 1997 150. Tuft SJ, Coster DJ: The corneal endothelium. Eye 4:389–424, 1990 151. O'Connor GR: Calcific band keratopathy. Trans Am Ophthalmol Soc 70:58–81, 1972 152. Vinores SA, Youssri AI, Luna JD et al: Upregulation of vascular endothelial growth factor in ischemic and non-ischemic
human and experimental retinal disease. Histol Histopathol 12:99–109, 1997 153. Gentile RC, Liebmann JM, Tello C et al: Ciliary body enlargement and cyst formation in uveitis. Br J Ophthalmol 80:895–899, 1996 154. Peiffer RL, Jr., Wilcock BP: Histopathologic study of uveitis in cats: 139 cases (1978-1988). J Am Vet Med Assoc 198:135–138, 1991 155. Kalina PH, Pach JM, Buettner H et al: Neovascularization of the disc in pars planitis. Retina 10:269–273, 1990 156. Phillips WBD, Bergren RL, McNamara JA: Pars planitis presenting with vitreous hemorrhage. Ophthalmic Surg 24:630–631, 1993 157. Gass J: Stereoscopic Atlas of Macular Diseases: Diagnosis and treatment. Vol 42, 4th
ed. St Louis: Mosby-Year Book, 1997 158. McMenamin PG, Forrester JV, Steptoe R et al: Ultrastructural pathology of experimental autoimmune uveitis in the rat. Autoimmunity 16:83–93, 1993 159. Dev S, Mieler WF, Pulido JS et al: Visual outcomes after pars plana vitrectomy for epiretinal membranes associated
with pars planitis. Ophthalmology 106:1086–1090, 1999 160. Gilbert C, Hiscott P, Unger W et al: Inflammation and the formation of epiretinal membranes. Eye 2:S140–S156, 1988 161. Rao NA, Wu GS: Free radical mediated photoreceptor damage in uveitis. Prog Retin Eye Res 19:41–68, 2000 162. Forster DJ, Cano MR, Green RL et al: Echographic features of the Vogt-Koyanagi-Harada syndrome. Arch Ophthalmol 108:1421–1426, 1990 163. Inomata H, Sakamoto T: Immunohistochemical studies of Vogt-Koyanagi-Harada disease with sunset
sky fundus. Curr Eye Res 9:35–40, 1990 164. Moorthy RS, Chong LP, Smith RE et al: Subretinal neovascular membranes in Vogt-Koyanagi-Harada syndrome. Am J Ophthalmol 116:164–170, 1993 165. Jampol LM, Orth D, Daily MJ et al: Subretinal neovascularization with geographic (serpiginous) choroiditis. Am J Ophthalmol 88:683–689, 1979 166. Halperin LS, Lewis H, Blumenkranz MS et al: Choroidal neovascular membrane and other chorioretinal complications of
acquired syphilis. Am J Ophthalmol 108:554–562, 1989 167. Frank KW, Weiss H: Unusual clinical and histopathological findings in ocular sarcoidosis. Br J Ophthalmol 67:8–16, 1983 168. Fine SL, Owens SL, Haller JA et al: Choroidal neovascularization as a late complication of ocular toxoplasmosis. Am J Ophthalmol 91:318–322, 1981 169. Korobelnik JF, Hannouche D, Marin F et al: Surgical treatment of retrofoveal choroid neovascularization in multifocal
choroiditis. J Fr Ophtalmol 21:146–151, 1998 170. Lampariello DA, Primo SA: Ocular toxocariasis: A rare presentation of a posterior pole granuloma
with an associated choroidal neovascular membrane. J Am Optom Assoc 70:245–252, 1999 171. Jampol LM, Sung J, Walker JD et al: Choroidal neovascularization secondary to Candida albicans chorioretinitis. Am J Ophthalmol 121:643–649, 1996 172. Maas S, Deutman AF, Bandhoe F et al: Surgical removal of subretinal neovascular membranes. Eur J Ophthalmol 5:48–55, 1995 173. Kosmorsky GS, Prayson R: Primary optic pathway sarcoidosis in a 38-year-old white man. J Neuroophthalmol 16:188–190, 1996 174. Kishi A, Nao-i N, Sawada A: Ultrasound biomicroscopic findings of acute angle-closure glaucoma in Vogt-Koyanagi-Harada
syndrome. Am J Ophthalmol 122:735–737, 1996 175. Gohdo T, Tsukahara S: Ultrasound biomicroscopy of shallow anterior chamber in Vogt-Koyanagi-Harada
syndrome. Am J Ophthalmol 122:112–114, 1996 176. Kawano Y, Tawara A, Nishioka Y et al: Ultrasound biomicroscopic analysis of transient shallow anterior chamber
in Vogt-Koyanagi-Harada syndrome. Am J Ophthalmol 121:720–723, 1996 177. Krohn J, Seland JH: Exudative retinal detachment in nanophthalmos. Acta Ophthalmol Scand 76:499–502, 1998 178. Falcon MG, Williams HP: Herpes simplex kerato-uveitis and glaucoma. Trans Ophthalmol Soc UK 98:101–104, 1978 179. Allen CB, Ulshafer RJ, Ellis EA et al: Scanning electron microscopic analysis of intraocular ossification in advanced
retinal disease. Scanning Microsc 1:233–239, 1987 180. Loewenstein JI, Hogan RN, Jakobiec FA: Osseous metaplasia in a preretinal
membrane. Arch Ophthalmol 115:117–9, 1997; published erratum
appears in Arch Ophthalmol 115:589, 1997 |