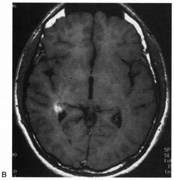
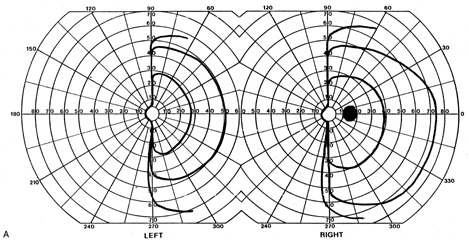
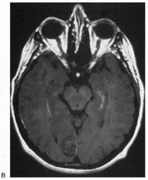
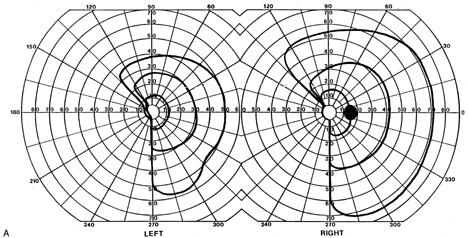
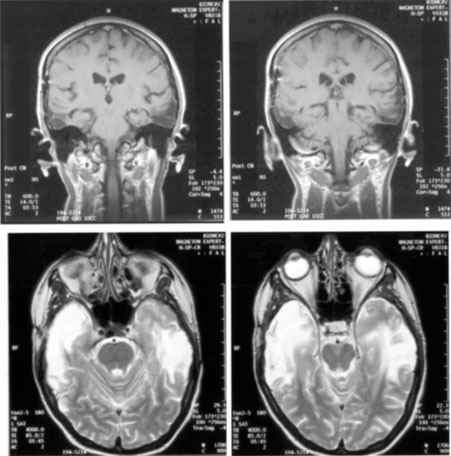
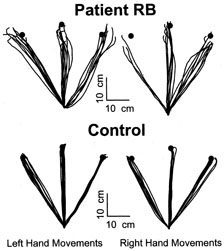
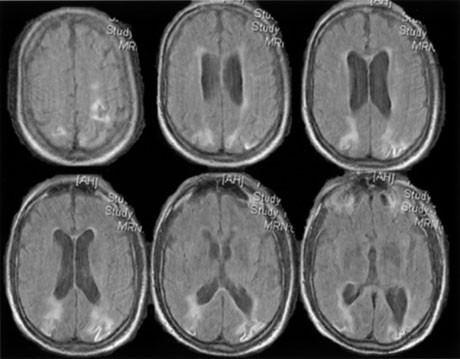
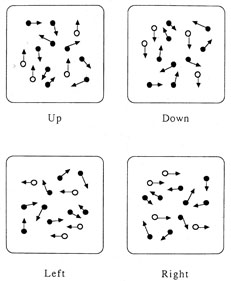
1. Hoyt W, Luis O: The primate chiasm: details of visual fiber organization studied by silver
impregnation techniques. Arch Ophthalmol 70:69, 1963 2. Tassinari G, Campara D, Balercia G, Chilosi M, Martignoni G: Magno- and parvocellular pathways are segregated in the human optic tract. Neuroreport 5:1425, 1994 3. Savino P, Paris M, Schatz N, Orr L, Corbett J: Optic tract syndrome. A review of 21 patients. Arch Ophthalmol 96:656, 1978 4. Newman S, Miller N: Optic tract syndrome. Neuro-ophthalmologic considerations. Arch Ophthalmol 101:1241, 1983 5. Frisèn L: The neurology of visual acuity. Brain 13:639, 1980 6. Paul T, Hoyt W: Funduscopic appearance of papilledema with optic tract atrophy. Arch Ophthalmol 94:467, 1976 7. Czarnecki J, Weingeist T, Burton J, Thompson H: “Twin peaks” papilledema: The appearance of papilledema with
optic tract atrophy. Can J Ophthalmol 11:279, 1976 8. Bell R, Thompson H: Relative afferent pupillary defect in optic tract hemianopias. Am J Ophthlamol 85:538, 1978 9. O'Connor P, Mein C, Hughes J, Dorwart R, Shacklett D: The Marcus Gunn pupil in incomplete optic tract hemianopias. J Clin Neuroophthalmol 2:227, 1982 10. Klein L, Fermaglich J, Kataah J, Luessenhop A: Cavernous hemangioma of optic chiasm, optic nerves and right optic tract: Case
report and review of the literature. Virchows Arch A Pathol Pathol Anat 383:225, 1979 11. Rosenblatt M, Behrens M, Zweufach P, et al: Magnetic resonance imaging of optic tract involvement in multiple sclerosis. Am J Ophthalmol 104:74, 1987 12. Youl B, Plant G, Stevens J, et al: Three cases of craniopharyngioma showing optic tract hypersignal on MRI. Neurology 40:1416, 1990 13. Tachibana O, Yamaguchi N, Yamashima T, Yamashita J: Radiation necrosis of the optic chiasm, optic tract, hypothalamus, and
upper pons after radiotherapy for pituitary adenoma, detected by gadolinium-enhanced, T1-weighted magnetic resonance imaging: case report. Neurosurgery 27:640, 1990 14. Beck R, Schatz N, Savino P: Involvement of the optic chiasm, optic tract, and geniculo-calcarine visual
system in multiple sclerosis. Bull Soc Belge Ophtalmol 208:159, 1983 15. Plant G, Kermode A, Turano G, et al: Symptomatic retrochiasmal lesions in multiple sclerosis. Clinical features, visual
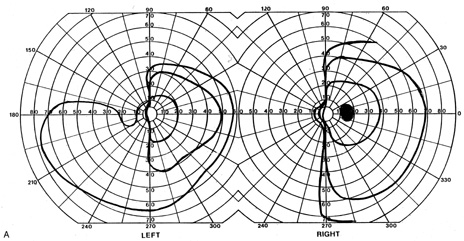
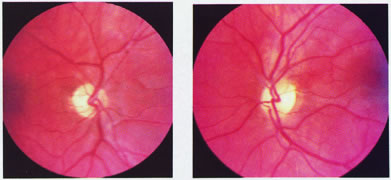
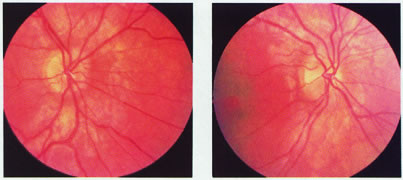
evoked potentials, and magnetic resonance imaging. Neurology 42:68, 1992 16. McLaurin E, Harrington D: Intracranial sarcoidosis with optic tract and temporal lobe involvement. Am J Ophthalmol 86:656, 1978 17. Zentner J, Grodd W, Hassler W: Cavernous angioma of the optic tract. J Neurol 236:117, 1989 18. Kupersmith M, Vargas M, Hoyt W, Berenstein A: Optic tract atrophy with cerebral arteriovenous malformations: direct and
transsynaptic degeneration. Neurology 44:80, 1994 19. Anderson D, Trobe J, Hood T, Gebarski S: Optic tract injury after anterior temporal lobectomy. Ophthalmology 96:1065, 1989 20. Manesis E, Petrou C, Brouzas D, Hadziyannis S: Optic tract neuropathy complicating low-dose interferon treatment. J Hepatol 21:474, 1994 21. Margo C, Hamed L, McCarty J: Congenital optic tract syndrome. Arch Ophthalmol 109:1120, 1991 22. Bender M, Bodis-Wollner I: Visual dysfunctions in optic tract lesions. Ann Neurol 3:187, 1978 23. Sherman S, Koch C: The control of retinogeniculate transmission in the mammalian lateral geniculate
nucleus. Exp Brain Res 63:1, 1986 24. Sillito A, Murphy P: The modulation of the retinal relay to the cortex in the dorsal lateral
geniculate nucleus. Eye 2(Suppl):S221, 1988 25. Harting J, Huerta M, Hashikawa T, van Lieshout D: Projection of the mammalian superior colliculus upon the dorsal lateral
geniculate nucleus: organization of the tectogeniculate pathways in nineteen
species. J Comp Neurol 304:275, 1991 26. Frisèn L, Holmegaard L, Rosenkrantz M: Sectoral optic atrophy and homonymous horizontal sectoranopia: a lateral
choroidal artery syndrome? J Neurol Neurosurg Psychiatry 41:374 , 1978 27. Frisèn L: Quadruple sectoranopia and sectorial optic atrophy. A syndrome of the distal
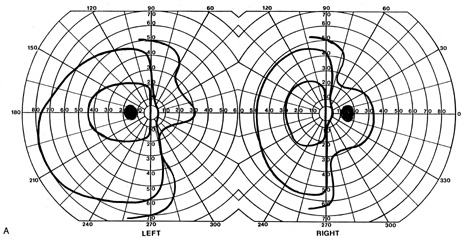
anterior choroidal artery. J Neurol Neurosurg Psychiatry 42:590, 1979 28. Helgason C, Caplan L, Goodwin J, Hedges T: Anterior choroidal artery-territory infarction. Report of cases and review. Arch Neurol 43:681, 1986 29. Donahue S, Kardon R, Thompson H. Hourglass-shaped visual fields as a sign of bilateral lateral geniculate
myelinolysis. Am J Ophthalmol 119:378, 1995 30. Barton J: Bilateral sectoranopia from osmotic demyelination. Neurology 57:2318, 2001 31. Greenfield D, Siatkowski R, Schatz N, Glaser J: Bilateral geniculitis associated with severe diarrhea. Am J Ophthalmol 122:280, 1996 32. Goldman J, Horoupian D: Demyelination of the lateral geniculate nucleus in central pontine myelinolysis. Ann Neurol 9:185, 1981 33. Jacobson D: The localizing value of a quadrantanopia. Arch Neurol 54:401, 1997 34. Carter J, O'Connor P, Shacklett D, Rosenberg M: Lesions of the optic radiations mimicking lateral geniculate nucleus visual
field defects. J Neurol Neurosurg Psychiatry 48:982, 1985 35. Smith C, Richardson W: The course and distribution of the arteries supplying the visual (striate) cortex. Am J Ophthalmol 61:1391, 1966 36. Horton J, Hoyt W: The representation of the visual field in human striate cortex: a revision
of the classis Holmes map. Arch Ophthalmol 109:816, 1991 37. Inouye T: Die Sehstorungen bei Schussverletzungen der kortikalen Sesphare. Leipzig: Engelmann, 1909 38. Holmes G, Lister W: Disturbances of vision from cerebral lesions with special reference to
the cortical representation of the macula. Brain 39:34, 1916 39. McFadzean R, Brosnahan D, Hadley D, Mutlukan E: Representation of the visual field in the occipital striate cortex. Br J Ophthalmol 78:185, 1994 40. Trauzettel-Klosinski S, Brendler K: Eye movements in reading with hemianopic field defects: the significance
of clinical parameters. Graefe's Arch Clin Exp Ophthalmol 236:91, 1998 41. Huber A: Homonymous hemianopia after occipital lobectomy. Am J Ophthalmol 54:623, 1962 42. Tootell R, Switkes E, Silverman M, Hamilton S: Functional anatomy of macaque striate cortex. II. Retinotopic organization. J Neurosci 8:1531, 1988 43. McAuley D, Russell R: Correlation of CAT scan and visual field defects in vascular lesions of
the posterior visual pathways. J Neurol Neurosurg Psychiatry 42:298, 1979 44. Gray L, Galetta S, Siegal T, Schatz N: The central visual field in homonymous hemianopia. Evidence for unilateral
foveal representation. Arch Neurol 54:312, 1997 45. Trauzettel-Klosinski S, Reinhard J: The vertical field border in hemianopia and its significance for fixation
and reading. Invest Ophthalmol Vis Sci 39:2177, 1998 46. Reinhard J, Trauzettel-Klosinski S: Nasotemporal overlap of retinal ganglion cells in humans: a functional
study. Invest Ophthalmol Vis Sci 44:1568, 2003 47. Heller-Bettinger I, Kepes J, Preskorn S, Wurster J: Bilateral altitudinal anopia caused by infarction of the calcarine cortex. Neurology 26:1176, 1976 48. Rush J: Nonbacterial thrombotic endocarditis and cortical blindness. Am J Ophthalmol 114:643, 1992 49. Horton JC, Hoyt WF: Quadrantic visual field defects. A hallmark of lesions in extrastriate (V2/V3) cortex. Brain 114:1703, 1991 50. Spalding J: Wounds of the visual pathway. Part II. The striate cortex. J Neurol Neurosurg Psychiatry 15:169, 1952 51. Benton S, Levy I, Swash M: Vision in the temporal crescent in occipital infarction. Brain 103:83, 1980 52. Chavis P, Al-Hazmi A, Clunie D, Hoyt W: Temporal crescent syndrome with magnetic resonance correlation. J Neuroophthalmol 17:151, 1997 53. Pessin M, Lathi E, Cohen M, et al: Clinical feature and mechanism of occipital lobe infarction. Ann Neurol 21:290, 1987 54. Hornstein S, Chamberlin W, Conomy J: Infarctions of the fusiform and calcarine regions: agitated delirium and
hemianopia. Trans Am Neurol Assoc 92:85, 1967 55. Medina J, Chokroverty S, Rubino F: Syndrome of agitated delirium and visual impairment: a manifestation of
medial temporo-occipital infarction. J Neurol Neurosurg Psychiatry 40:861, 1977 56. Vaphiades M, Celesia G, Brigell M: Positive spontaneous visual phenomena limited to the hemianopic field in
lesions of central visual pathways. Neurology 47:408, 1996 57. Symonds C, McKenzie I: Bilateral loss of vision from cerebral infarction. Brain 80:415, 1957 58. Bougousslavsky J, van Melle G: Unilateral occipital infarction: evaluation of the risk of developing bilateral
loss of vision. J Neurol Neurosurg Psychiatry 46:78, 1983 59. Halpern J, Sedler R: Traumatic bilateral homonymous hemianopic scotomas. Ann Ophthalmol 12:1022, 1980 60. Aldrich M, Alessi A, Beck R, Gilman S: Cortical blindness: etiology, diagnosis and prognosis. Ann Neurol 21:149, 1987 61. Chisholm I: Cortical blindness in cranial arteritis. Br J Ophthalmol 59:332, 1995 62. Naito H, Kurokawa K, Kanno T, Toya S, Osano M: Status epilepticus and cortical blindness due to subclavian steal syndrome
in a girl with Blalock's operation. Surg Neurol 1:46, 1973 63. Carney A, Anderson E: Cortical blindness and tourniquet subclavian steal. JAMA 245:572, 1981 64. Wells T, Graham C, Moss M, Kearns G: Nifedipine poisoning in a child. Pediatrics 86:91, 1990 65. Morton C, Hickey-Dwyer M: Cortical blindness after nifedipine treatment. BMJ 305:693, 1992 66. Lawrence-Friedl D, Bauer K: Bilateral cortical blindness: an unusual presentation of bacterial endocarditis. Ann Emerg Med 21:1502, 1992 67. Morgan R, Nugent B, Harrison J, O'Connor P: Voluntary alteration of pattern visual evoked responses. Ophthalmology 92:1356, 1985 68. Spehlmann R, Gross R, Ho S, Leetsma J, Norcross K: Visual evoked potentials and postmortem findings in a case of cortical
blindness. Ann Neurol 2:531, 1977 69. Celesia G, Archer C, Kuroiwa Y, Goldfader P: Visual function of the extra-geniculo-calcarine system in man: relationship
to cortical blindness. Arch Neurol 37:704, 1980 70. Frank Y, Torres F: Visual evoked potentials in the evaluation of “cortical blindness” in
children. Ann Neurol 6:126, 1979 71. Wong V: Cortical blindness in children. A study of etiology and prognosis. Pediatr Neurol 7:178, 1991 72. Gjerris F, Mellemgaard L: Transitory cortical blindness in head injury. Acta Neurol Scand 45:623, 1969 73. Griffith J, Dodge P: Transient blindness following head injury in children. N Engl J Med 278:648, 1968 74. Drubach D, Carmona S, Meyerrose G, Peralta L, Sostre S: Brain SPECT in a case of cortical blindness. Stroke 25:1061, 1994 75. Kooi K, Sharbrough F: Electrophyiological findings in cortical blindness. Electroencephalogr Clin Neurophysiol 20:260, 1966 76. Duchowny M, Weiss I, Majlessi H, Barnet A: Visual evoked responses in childhood cortical blindness after head trauma
and meningitis. A longitudinal study of six cases. Neurology 24:933, 1974 77. Tepperberg J, Nussbaum D, Feldman F: Cortical blindness following meningitis due to Hemophilus influenzae type
B. J Pediatr 91:434, 1977 78. Ramani V: Cortical blindness following ictal nystagmus. Arch Neurol 42:191, 1985 79. Skolnik S, Mizen T, Burde R: Transient post-ictal cortical blindness. J Clin Neuroophthalmol 7:151, 1987 80. Joseph J, Louis S: Transient ictal cortical blindness during middle age. A case report and
review of the literature. J Neuroophthalmol 15:39, 1995 81. Miyata Y, Motomura S, Tsuji Y, Koga S: Hepatic encephalopathy and reversible cortical blindness. Am J Gastroenterol 83:780, 1988 82. Kupferschmidt H, Bont A, Schnorf H, et al: Transient cortical blindness and bioccipital brain lesions in two patients
with acute intermittent porphyria. Ann Intern Med 123:598, 1995 83. Mukamel M, Weitz R, Nissenkorn I, Yassur I, Varsano I: Acute cortical blindness associated with hypoglycemia. J Pediatr 98:583, 1981 84. Marra T, Shah M, Mikus M: Transient cortical blindness due to hypertensive encephalopathy. Magnetic
resonance imaging correlation. J Clin Neuroophthalmol 13:35, 1993 85. Tychsen L, Hoyt W: Hydrocephalus and transient cortical blindness. Am J Ophthalmol 98:819, 1984 86. Greenblatt S: Post-traumatic cerebral blindness: association with migraine and seizure
diathesis. JAMA 225:1073, 1973 87. Patronas N, Argyropoulu M: Intravascular thrombosis as a possible cause of transient cortical brain
lesions. CT and MRI. J Comput Assist Tomogr 16:849, 1992 88. Berman I, Mann M: Seizures and transient cortical blindness associated with cisplatinum (II) diamminedichloride (PPD) therapy in a thirty-year-old
man. Cancer 45:764, 1980 89. Philip P, Carmichael J, Harris A: Convulsions and transient cortical blindness after cisplatin. BMJ 302:416, 1991 90. Rubin A: Transient cortical blindness and occipital seizures with cyclosporine toxicity. Transplantation 47:572, 1989 91. Byrd R, Rohrbaugh T, Raney R, Norris D: Transient cortical blindness secondary to vincristine therapy in childhood
malignancies. Cancer 47:37, 1981 92. Shutter L, Green J, Newman N, Hooks M, Gordon R: Cortical blindness and white matter lesions in a patient receiving FK506 after
liver transplantation. Neurology 43:2417, 1993 93. Studdard W, Davis D, Young S: Cortical blindness after cerebral angiography. Case report. J Neurosurg 54:240, 1981 94. Parry R, Rees J, Wilde P: Transient cortical blindness after coronary angiography. Br Heart J 70:563, 1993 95. Lantos G: Cortical blindness due to osmotic disruption of the blood–brain barrier
by angiographic contrast material: CT and MRI studies. Neurology 39:567, 1989 96. Eldridge P, Punt J: Transient traumatic cortical blindness in children. Lancet 1:815, 1988 97. Liebowitz H, Hall P: Cortical blindness as a complication of eclampsia. Ann Emerg Med 13:365, 1984 98. Cunningham F, Fernandez C, Hernandez C: Blindness associated with preeclampsia and eclampsia. Am J Obstet Gynecol 172:1291, 1995 99. Gospe S: Transient cortical blindness in an infant exposed to methamphetamine. Ann Emerg Med 26:380, 1995 100. DeSousa A, Kleiman M, Mealey J: Quadriplegia and cortical blindness in Hemophilus influenzae meningitis. J Pediatr 93:253, 1978 101. Woodward G: Posttraumatic cortical blindness: are we missing the diagnosis in children? Ped Emerg Care 6:289, 1990 102. Barnet A, Manson J, Wilner E: Acute cerebral blindness in childhood. Neurology 20:1147, 1970 103. Moel D, Kwun Y: Cortical blindness as a complication of hemodialysis. J Pediatr 93:890, 1978 104. Hochstetler K, Beals R: Transient cortical blindness in a child. Ann Emerg Med 16:218, 1987 105. Rodriguez A, Lozano J, del Pozo D, Paez J: Post-traumatic transient cortical blindness. Int Ophthalmol 17:277, 1993 106. Carmola J, Harris B: Transient cortical blindness: still an overlooked syndrome? N Engl J Med 282:1325, 1970 107. Kaye E, Herskowitz J: Transient post-traumatic cortical blindness: brief versus prolonged syndromes
in childhood. J Child Neurol 1:206, 1986 108. Makino A, Soga T, Obayashi M, et al: Cortical blindness causes by acute general cerebral swelling. Surg Neurol 29:393, 1989 109. Lambert S, Hoyt C, Jan J, Barkovitch J, Flodmark O: Visual recovery from hypoxic cortical blindness during childhood. Computed
tomographic and magnetic resonance predictors. Arch Ophthalmol 105:1371, 1988 110. Goodlin R, Strieb E, Sun S, Cox T, Williams N: Cortical blindness as the initial symptom in severe preeclampsia. Am J Ophthalmol 147:841, 1983 111. Seaward G, England M, Nagar A, van Gelderen C: Transient post-partum amaurosis. A report of four cases. J Reprod Med 34:253, 1989 112. Duncan R, Hadley D, Bone I, et al: Blindness in eclampsia: CT and MR imaging. J Neurol Neurosurg Psychiatry 52:899, 1989 113. Lau S, Chan F, Yu Y, Woo E, Huang C: Cortical blindness in toxemia of pregnancy: findings on computed tomography. Br J Radiol 60:347, 1987 114. Herzog T, Angel O, Karram M, Evertson L: Use of magnetic resonance imaging in the diagnosis of cortical blindness
in pregnancy. Obstet Gynecol 76:980, 1990 115. Beal M, Chapman P: Cortical blindness and homonymous hemianopia in the postpartum period. JAMA 244:2085, 1980 116. Stiller R, Leone-Tomaschoff S, Cuteri J, Beck L: Post-partum pulmonary embolus as an unusual cause of cortical blindness. Am J Obstet Gynecol 162:696, 1990 117. Branch D, Andres R, Digre K, Rote N, Scott J: The association of antiphospholipid antibodies with severe eclampsia. Obstet Gynecol 73:541, 1989 118. Carpenter E, Kava H, Plotkin D: The development of total blindness as a complication of pregnancy. Am J Obstet Gynecol 66:641, 1953 119. Beck R, Gamel J, Willcourt R, Berman G: Acute ischemic optic neuropathy in severe eclampsia. Am J Ophthalmol 90:342, 1980 120. Sandifer PH: Anosognosia and disorders of body scheme. Brain 69:122, 1946 121. McDaniel K, McDaniel L: Anton's syndrome in a patient with posttraumatic optic neuropathy
and bifrontal contusions. Arch Neurol 48:101, 1991 122. Hess R, Pointer J: Spatial and temporal contrast sensitivity in hemianopia. Brain 112:871, 1989 123. Rizzo M, Robin D: Bilateral effects of unilateral occipital lobe lesions in humans. Brain 119:951, 1996 124. Barton JJ, Sharpe JA: Smooth pursuit and saccades to moving targets in blind hemifields. A comparison
of medial occipital, lateral occipital and optic radiation lesions. Brain 120:681, 1997 125. Ball K, Owsley C, Sloane M, Roenker D, Bruni J: Visual attention problems as a predictor of vehicle crashes in older drivers. Invest Ophthalmol Vis Sci 34:3110, 1993 126. Rizzo M, Reinach S, McGehee D, Dawson J. Simulated car crashes and crash predictors in drivers with Alzheimer's
disease. Arch Neurol 54:545, 1997 127. Zihl J, von Cramon D: Visual field recovery from scotoma in patients with post-geniculate damage. Brain 108:335, 1985 128. Parisi J, Bell R, Yassein H: Homonymous hemianopic field defects and driving in Canada. Can J Ophthalmol 26:252, 1991 129. Szlyk J, Brigell M, Seiple W: Effects of age and hemianopic visual field loss on driving. Optom Vision Sci 70:1031, 1993 130. Pommerenke K, Markowitsch J: Rehabilitation training of homonymous visual field defects in patients
with postgeniculate damage of the visual system. Restor Neurol Neurosci 1:47, 1989 131. Kerkoff G, Munssinger U, Meier E: Neurovisual rehabilitation in cerebral blindness. Arch Neurol 51:474, 1994 132. Kasten E, Sabel B: Visual field enlargement after computer training in brain-damaged patients
with homonymous deficits: an open pilot trial. Restor Neurol Neurosci 8:113, 1995 133. Kasten E, Wüst S, Behrens-Baumann W, Sabel B: Computer-based training for the treatment of partial blindness. Nature Med 4:1083, 1998 134. Schlageter K, Gray B, Hall K, Shaw R, Sammet R: Incidence and treatment of visual dysfunction in traumatic brain injury. Brain Injury 7:439, 1993 135. Behrmann M, Watt S, Black S, Barton J: Impaired visual search in patients with unilateral neglect: an oculographic
analysis. Neuropsychologia 35:1445, 1997 136. Barton J, Behrmann M, Black S: Ocular search during line bisection. The effects of hemineglect and hemianopia. Brain 121:1117, 1998 137. Kerkhoff G, Münssinger U, Eberle-Strauss G, et al: Rehabilitation of hemianopic alexia in patients with post-geniculate visual
field disorders. Neuropsychol Rehab 2:21, 1992 138. Rossi P, Kheyfets S, Reding M: Fresnel prisms improve visual perception in stroke patients with homonymous
hemianopia or unilateral visual neglect. Neurology 40:1597, 1990 139. Barbur J, Ruddock K, Waterfield V: Human visual responses in the absence of the geniculo-calcarine projection. Brain 103:905, 1980 140. Barbur J, Watson J, Frackowiak R, Zeki S: Conscious visual perception without V1. Brain 116:1293, 1993 141. Blythe I, Kennard C, Ruddock K: Residual vision in patients with retrogeniculate lesions of the visual
pathways. Brain 110:887, 1987 142. Sanders MD, Warrington E, Marshall J, Weiskrantz L: Blindsight: vision in a field defect. Lancet April 20:707, 1974 143. Stoerig P, Cowey A: Blindsight in man and monkey. Brain 120:535, 1997 144. Weiskrantz L: Residual vision in a scotoma: a follow-up study of “form” discrimination. Brain 110:77, 1987 145. Marcel AJ: Blindsight and shape perception: deficit of visual consciousness or of
visual function? Brain 121:1565, 1998 146. Sahraie A, Weiskrantz L, Barbur J, et al: Pattern of neuronal activity associated with conscious and unconscious
processing of visual signals. Proc Natl Acad Sci USA 94:9406, 1997 147. Weiskrantz L, Barbur JL, Sahraie A: Parameters affecting conscious versus unconscious visual discrimination
with damage to the visual cortex (V1). Proc Natl Acad Sci USA 92:6122, 1995 148. Pöppel E, Held R, Frost D: Residual visual function after brain wounds involving the central visual
pathways in man. Nature 243:295, 1973 149. Perenin M-T, Jeannerod M: Residual vision in cortically blind hemifields. Neurosychologia 13:1, 1975 150. Weiskrantz L, Warrington E, Sanders M, Marshall J: Visual capacity in the hemianopic field following a restricted occipital
ablation. Brain 97:709, 1974 151. Blythe IM, Bromley JM, Kennard C, Ruddock KH: Visual discrimination of target displacement remains after damage to the
striate cortex in humans. Nature 320:619, 1986 152. Barton J, Sharpe J: Smooth pursuit and saccades to moving targets in blind hemifields. A comparison
of medial occipital, lateral occipital, and optic radiation lesions. Brain 120:681, 1997 153. Meienberg O, Zangemeister WH, Rosenberg M, et al: Saccadic eye movement stategies in patients with homonymous hemianopia. Ann Neurol 9:537, 1981 154. Corbetta M, Marzi C, Tassinari G, Aglioti S: Effectiveness of different task paradigms in revealing blindsight. Brain 113:603, 1990 155. Perenin M-T, Ruel J, Hécaen H: Residual visual capacities in a case of cortical blindness. Cortex 6:605, 1980 156. Perenin M-T, Jeannerod M: Visual functions within the hemianopic field following early cerebral hemidecortication
in man-I. Spatial localization. Neuropsychologia 16:1, 1978 157. Bridgeman B, Staggs D: Plasticity in human blindsight. Vision Res 22:1199, 1982 158. Ptito A, Lepore F, Ptiito M, Lassonde M: Target detection and movement discrimination in the blind field of hemispherectomized
patients. Brain 114:497, 1991 159. Riddoch G: Dissociation of visual perception due to occipital injuries with especial
reference to appreciation of movement. Brain 40:15, 1917 160. Morland AB, Jones SR, Finlay AL, et al: Visual perception of motion, luminance and color in a human hemianope. Brain 122:1183, 1999 161. Perenin M-T: Discrimination of motion direction in perimetrically blind fields. NeuroReport 2:397, 1991 162. Heide W, Koenig E, Dichgans J: Optokinetic nystagmus, self-motion sensation and their aftereffects in
patients with occipit-parietal lesions. Clin Vision Sci 5:145, 1990 163. Barton JJS, Sharpe JA: Motion direction discrimination in blind hemifields. Ann Neurol 41:255, 1997 164. Azzopardi P, Cowey A: Motion discrimination in cortically blind patients. Brain 124:30, 2001 165. ter Braak J, Schenk V, van Vliet A: Visual reactions in a case of long-lasting cortical blindness. J Neurol Neurosurg Psychiatry 34:140, 1971 166. Verhagen W, Huygen P, Mulleners W: Lack of optokinetic nystagmus and visual motion perception in acquired
cortical blindness. Neuroophthalmology 17:211, 1997 167. Mestre D, Brouchon M, Ceccaldi M, Poncet M: Perception of optical flow in cortical blindness: a case report. Neuropyschologia 30:783, 1992 168. Perenin MT, Rossetti Y: Grasping without form discrimination in a hemianopic field. Neuroreport 7:793, 1996 169. Morland A, Ogilvie J, Ruddock K, Wright J: Orientation discrimination is impaired in the absence of the striate cortical
contribution to human vision. Proc R Soc Lond B Biol Sci 263:633, 1996 170. Jackson SR: Pathological perceptual completion in hemianopia extends to the control
of reach-to-grasp movements. Neuroreport 10:2461, 1999 171. Milner A, Goodale M: The Visual Brain in Action. Oxford: Oxford University Press, 1995 172. Stoerig P: Chromaticity and achromaticity. Evidence for a functional differentiation
in visual field defects. Brain 110:869, 1987 173. Stoerig P, Cowey A: Increment-threshold spectral sensitivity in blindsight. Evidence for color
opponency. Brain 114:1487, 1991 174. Guo K, Benson PJ, Blakemore C: Residual motion discrimination using color information without primary
visual cortex. Neuroreport 9:2103, 1998 175. Morland AB, Ruddock KH: Retinotopic organisation of cortical mechanisms responsive to color: evidence
from patient studies. Acta Psychol 97:7, 1997 176. Barbur JL, Harlow AJ, Weiskrantz L: Spatial and temporal response properties of residual vision in a case of
hemianopia. Phil Trans R Soc Lond B Biol Sci 343:157, 1994 177. Hess RF, Pointer JS: Spatial and temporal contrast sensitivity in hemianopia. Brain 112:871, 1989 178. Weiskrantz L, Harlow A, Barbur JL: Factors affecting visual sensitivity in a hemianopic subject. Brain 114:2269, 1991 179. Weiskrantz L, Cowey A, Le Mare C: Learning from the pupil: a spatial visual channel in the absence of V1 in
monkey and human. Brain 121:1065, 1998 180. Weiskrantz L, Cowey A, Barbur JL: Differential pupillary constriction and awareness in the absence of striate
cortex. Brain 122:1533, 1999 181. Barbur JL, Weiskrantz L, Harlow JA: The unseen color aftereffect of an unseen stimulus: insight from blindsight
into mechanisms of color afterimages. Proc Natl Acad Sci USA 96:11637, 1999 182. Morris JS, Ohman A, Dolan RJ: A subcortical pathway to the right amygdala mediating “unseen” fear. Proc Natl Acad Sci USA 96:1680, 1999 183. de Gelder B, Vroomen J, Pourtois G, Weiskrantz L: Non-conscious recognition of affect in the absence of striate cortex. Neuroreport 10:3759, 1999 184. Morris JS, DeGelder B, Weiskrantz L, Dolan RJ: Differential extrageniculostriate and amygdala responses to presentation
of emotional faces in a cortically blind field. Brain 124:1241, 2001 185. Hamm AO, Weike AI, Schupp HT, et al: Affective blindsight: intact fear conditioning to a visual cue in a cortically
blind patient. Brain 126:267, 2003 186. Marzi C, Tassinari G, Agliotti S, Lutzemberger L: Spatial summation across the vertical meridian in hemianopics: a test of
blindsight. Neuropsychologia 24:749, 1986 187. Intriligator JM, Xie R, Barton JJ: Blindsight modulation of motion perception. J Cogn Neurosci 14:1174, 2002 188. Ward R, Jackson SR: Visual attention in blindsight: sensitivity in the blind field increased
by targets in the sighted field. Neuroreport 13:301, 2002 189. Rafal R, Smith J, Krantz J, Cohen A, Brennan C: Extrageniculate vision in hemianopic humans: saccade inhibition by signals
in the blind field. Science 250:118, 1990 190. Walker R, Mannan S, Maurer D, Pambakian AL, Kennard C: The oculomotor distractor effect in normal and hemianopic vision. Proc R Soc Lond B Biol Sci 267:431, 2000 191. Danziger S, Fendrich R, Rafal RD: Inhibitory tagging of locations in the blind field of hemianopic patients. Conscious Cogn 6:291, 1997 192. Danckert J, Maruff P, Kinsella G, de Graaff S, Currie J: Investigating form and color perception in blindsight using an interference
task. Neuroreport 9:2919, 1998 193. Braddick O, Atkinson J, Hood B, et al: Possible blindsight in infants lacking one cerebral hemisphere. Nature 360:461, 1992 194. Ptito A, Lassonde M, Lepore F, Ptito M: Visual discrimination in hemispherectomized patients. Neuropsychologia 25:869, 1987 195. Tomaiuolo F, Ptito M, Marzi CA, Paus T, Ptito A: Blindsight in hemispherectomized patients as revealed by spatial summation
across the vertical meridian. Brain 120:795, 1997 196. Wessinger CM, Fendrich R, Gazzaniga MS, Ptito A, Villemure J-G: Extrageniculostriate vision in humans: investigations with hemispherectomy
patients. Prog Brain Res 112:405, 1996 197. Wessinger C, Fendrich R, Ptito A, Villemure J, Gazzaniga M: Residual vision with awareness in the field contralateral to a partial
or complete functional hemispherectomy. Neuropsychologia 34:1129, 1996 198. Stoerig P, Faubert J, Ptito M, Diaconu V, Ptito A: No blindsight following hemidecortication in human subjects? Neuroreport 7:1990, 1996 199. Faubert J, Diaconu V, Ptito M, Ptito A: Residual vision in the blind field of hemidecorticated humans predicted
by a diffusion scatter model and selective spectral absorption of the
human eye. Vision Res 39:149, 1999 200. King S, Azzopardi P, Cowey A, Oxbury J, Oxbury S: The role of light scatter in the residual visual sensitivity of patients
with complete cerebral hemispherectomy. Visual Neurosci 13:1, 1996 201. Faubert J, Diaconu V: From visual consciousness to spectral absorption in the human retina. Progr Brain Res 134:399, 2001 202. Balliet R, Blood KM, Bach-y-Rita P: Visual field rehabilitation in the cortically blind? J Neurol Neurosurg Psychiatry 48:1113, 1985 203. Campion J, Latto R, Smith YM: Is blindsight an effect of scattered light, spared cortex, and near-threshold
vision? Behav Brain Sci 6:423, 1983 204. Fendrich R, Wessinger CM, Gazzaniga MS: Residual vision in a scotoma: implications for blindsight. Science 258:1489, 1992 205. Kasten E, Wuest S, Sabel B: Residual vision in transition zones in patients with cerebral blindness. J Clin Exp Neuropsychol 20:581, 1998 206. Scharli H, Harman A, Hogben J: Blindsight in subjects with homonymous visual field defects. J Cogn Neurosci 11:52, 1999 207. Scharli H, Harman AM, Hogben JH: Residual vision in a subject with damaged visual cortex. J Cogn Neurosci 11:502, 1999 208. Meeres SL, Graves RE: Localization of unseen stimuli by humans with normal vision. Neuropsychologia 28:1231, 1990 209. Kolb FC, Braun J: Blindsight in normal observers. Nature 377:336, 1995 210. Azzopardi P, Cowey A: Is blindsight like normal, near-threshold vision? Proc Natl Acad Sci USA 94:14190, 1997 211. Stoerig P, Hébner M, Pöppel E: Signal detection analysis of residual vision in a field defect due to a
post-geniculate lesion. Neuropsychologia 23:589, 1985 212. Azzopardi P, Cowey A: Blindsight and visual awareness. Conscious Cogn 7:292, 1998 213. Dodds C, Machado L, Rafal R, Ro T: A temporal/nasal asymmetry for blindsight in a localisation task: evidence
for extrageniculate mediation. Neuroreport 13:655, 2002 214. Bittar RG, Ptito M, Faubert J, Dumoulin SO, Ptito A: Activation of the remaining hemisphere following stimulation of the blind
hemifield in hemispherectomized subjects. Neuroimage 10:339, 1999 215. Stoerig P, Kleinschmidt A, Frahm J: No visual responses in denervated V1: high-resolution functional magnetic
resonance imaging of a blindsight patient. Neuroreport 9:21, 1998 216. Cowey A, Stoerig P: The neurobiology of blindsight. Trends Neurosci 14:140, 1995 217. Rodman H, Gross C, Albright T: Afferent basis of visual response properties in area MT of the macaque. I. Effects
of striate cortex removal. J Neurosci 9:2033, 1989 218. Girard P, Salin P, Bullier J: Response selectivity of neurons in area MT of the macaque monkey during
reversible inactivation of area V1. J Neurophysiol 67:1437, 1992 219. Rosa MG, Tweedale R, Elston GN: Visual responses of neurons in the middle temporal area of new world monkeys
after lesions of striate cortex. J Neurosci 20:5552, 2000 220. Girard P, Salin P, Bullier J: Visual activity in areas V3a and V3 during reversible inactivation of area
V1 in the macaque monkey. J Neurophysiol 66:1493, 1991 221. Gross C: Contributions of striate cortex and the superior colliculus to visual functions
in area MT, the superior temporal polysensory area and inferior
temporal cortex. Neuropsychologia 29:497, 1991 222. Payne BR, Lomber SG, Macneil MA, Cornwell P: Evidence for greater sight in blindsight following damage of primary visual
cortex early in life. Neuropsychologia 34:741, 1996 223. Sorenson KM, Rodman HR: A transient geniculo-extrastriate pathway in macaques? Implications for ‘blindsight.”rsquo; Neuroreport 10:3295, 1999 224. ffytche D, Guy C, Zeki S: The parallel visual motion inputs into areas V1 and V5 of human cerebral
cortex. Brain 118:1375, 1995 225. Beckers G, Zeki S: The consequences of inactivating areas V1 and V5 in visual motion perception. Brain 118:49, 1995 226. Hotson J, Braun D, Herzberg W, Boman D: Transcranial magnetic stimulation of extrastriate cortex degrades human
motion direction discrimination. Vision Res 34:2115, 1994 227. Holliday IE, Anderson SJ, Harding GF: Magnetoencephalographic evidence for non-geniculostriate visual input to
human cortical area V5. Neuropsychologia 35:1139, 1997 228. ffytche DH, Guy CN, Zeki S: Motion specific responses from a blind hemifield. Brain 119:1971, 1996 229. Magnussen S, Mathiesen T: Detection of moving and stationary gratings in the absence of striate cortex. Neuropsychologia 27:725, 1989 230. Moore T, Rodman H, Repp A, Gross C, Mezrich R: Greater residual vision in monkeys after striate damage in infancy. J Neurophysiol 76:3928, 1996 231. Zihl J: “Blindsight”: improvement of visually guided eye movements
by systematic practice in patients with cerebral blindness. Neuropsychologia 18:71, 1980 232. Zihl J, Werth R: Contributions to the study of “blindsight.” I. Can stray light
account for saccadic localization in patients with postgeniculate
visual field defects? Neuropsychologia 22:1, 1984 233. Zihl J, Werth R: Contributions to the study of “blindsight.” II. The role of
specific practice for saccadic localization in patients with postgeniculate
visual field defects. Neuropsychologia 22:13, 1984 234. Albert ML, Reches A, Silverberg R: Hemianopic color blindness. J Neurol Neurosurg Psychiatry 38:546, 1975 235. Paulson HL, Galetta SL, Grossman M, Alavi A: Hemiachromatopsia of unilateral occipitotemporal infarcts. Am J Ophthalmol 118:518, 1994 236. MacKay G, Dunlop JC: The cerebral lesions in a case of complete acquired color-blindness. Scott Med Surg J 5:503, 1899 237. Pallis CA: Impaired identification of faces and places with agnosia for colors. J Neurol Neurosurg Psychiatry 18:218, 1955 238. Meadows JC: Disturbed perception of colors associated with localized cerebral lesions. Brain 97:615, 1974 239. Rizzo M, Smith V, Pokorny J, Damasio A: Color perception profiles in central achromatopsia. Neurology 43:995, 1993 240. Rizzo M, Smith V, Pokorny J, Damasio AR: Color perception profiles in central achromatopsia. Neurology 43:995, 1993 241. Damasio A, Yamada T, Damasio H, Corbett J, McKee J: Central achromatopsia: Behavioral, anatomic and physiologic aspects. Neurology 30:1064, 1980 242. Critchley M: Acquired anomalies of color perception of central origin. Brain 88:711, 1965 243. Sacks O: The case of the color-blind painter. An Anthropologist on Mars. New York: Alfred A Knopf, 1995 244. Land E: Recent advances in retinex theory. Vision Res 26:7, 1986 245. Zeki SM: A century of cerebral achromatopsia. Brain 113:1721, 1990 246. Land E, Hubel D, Livingstone M, Perry S, Burns M: Color-generating interactions across the corpus callosum. Nature 303:616, 1983 247. Kennard C, Lawden M, Morland AB, Ruddock KH: Color identification and color constancy are impaired in a patient with
incomplete achromatopsia associated with prestriate lesions. Proc R Soc Lond B 260:169, 1995 248. Clarke S, Walsh V, Schoppig A, Assal G, Cowey A: Color constancy impairments in patients with lesions of the prestriate
cortex. Exp Brain Res 123:154, 1998 249. Hurlbert AC, Bramwell DI, Heywood C, Cowey A: Discrimination of cone contrast changes as evidence for color constancy
in cerebral achromatopsia. Exp Brain Res 123:136, 1998 250. D'Zmura MD, Knoblauch K, Henaff M-A, Michel F: Dependence of color on context in a case of cortical color vision deficiency. Vision Research 38:3455, 1998 251. Heywood CA, Cowey A, Newcombe F: Chromatic discrimination in a cortically color blind observer. Eur J Neurosci 3:802, 1991 252. Heywood CA, Nicholas JJ, Cowey A: Behavioural and electrophysiological chromatic and achromatic contrast
sensitivity in an achromatopsic patient. J Neurol Neurosurg Psychiatry 61:638, 1996 253. Adachi-Usami E, Tsukamoto M, Shimada Y: Color vision and color pattern evoked cortical potentials in a patient
with acquired cerebral dyschromatopsia. Doc Ophthalmol 90:259, 1997 254. Pearlman AL, Birch J, Meadows JC: Cerebral color blindness: an acquired defect in hue discrimination. Ann Neurol 5:253, 1979 255. Heywood CA, Kentridge RW, Cowey A: Form and motion from color in cerebral achromatopsia. Exp Brain Res 123:145, 1998 256. Cole G, Heywood C, Kentridge R, Fairholm I, Cowey A: Attentional capture by color and motion in cerebral achromatopsia. Neuropsychologia 41:1837, 2003 257. Cavanagh P, Hénaff M-A, Michel F, et al: Complete sparing of high-contrast color input to motion perception in cortical
color blindness. Nature Neurosci 1:242, 1998 258. Green GJ, Lessell S: Acquired cerebral dyschromatopsia. Arch Ophthalmol 95:121, 1977 259. Mendola J, Corkin S: Visual discrimination and attention after bilateral temporal-lobe lesions: a
case study. Neuropsychologia 37:91, 1999 260. Ogden JA: Visual object agnosia, prosopagnosia, achromatopsia, loss of visual imagery, and
autobiographic amnesia following recovery from cortical blindness: case
M.H. Neuropsychologia 31:571, 1993 261. Verrey D: Hemiachromatopsie droite absolue. Arch Ophthalmol (Paris) 8:289, 1888 262. Victor J, Maiese K, Shapley R, Sitdis J, Gazzaniga M: Acquired central dyschromatopsia: analysis of a case with preservation
of color discrimination. Clin Vision Sci 4:183, 1989 263. Damasio H, Frank R: Three-dimensional in vivo mapping of brain lesions in humans. Arch Neurol 49:137, 1992 264. Dean P: Visual cortex ablation and thresholds for successively presented stimuli
in Rhesus monkeys. II. Hue. Exp Brain Res 35:69, 1979 265. Wild HM, Butler SR, Carden D, Kulikowski JJ: Primate cortical area V4 important for color constancy but not wavelength
discrimination. Nature 313:133, 1985 266. Heywood CA, Cowey A: On the role of cortical area V4 in the discrimination of hue and pattern
in macaque monkeys. J Neurosci 7:2601, 1987 267. Heywood CA, Gadotti A, Cowey A: Cortical area V4 and its role in the perception of color. J Neurosci 12:4056, 1992 268. Walsh V, Kulikowski JJ, Butler SR, Carden D: The effects of lesions of area V4 on the visual abilities of macaques: color
categorization. Behav Brain Res 7:1, 1992 269. Schiller P: The effects of V4 and middle temporal (MT) lesionson visual performance
in the rhesus monkey. Vis Neurosci 10:717, 1993 270. Cowey A, Heywood C, Irving-Bell L: The regional cortical basis of achromatopsia: a study on macaque monkeys
and an achromatopsic patient. Eur J Neurosci 14:1555, 2001 271. Heywood C, Gaffan D, Cowey A: Cerebral achromatopsia in monkeys. Eur J Neurosci 7:1064, 1995 272. Hadjikhani N, Liu A, Dale A, Cavanagh P, Tootell R: Retinotopy and color selectivity in human cortical visual area V8. Nature Neurosci 1:235, 1998 273. Bartels A, Zeki S: The architecture of the color centre in the human visual brain: new results
and a review. Eur J Neurosci 12:172, 2000 274. Gulyás B, Roland P: Cortical fields participating in form and color discrimination in the human
brain. Neuroreport 2:585, 1991 275. Gulyás B, Heywood C, Popplewell D, Roland P, Cowey A: Visual form discrimination from color or motion cues: functional anatomy
by positron emission tomography. Proc Natl Acad Sci USA 91:9965, 1994 276. Beauchamp M, Haxby J, Jennings J, DeYoe E: An fMRI version of the Farnsworth-Munsell 100-Hue test reveals multiple
color-selective areas in human ventral occipitotemporal cortex. Cereb Cortex 9:257, 1999 277. Merigan W: Human V4? Curr Biol 3:226, 1993 278. Wandell B, Wade A: Functional imaging of the visual pathways. Neurol Clin 21:417, 2003 279. Orrell R, James-Galton M, Stevens J, Rossor M: Cerebral achromatopsia as a presentation of Trousseau's syndrome. Postgrad Med J 71:44, 1995 280. Aldrich M, Vanderzant C, Alessi A, Abou-Khalil B, Sackellares J: Ictal cortical blindness with permanent visual loss. Epilepsia 30:116, 1989 281. Freedman L, Costa L: Pure alexia and right hemiachromatopsia in posterior dementia. J Neurol Neurosurg Psychiatr 55:500, 1992 282. Lawden M, Cleland P: Achromatopsia in the aura of migraine. J Neurol Neurosurg Psychiatr 56:708, 1993 283. Fine R, Parker G: Disturbance of central vision after carbon monoxide poisoning. Aust NZ J Ophthalmol 24:137, 1996 284. Kölmel HW: Pure homonymous hemiachromatopsia. Findings with neuroophthalmologic examination
and imaging procedures. Eur Arch Psychiatr Neurol Sci 237:237, 1988 285. Linksz A: An Essay on Color Vision and Clinical Color Vision Tests. New York: Grune and Stratton, 1964 286. Working Group 41 N-NCoV: Procedures for Testing Color Vision. Washington, DC: National Academy Press, 1981 287. Wyszecki G, Stiles W: Color Science: Concepts and Methods, Quantitative Data and Formulae. New York: Wiley, 1982 288. Hardy L, Rand G, Rittler M: AO-HRR pseudoisochromatic plates. Buffalo, NY: American Optical Co, 1957 289. Ichikawa K, Hukame H, Tanabe S: Detection of acquired color vision defects by standard pseudoisochromatic
plates, part 2. Doc Ophthalmol Proc 46:133, 1987 290. Frisèn L, Kalm P: Sahlgren's saturation test for detecting and grading acquired dyschromatopsia. Am J Ophthalmol 92:252, 1981 291. Verriest G, Uvijls A, Aspinall P, et al: The lightness discrimination test. Bull Soc Belge Ophtalmol 183:162, 1979 292. Pinckers A, Verriest G: Results of shorthand lightness discrimination test. In: Verriest G (ed): Color Vision Deficiencies VII. Dordrecht, The Netherlands: Martinus Nijhoff/Dr. W Junk, 1987:163 293. Heywood CA, Wilson B, Cowey A: A case study of cortical color ‘blindness’ with relatively
intact achromatic discrimination. J Neurol Neurosurg Psychiatr 50:22, 1987 294. Beauchamp M, Haxby J, Rosen A, DeYoe E: A functional MRI case study of acquired cerebral dyschromatopsia. Neuropsychologia 38:1170, 2000 295. Holmes G: Pure word blindness. Folia Psychiatr Neurol Neurochir Neerl 53:279, 1950 296. Geschwind N, Fusillo M: Color-naming defects in association with alexia. Arch Neurol 15:137, 1966 297. Oxbury JM, Oxbury SM, Humphrey NK: Varieties of color anomia. Brain 92:847, 1969 298. de Vreese LP: Two systems for color-naming defects: verbal disconnection versus color
imagery disorder. Neuropsychologia 29:1, 1991 299. Kinsbourne M, Warrington EK: Observations on color agnosia. J Neurol Neurosurg Psychiatry 27:296, 1964 300. Luzzatti C, Davidoff J: Impaired retrieval of object-color knowledge with preserved color naming. Neuropsychologia 32:933, 1994 301. Miceli G, Fouch E, Capasso R, et al: The dissociation of color from form and function knowledge. Nat Neurosci 4:662, 2001 302. Farah M: Visual Agnosia: Disorders of Visual Recognition and What They Tell Us about
Normal Vision. Cambridge, MA: MIT Press, 1990 303. Riddoch M, Humphreys G: Visual agnosia. Neurol Clin 21:501, 2003 304. Teuber HL: Alteration of perception and memory in man. In: Weiskrantz L (ed): Analysis of Behavioral Change. New York: Harper & Row, 1968 305. Lissauer H: Einfall von Seelenblindheit nebst einem Bintrag zur Theorie derselben. Arch Psychiatr Nervenkr 2:22, 1890 306. Tranel D: Assessment of higher-order visual function. Curr Opin Ophthalmol 5:29, 1994 307. Anderson S, Rizzo M: Recovery and rehabilitation of visual cortical dysfunction. Neurorehabilitation 5:129, 1995 308. Lezak M: Neuropsychological Assessment. New York: Oxford University Press, 1995 309. Spreen O, Strauss E: A Compendium of Neuropsychological Tests. New York: Oxford University Press, 1991 310. Benton A, Van Allen M: Visuoperceptual, visuospatial, and visuoconstructive disorders. In Heilman K, Valenstein E (eds): Clinical Neuropsychology. London: Oxford University Press, 1985:161 311. Benton A, Hamsher J, Varney N, et al: Contributions to Neuropsychological Assessment. New York: Oxford University Press, 1983 312. Goodglass H, Kaplan E: The Assessment of Aphasia and Related Disorders. Philadelphia: Lea & Febiger, 1983 313. Hooper H: The Hooper Visual Organization Test Manual. Los Angeles: Western Psychological Services, 1958 314. Mooney C, Ferguson G: A new closure test. Can J Psychol 5:129, 1951 315. Newcombe F: Selective deficits after focal cerebral injury. In Dimond S, Beanmont J (eds): Hemisphere Function in the Human Brain. New York: Halsted Press, 1974:311 316. Bodamer J: Prosopagnosie. Arch Psychiatr Nervenkr 179:6, 1947 317. Tejeria L, Harper RA, Artes PH, Dickinson CM: Face recognition in age related macular degeneration: perceived disability, measured
disability, and performance with a bioptic device. Br J Ophthalmol 86:1019, 2002 318. Mendez M, Martin R, Smyth K, Whitehouse P: Disturbances of person identification in Alzheimer's disease. A retrospective
study. J Nerv Ment Dis 180:94, 1992 319. Roudier M, Marcie P, Grancher A, et al: Discrimination of facial identity and of emotions in Alzheimer's disease. J Neurol Sci 154:151, 1998 320. Cronin-Golomb A, Cronin-Golomb M, Dunne TE, et al: Facial frequency manipulation normalizes face discrimination in AD. Neurology 54:2316, 2000 321. Janati A: Kluver-Bucy syndrome in Huntington's chorea. J Nerv Ment Dis 173:632, 1985 322. Dewick H, Hanley J, Davies A, Playfer J, Turnbull C: Perception and memory for faces in Parkinson's disease. Neuropsychologia 29:785, 1991 323. Cousins R, Hanley JR, Davies AD, Turnbull CJ, Playfer JR: Understanding memory for faces in Parkinson's disease: the role of
configural processing. Neuropsychologia 38:837, 2000 324. Young A, Ellis H: Childhood prosopagnosia. Brain Cognit 9:16, 1989 325. Kracke I: Developmental prosopagnosia in Asperger syndrome: presentation and discussion
of an individual case. Dev Med Child Neurol 36:873, 1994 326. Takahashi N, Kawamura M, Hirayama K, Shiota J, Isono O: Prosopagnosia: a clinical and anatomic study of four patients. Cortex 31:317, 1995 327. Tranel D, Damasio A. Knowledge without awareness: an autonomic index of facial recognition by
prosopagnosics. Science 228:1453, 1985 328. Young A, Aggleton J, Hellawell D, et al: Face processing impairments after amygdalotomy. Brain 118:15, 1995 329. de Haan E, Campbell R: A fifteen year follow-up of a case of developmental prosopagnosia. Cortex 27:489, 1991 330. Campbell R, Heywood C, Cowey A, Regard M, Landis T: Sensitivity to eye gaze in prosopagnosic patients and monkeys with superior
temporal sulcus ablation. Neuropsychologia 28:1123, 1990 331. Bruyer R, Laterre C, Seron X, et al: A case of prosopagnosia with some preserved covert remembrance of familiar
faces. Brain Cognit 2:257, 1983 332. Sergent J, Villemure J-G: Prosopagnosia in a right hemispherectomized patient. Brain 112:975, 1989 333. Sergent J, Poncet M: From covert to overt recognition of faces in a prosopagnosic patient. Brain 113:989, 1990 334. Evans J, Heggs A, Antoun N, Hodges J: Progressive prosopagnosia associated with selective right temporal lobe
atrophy. Brain 118:1, 1995 335. Tranel D, Damasio AR, Damasio H: Intact recognition of facial expression, gender, and age in patients with
impaired recognition of face identity. Neurology 38:690, 1988 336. Perrett D, Hietanen J, Oram M, Benson P: Organization and functions of cells responsive to faces in the temporal
cortex. Phil Trans R Soc Lond B 335:23, 1992 337. Gauthier I, Logothetis N: Is face recognition not so unique after all? Cogn Neuropsychol 17:125, 2000 338. Haxby JV, Gobbini MI, Furey ML, et al: Distributed and overlapping representations of faces and objects in ventral
temporal cortex. Science 293:2425, 2001 339. Bruyer R: Covert facial recognition in prosopagnosia: a review. Brain Cogn 15:223, 1991 340. Young A: Covert recognition. In Farah M, Ratcliff G (eds): The Neuropsychology of High-Level Vision. Hillsdale, NJ: LEA, 1994 341. Bauer R: Autonomic recognition of names and faces in prosopagnosia: a neuropsychological
application of the guilty knowledge test. Neuropsychologia 22:457, 1984 342. Bauer R, Verfaellie M: Electrodermal discrimination of familiar but not unfamiliar faces in prosopagnosia. Brain Cogn 8:240, 1988 343. Renault B, Signoret J-L, DeBruille B, Breton F, Bolgert F: Brain potentials reveal covert facial recognition in prosopagnosia. Neuropsychologia 27:905, 1989 344. McNeil J, Warrington E: Prosopagnosia: a re-classification. Q J Exp Psychol 43A:267, 1991 345. Sergent J, Signoret J-L: Implicit access to knowledge derived from unrecognized faces. Cereb Cortex 2:389, 1992 346. Barton JJ, Cherkasova M, O'Connor M: Covert recognition in acquired and developmental prosopagnosia. Neurology 57:1161, 2001 347. Schweinberger S, Klos T, Sommer W: Covert face recognition in prosopagnosia: a dissociable function? Cortex 31:517, 1995 348. Rizzo M, Hurtig R, Damasio A: The role of scanpaths in facial recognition and learning. Ann Neurol 22:41, 1987 349. de Haan E, Young A, Newcombe F: Faces interfere with name classification in a prosopagnosic patient. Cortex 23:309, 1987 350. Young A, Hellawell D, de Haan E: Cross-domain semantic priming in normal subjects and a prosopagnosic patient. Q J Exp Psychol 40A:561, 1988 351. Farah M, O'Reilly R, Vecera S: Dissociated overt and covert recognition as an emergent property of a lesioned
neural network. Psychol Rev 100:571, 1993 352. Young A, Burton A: Simulating face recognition: implications for modelling cognition. Cogn Neuropsychol 16:1, 1999 353. O'Reilly R, Farah M: Simulation and explanation in neuropsychology and beyond. Cogn Neuropsychol 16:49, 1999 354. Barton JJ, Cherkasova M: Face imagery and its relation to perception and covert recognition in prosopagnosia. Neurology 61:220, 2003 355. Kanwisher N: Domain specificity in face perception. Nat Neurosci 3:759, 2000 356. Tarr MJ, Gauthier I: FFA: a flexible fusiform area for subordinate-level visual processing automatized
by expertise. Nat Neurosci 3:764, 2000 357. Lhermitte F, Chain F, Escourolle R, Ducarne B, Pillon B: Étude anatomo-clinique d'un cas de prosopagnosie. Rev Neurol 126:329, 1972 358. Whiteley A, Warrington E: Prosopagnosia: a clinical, psychological, and anatomical study of three
patients. J Neurol Neurosurg Psychiatry 40:395, 1977 359. Damasio A, Damasio H, van Hoessen G: Prosopagnosia: anatomic basis and behavioral mechanisms. Neurology 32:331, 1982 360. de Renzi E: Prosopagnosia in two patients with CT scan evidence of damage confined
to the right hemisphere. Neuropsychologia 24:385, 1986 361. McNeil J, Warrington E: Prosopagnosia: a face-specific disorder. Q J Exp Psychol 46A:1, 1993 362. Henke K, Schweinberger S, Grigo A, Klos T, Sommer W: Specificity of face recognition: recognition of exemplars of non-face objects
in prosopagnosia. Cortex 34:289, 1998 363. Farah M, Levinson K, Klein K: Face perception and within-category discrimination in prosopagnosia. Neuropsychologia 33:661, 1995 364. Gauthier I, Behrmann M, Tarr M: Can face recognition really be dissociated from object recognition? J Cogn Neurosci 11:349, 1999 365. Barton J, Cherkasova M, Press D, Intriligator J, O'Connor M: Perceptual function in prosopagnosia. J Neuroophthalmol 2004 366. Albert M, Butters N, Levin J: Temporal gradients in retrograde amnesia of patients with alcoholic Korsakoff's
disease. Arch Neurol 36:211, 1979 367. Warrington E: Warrington Recognition Memory Test. Los Angeles: Western Psychological Services, 1984 368. Benton A, van Allen M: Prosopagnosia and facial discrimination. J Neurol Sci 15:167, 1972 369. Parry F, Young A, Saul J, Moss A: Dissociable face processing impairments after brain injury. J Clin Exp Neuropsychol 13:545, 1991 370. Bruce V, Young A: Understanding face recognition. Br J Psychol 77:305, 1986 371. Damasio A, Tranel D, Damasio H: Face agnosia and the neural substrates of memory. Ann Rev Neurosci 13:89, 1990 372. de Renzi E, Faglioni P, Grossi D, Nichelli P: Apperceptive and associative forms of prosopagnosia. Cortex 27:213, 1991 373. Levine D, Calvanio R: Prosopagnosia: a defect in visual configural processing. Brain Cogn 10:149, 1989 374. Rentschler I, Treutwein B, Landis T: Dissociation of local and global processing in visual agnosia. Vision Res 34:963, 1994 375. de Renzi E, Faglioni P, Spinnler H: The performance of patients with unilateral brain damage on face recognition
tasks. Cortex 4:17, 1968 376. Carlesimo G, Caltagirone C: Components in the visual processing of known and unknown faces. J Clin Exp Neuropsychol 17:691, 1995 377. Barton JJ, Press DZ, Keenan JP, O'Connor M: Lesions of the fusiform face area impair perception of facial configuration
in prosopagnosia. Neurology 58:71, 2002 378. Joubert S, Felician O, Barbeau E, et al: Impaired configurational processing in a case of progressive prosopagnosia
associated with predominant right temporal lobe atrophy. Brain 126:2537, 2003 379. Leder H, Bruce V: Local and relational aspects of face distinctiveness. Q J Exp Psychol 51A:449, 1998 380. Barton J, Keenan J, Bass T: Discrimination of spatial relations and features in faces: effects of inversion
and viewing duration. Br J Psychol 92(Pt 3):527-49, 2001 381. Ellis H, Young A, Critchley E: Loss of memory for people following temporal lobe damage. Brain 112(pt 6):1469, 1989 382. Hanley J, Young A, Pearson N: Defective recognition of familiar people. Cogn Neuropsychol 6:179, 1989 383. Gorno-Tempini ML, Price CJ: Identification of famous faces and buildings: a functional neuroimaging
study of semantically unique items. Brain 124:2087, 2001 384. Leveroni C, Seidenberg M, Mayer A, et al: Neural systems underlying the recognition of familiar and newly learned
faces. J Neurosci 20:878, 2000 385. Levine D, Warach J, Farah M: Two visual systems in mental imagery: dissociation of “what” and “where” in imagery disorders due to bilateral posterior
cerebral lesions. Neurology 35:1010, 1985 386. Landis T, Cummings J, Christen L, Bogen J, Imhof H-G: Are unilateral right posterior lesions sufficient to cause prosopagnosia ? Clinical
and radiological findings in six additional patients. Cortex 22:243, 1986 387. de Haan E, Young A, Newcombe F: Face recognition without awareness. Cogn Neuropsychol 4:385, 1987 388. Meadows J: The anatomical basis of prosopagnosia. J Neurol Neurosurg Psychiatr 37:489, 1974 389. McCarthy G, Puce A, Gore J, Allison T: Face-specific processing in the human fusiform gyrus. J Cogn Neurosci 9:605, 1997 390. Kanwisher N, McDermott J, Chun M: The fusiform face area: a module in human extrastriate cortex specialized
for face perception. J Neurosci 17:4302, 1997 391. Haxby J, Hoffman E, Gobbini M: The distributed human neural system for face perception. Trends Cogn Sci 4:223, 2000 392. Michel F, Perenin M-T, Sieroff E: Prosopagnosie sans hémianopsie après lésion unilatérale
occipito-temporale droite. Rev Neurol 142:545, 1986 393. Barton JJ, Zhao J, Keenan JP: Perception of global facial geometry in the inversion effect and prosopagnosia. Neuropsychologia 41:1703, 2003 394. Malone D, Morris H, Kay M, Levin H: Prosopagnosia: a double dissociation between the recognition of familiar
and unfamiliar faces. J Neurol Neurosurg Psychiatr 45:820, 1982 395. Tyrell P, Warrington E, Frackowiak R, Rossor M: Progressive degeneration of the right temporal lobe studied with positron
emission tomography. J Neurol Neurosurg Psychiatr 53:1048, 1990 396. Martins IP, Cunha e Sa M: Loss of topographic memory and prosopagnosia during migraine aura. Cephalalgia 19:841, 1999 397. McConachie H: Developmental prosopagnosia: a single case report. Cortex 12:76, 1976 398. Ariel R, Sadeh M: Congenital visual agnosia and prosopagnosia in a child: a case report. Cortex 32:221, 1996 399. Barton JJ, Cherkasova MV, Press DZ, Intriligator JM, O'Connor M: Developmental prosopagnosia: a study of three patients. Brain Cogn 51:12, 2003 400. Polster M, Rapcsak S: Representations in learning new faces: evidence from prosopagnosia. J Int Neuropsychol Soc 2:240, 1996 401. Young A, Newcombe F, de Haan E, Small M, Hay D: Face perception after brain injury. Brain 116:941, 1993 402. de Renzi E, Bonacini M, Faglioni P: Right posterior brain-damaged patients are poor at assessing the age of
a face. Neuropsychologia 27:839, 1989 403. Young A, Flude B, Hay D, Ellis A: Impaired discrimination of familiar from unfamiliar faces. Cortex 29:65, 1993 404. Rapcsak S, Polster M, Comer J, Rubens A: False recognition and misidentification of faces following right hemisphere
damage. Cortex 30:565, 1994 405. Rapcsak SZ, Nielsen L, Littrell LD, et al: Face memory impairments in patients with frontal lobe damage. Neurology 57:1168, 2001 406. Rapcsak S, Polster M, Glisky M, Comer J: False recognition of unfamiliar faces following right hemisphere damage: neuropsychological
and anatomical observations. Cortex 32:593, 1996 407. Binder J, Mohr J: The topography of callosal reading pathways. A case control analysis. Brain 115:1807, 1992 408. Horikoshi T, Asari Y, Watanabe A, et al: Music alexia in a patient with mild pure alexia: disturbed visual perception
of non-verbal meaningful figures. Cortex 33:187, 1997 409. Beversdorf D, Heilman K: Progressive ventral posterior cortical degeneration presenting as alexia
for music and words. Neurology 50:657, 1998 410. Black S, Behrmann M: Localization in alexia. In Localization and Neuroimaging in Neuropsychology. New York: Academic Press, 1994 411. Bub D, Black S, Howell J: Word recognition and orthographic context effects in a letter-by-letter
reader. Brain Lang 36:357, 1989 412. Coslett H, Saffran E, Greenbaum S, Schwartz H: Reading in pure alexia. Brain 116:21, 1993 413. Albert M, Yamadori A, Gardner H, Howes D: Comprehension in alexia. Brain 96:317, 1973 414. Coslett H, Saffran E: Evidence for preserved reading in pure alexia. Brain 112:327, 1989 415. Caplan L, Hedley-White T: Cuing and memory dysfunction in alexia without agraphia: a case report. Brain 97:251, 1974 416. Feinberg T, Dyckes-Berke D, Miner C, Roane D: Knowledge, implicit knowledge and metaknowledge in visual agnosia and pure
alexia. Brain 118:789, 1995 417. Damasio A, Damasio H: The anatomic basis of pure alexia. Neurology 33:1573, 1983 418. Lepore F: Visual deficits in alexia without agraphia. Neuroophthalmology 19:1, 1998 419. de Renzi E, Zambolin A, Crisi G: The pattern of neuropsychological impairment associated with left posterior
cerebral artery infarcts. Brain 110:1099, 1987 420. Greenblatt S: Alexia without agraphia or hemianopia. Brain 96:307, 1973 421. Vincent F, Sadowsky C, Saunders R, Reeves A: Alexia without agraphia, hemianopia, or color-naming defect: a disconnection
syndrome. Neurology 27:689, 1977 422. Uitti R, Donat J, Romanchuk K: Pure alexia without hemianopia. Arch Neurol 41:1130, 1984 423. Ajax E: Dyslexia without agraphia. Arch Neurol 17:645, 1967 424. Bub D, Arguin M: Visual word activation in pure alexia. Brain Lang 49:77, 1995 425. Henderson V, Friedman R, Teng E, Weiner J: Left hemisphere pathways in reading: inferences from pure alexia without
hemianopia. Neurology 35:962, 1985 426. Erdem S, Kansu T: Alexia without either agraphia or hemianopia in temporal lobe lesion due
to herpes simplex encephalitis. J Neuroophthalmol 15:102, 1995 427. Jonsdóttir M, Magnússon T, Kjartansson O: Pure alexia and word-meaning deafness in a patient with multiple sclerosis. Arch Neurol 55:1473, 1998 428. Freedman L, Selchen D, Black S, Garnett E, Nahmias C: Posterior cortical dementia with alexia: neurobehavioural, MRI and PET
findings. J Neurol Neurosurg Psychiatry 54:443, 1991 429. Dejerine J: Contributions a l'étude anatomopathologique et clinique des
differentes varietes de cecite verbale. Memoires de la Societé Biologique 44:61, 1892 430. Lanzinger S, Weder B, Oettli R, Fretz C: Neuroimaging findings in a patient recovering from global alexia to spelling
dyslexia. J Neuroimaging 9:48, 1999 431. Silver F, Chawluk J, Bosley T, et al: Resolving metabolic abnormalities in a case of pure alexia. Neurology 38:731, 1988 432. Stommel E, Friedman R, Reeves A: Alexia without agraphia associated with spleniogeniculate infarction. Neurology 41:587, 1991 433. Behrmann M, Nelson J, Sekuler A: Visual complexity in letter-by-letter reading: “pure” alexia
is not pure. Neuropsychologia 36:1115, 1998 434. Warrington E, Shallice T: Word-form dyslexia. Brain 103:99, 1980 435. Shallice T, Warrington. E: The possible role of selective attention in acquired dyslexia. Neuropsychologia 15:31, 1977 436. Levine D, Calvanio R: A study of the visual defect in verbal alexia-simultanagnosia. Brain 101:65, 1978 437. Mendez M, Cherrier M: The evolution of alexia and simultanagnosia in posterior cortical atrophy. Neuropsychiatry, Neuropsychol, Behav Neurol 11:76, 1998 438. Vaina L, Grzywacz N, Kikinis R: Segregation of computations underlying perception of motion discontinuity
and coherence. Neuroreport 5:2289, 1994 439. Chanoine V, Ferreira C, Demonet J, Nespoulos J, Poncet M: Optic aphasia with pure alexia: a mild form of visual associative agnosia? A
case study. Cortex 34:437, 1998 440. Moscovitch M, Winocur G, Behrmann M: What is special about face recognition? Nineteen experiments on a person
with visual object agnosia and dyslexia but normal face recognition. J Cogn Neurosci 9:555, 1997 441. Beversdorf D, Ratcliffe N, Rhodes C, Reeves A: Pure alexia: clinical-pathologic evidence for a lateralized visual language
association cortex. Clin Neuropathol 16:328, 1997 442. Benito-León J, Sanchez-Suarez C, Diaz-Guzman J, Martinez-Salio A: Pure alexia could not be a disconnection syndrome. Neurology 49:305, 1997 443. Beeson P, Insalaco D: Acquired alexia: lessons from successful treatment. J Int Neuropsychol Soc 4:621, 1998 444. Maher L, Clayton M, Barrett A, Schober-Peterson D, Rothi L: Rehabilitation of a case of pure alexia: exploiting residual reading abilities. J Int Neuropsychol Soc 4:636, 1998 445. Conway T, Heilman P, Rothi L, et al: Treatment of a case of phonologic alexia with agraphia using the Auditory
Discrimination in Depth (ADD) program. J Int Neuropsychol Soc 4:608, 1998 446. Nitzberg Lott S, Friedman R: Can treatment for pure alexia improve letter-by-letter reading speed without
sacrificing accuracy? Brain Language 67:188, 1999 447. Geschwind N: Disconnexion syndromes in animals and man. Brain 88:17, 1965 448. Gazzaniga M, Freedman H: Observations on visual processes after posterior callosal section. Neurology 23:1126, 1973 449. Molko N, Cohen L, Mangin J, et al: Visualizing the neural bases of a disconnection syndrome with diffusion
tensor imaging. J Cogn Neurosci 14:629, 2002 450. Castro-Caldas A, Salgado V: Right hemifield alexia without hemianopia. Arch Neurol 41:84, 1984 451. Binder J, Lazar R, Tatemichi T, et al: Left hemiparalexia. Neurology 42:562, 1992 452. Benson D: Alexia. In Bruyn G, Klawans H, Vinken P (eds): Handbook of Clinical Neurology. New York: Elsevier, 1985 453. Kawahata N, Nagata K: Alexia with agraphia due to the left posterior inferiortemporal lobe lesion. Neuropsychological
analysis and its pathogenetic mechanisms. Brain Lang 33:296, 1988 454. Ardila A, Rosselli M, Arvizu L, Kuljis R: Alexia and agraphia in posterior cortical atrophy. Neuropsychiatry, Neuropsychol, Behav Neurol 10:52, 1997 455. Ferracci F, Conte F, Gentile M, et al: Marchiafava-Bignami disease. Computer tomographic scan, HMPAO-SPECT, and
FLAIR MRI findings in a patient with subcortical aphasia, alexia, bilateral
agraphia, and left-handed deficit of constructional ability. Arch Neurol 56:107, 1999 456. Benson D, Brown J, Tomlinson E: Varieties of alexia. Word and letter blindness. Neurology 21:951, 1971 457. Benson D: The third alexia. Arch Neurol 34:327, 1977 458. Kirkham T: The ocular symptomology of pituitary tumors. Proc R Soc Med 65:517, 1972 459. de Luca M, Spinelli D, Zoccolotti P: Eye movement patterns in reading as a function of visual field defects
and contrast sensitivity loss. Cortex 32:491, 1996 460. Zihl J: Eye movement patterns in hemianopic dyslexia. Brain 118:891, 1995 461. Behrmann M, Moscovitch M, Black S, Mozer M: Perceptual and conceptual factors in neglect dyslexia: two contrasting
case studies. Brain 113:1163, 1990 462. Patterson K, Wilson B: A rose is a nose: a deficit in initial letter identification. Cogn Neuropsychol 13:447, 1990 463. Husain M, Stein J: Rezsö Bálint and his most celebrated case. Arch Neurol 45:89, 1988 464. Holmes G: Disturbances of visual orientation. Br J Ophthalmol 2:449, 1918 465. Pierrot-Deseilligny C, Gray F, Brunet P: Infarcts of both inferior parietal lobules with impairment of visually
guided eye movements, peripheral inattention and optic ataxia. Brain 109:81, 1986 466. Baylis G, Driver J, Baylis L, Rafal R: Reading of letters and words in a patient with Balint's syndrome. Neuropsychologia 32:1273, 1994 467. Friedman D, Jankovic J, McCrary J: Neuro-ophthalmic findings in progressive supranuclear palsy. J Clin Neuroophthalmol 12:104, 1992 468. Beauvois M, Dérouesné J: Phonologic alexia: three dissociations. J Neurol Neurosurg Psychiatry 42:1115, 1979 469. Funnell E. Phonologic processes in reading: new evidence from acquired dyslexia. Br J Psychol 74:159, 1983 470. Friedman R, Kohn S: Impaired activation of the phonologic lexicon: effects upon oral reading. Brain Lang 38:278, 1990 471. Friedman R: Two types of phonologic alexia. Cortex 31:397, 1995 472. Shallice T, Warrington E, McCarthy R: Reading without semantics. Q J Exp Psychol 35A:111, 1983 473. Patterson K, Morton J: From orthograph to phonology: an attempt at an old interpretation. In Patterson K, Marshall J, Coltheart M (eds): Surface Dyslexia. London: LEA, 1985:335 474. Cummings J, Houlihan J, Hill M: The pattern of reading deterioration in dementia of the Alzheimer type. Brain Lang 29:315, 1986 475. Friedman R, Ferguson S, Robinson S, Sunderland T: Dissociation of mechanisms of reading in Alzheimer's disease. Brain Lang 43:400, 1992 476. Coltheart M: Deep dyslexia, a review of the syndrome. In Coltheart M, Patterson K, Marshall J (eds): Deep Dyslexia. London: Routledge & Kegan Paul, 1980 477. Aguirre G, D'Esposito M: Topographical disorientation: a synthesis and taxonomy. Brain 122:1613, 1999 478. Takahashi N, Kawamura M: Pure topographical disorientation: the anatomical basis of landmark agnosia. Cortex 38:717, 2002 479. Pai M: Topographic disorientation: two cases. J Formos Med Assoc 96:660, 1997 480. McCarthy R, Evans J, Hodges J: Topographic amnesia: spatial memory disorder, perceptual dysfunction, or
category specific semantic memory impairment? J Neurol Neurosurg Psychiatry 60:318, 1996 481. O'Craven KM, Kanwisher N: Mental imagery of faces and places activates corresponding stimulus-specific
brain regions. J Cogn Neurosci 12:1013, 2000 482. de Renzi E, Faglioni P, Villa P: Topographical amnesia. J Neurol Neurosurg Psychiatry 40:498, 1977 483. Takahashi N, Kawamura M, Shiota J, Kasahata N, Hirayama K: Pure topographic disorientation due to right retrosplenial lesion. Neurology 49:464, 1997 484. Sato K, Sakajiri K, Komai K, Takamori M: A patient with amnesic syndrome with defective route finding due to left
posterior cerebral artery territory infarction. No To Shinkei 50:69, 1998 485. Habib M, Sirigu A: Pure topographical disorientation: a definition and anatomical basis. Cortex 23:73, 1987 486. Bálint R. Seelenlahmung des ‘Schauens,’ optische Ataxie, räumliche
Storung der Aufmerksamkeit. Monatschrift für Psychiatrie und Neurologie 25:51, 1909 487. Hécaen H, de Ajuriaguerra J: Balint's syndrome (psychic paralysis of visual fixation) and
its minor forms. Brain 77:373, 1954 488. Coslett H, Saffran E: Simultanagnosia. To see but not two see. Brain 114:1523, 1991 489. Rizzo M, Robin DA: Simultanagnosia: a defect of sustained attention yields insights on visual
information processing. Neurology 40:447, 1990 490. Wolpert T: Die Simultanagnosie. Zeitschrift für Gesamte Neruologie und Psychiatrie 93:397, 1924 491. Luria AR, Pravdina-Vinarskaya EN, Yarbus AL: Disturbances of ocular movement in a case of simultanagnosia. Brain 86:219, 1962 492. Holmes G: Disturbances of vision caused by cerebral lesions. Br J Ophthalmol 2:353, 1918 493. Perenin M, Vighetto A: Optic ataxia: a specific disruption in visuomotor mechanisms. I. Different
aspects of the deficit in reaching for objects. Brain 111:643, 1988 494. Jakobson L, Archibald Y, Carey D, Goodale M: A kinematic analysis of reaching and grasping movements in a patient recovering
from optic ataxia. Neuropsychologia 29:803, 1991 495. Rizzo M, Rotella D, Darling W: Troubled reaching after right occipito-temporal damage. Neuropsychologia 30:711, 1992 496. Cogan DG: Congenital ocular motor apraxia. Can J Ophthalmol 1:253, 1965 497. Holmes G: Spasm of fixation. Trans Ophthalmol Soc UK 50:253, 1930 498. Johnston JL, Sharpe JA, Morrow MJ: Spasm of fixation: a quantitative study. J Neurol Sci 107:166, 1992 499. Mackworth NH: The breakdown of vigilance during prolonged visual search. Q J Exp Psychol 1:6, 1948 500. Mackworth NH, Kaplan IT, Matlay W: Eye movements during vigilance. Percept Mot Skills 18:397, 1964 501. Broadbent DE: Perception and Communication. New York: Pergamon Press, 1958 502. Luria AR: Disorders of simultaneous perception in a case of bilateral occipito-parietal
brain injury. Brain 82:437, 1959 503. Castaigne P, Rondot P, Dumas J, Tempier P: Ataxie optique localisee au cote gauche dans les deux hemichamps visuels
homonymes gauches. Rev Neurol (Paris) 131:23, 1975 504. Hecaen H, de Ajuriaguerra J: Bálint's syndrome (psychic paralysis of visual fixation) and
its minor forms. Brain 77:373, 1954 505. Battaglia Mayer A, Ferraina S, Marconi B, et al: Early motor influences on visuomotor transformations for reaching: a positive
image of optic ataxia. Exp Brain Res 123:172, 1998 506. Boller F, Cole M, Kim Y, Mack J, Patawaran C: Optic ataxia: clinical-radiological correlations with the EMI scan. J Neurol Neurosurg Psychiatry 38:954, 1975 507. Nagaratnam N, Grice D, Kalouche H: Optic ataxia following unilateral stroke. J Neurol Sci 155:204, 1998 508. Andersen R, Brotchie P, Mazzoni P: Evidence for the lateral intraparietal area as the parietal eye field. Curr Biol 2:840, 1992 509. Holmes G, Horrax G: Disturbances of spatial orientation and visual attention, with loss of
stereoscopic vision. Arch Neurol Psychiatry 1:385, 1919 510. Rizzo M: Bálint's syndrome and associated visuospatial disorders. In Kennard C (ed):Bailliere's International Practice and Research. Philadelphia: WB Saunders, 1993:415 511. Kertesz A: Visual agnosia: the dual deficit of perception and recognition. Cortex 15:403, 1979 512. Onofrj M, Fulgente T, Thomas A: Event related potentials recorded in Dorsal Simultanagnosia. Brain Res Cogn Brain Res 3:25, 1995 513. Jarry D, Rigolet M-H, Rivaud S, Bakshine S: Diagnostic électrophysiologique de deux syndromes psycho-visuels: syndrome
de Balint et cécité corticale. J Fr Ophtalmol 22:876, 1999 514. Auerbach S, Alexander M: Pure agraphia and unilateral optic ataxia associated with a left superior
parietal lobule lesion. J Neurol Neurosurg Psychiatry 44:430, 1981 515. Ando S, Moritake K: Pure optic ataxia associated with a right parieto-occipital tumour. J Neurol Neurosurg Psychiatry 53:805, 1990 516. Ogren MP, Mateer CA, Wyler AR: Alterations in visually related eye movements following left pulvinar damage
in man. Neuropsychologia 22:187, 1984 517. Hijdra A, Meerwaldt J: Balint's syndrome in a man with borderzone infarcts caused by atrial
fibrillation. Clin Neurol Neurosurg 86:51, 1984 518. Montero J, Pena J, Genis D, et al: Balint's syndrome: report of four cases with watershed parieto-occipital
lesions from vertebrobasilar ischemia or systemic hypotension. Acta Neurol Belg 82:270, 1982 519. Graff-Radford N, Bolling J, Earnest Ft, et al: Simultanagnosia as the initial sign of degenerative dementia. Mayo Clin Proc 68:955, 1993 520. Hof P, Bouras C, Constandinidis J, et al: Selective disconnection of specific visual association pathways in cases
of Alzheimer's disease presenting the Balint's syndrome. J Neuropathol Exp Neurol 49:168, 1990 521. Iizuka O, Soma Y, Otsuki M, et al: Posterior cortical atrophy with incomplete Balint's syndrome. No To Shinkei 49:841, 1997 522. Perez FM, Tunkel RS, Lachman EA, Nagler W: Balint's syndrome arising from bilateral posterior cortical atrophy
or infarction: rehabilitation strategies and their limitation. Disabil Rehabil 18:300, 1996 523. Trobe J, Bauer R: Seeing but not recognizing. Surv Ophthalmol 30:328, 1986 524. Truffert A, Dumas J, Dandelot J: Dysconnexion interhemispherique, syndrome de Balint et troubles arthriques
persistants: maladie de Marchiafava-Bignami avec hemorrhagie de la
substance blanche. Rev Neurol (Paris) 152:174, 1996 525. Paytubi Gari C, Lopez-Balaguer J, Balmana J, Cadafalch J: Sindrome de Balint secundario a leucoencefalopatia multifocal progresiva
en una paciente con sindrome de inmunodeficiencia adquirida. Med Clin (Barc) 111:357, 1998 526. Schnider A, Landis T, Regard M: Balint's syndrome in subacute HIV encephalitis. J Neurol Neurosurg Psychiatry 54:822, 1991 527. Shah P, Nafee A: Migraine aura masquerading as Balint's syndrome. J Neurol Neurosurg Psychiatry 67:554, 1999 528. Zihl J, von Cramon D, Mai N: Selective disturbance of movement vision after bilateral brain damage. Brain 106:313, 1983 529. Zihl J, von Cramon D, Mai N, Schmid C: Disturbance of movement vision after bilateral posterior brain damage. Further
evidence and follow-up observations. Brain 114:2235, 1991 530. Hess R, Baker CJ, Zihl J: The “motion-blind” patient: low-level spatial and temporal
filters. J Neurosci 9:1628, 1989 531. Baker CJ, Hess R, Zihl J: Residual motion perception in a “motion-blind” patient, assessed
with limited-lifetime random dot stimuli. J Neurosci 11:454, 1991 532. Rizzo M, Nawrot M, Zihl J: Motion and shape perception in cerebral akinetopsia. Brain 118:1105, 1995 533. Marcar V, Zihl J, Cowey A: Comparing the visual deficits of a motion blind patient with the visual
deficits of monkeys with area MT removed. Neuropsychologia 35:1459, 1997 534. Campbell R, Zihl J, Massaro D, Munhall K, Cohen M: Speechreading in the akinetopsic patient, L.M. Brain 120:1793, 1997 535. McLeod P, Heywood C, Driver J, Zihl J: Selective deficit of visual search in moving displays after extrastriate
damage. Nature 339:466, 1989 536. Shipp S, de Jong B, Zihl J, Frackowiak R, Zeki S: The brain activity related to residual motion vision in a patient with
bilateral lesions of V5. Brain 117:1023, 1994 537. Vaina L, Cowey A: Impairment of the perception of second order motion but not first order
motion in a patient with unilateral focal brain damage. Proc R Soc Lond B 263:1225, 1996 538. Vaina L, Makris N, Kennedy D, Cowey A: The selective impairment of the perception of first-order motion by unilateral
cortical brain damage. Visual Neurosci 15:333, 1998 539. Barton J, Sharpe J, Raymond J: Directional defects in pursuit and motion perception in humans with unilateral
cerebral lesions. Brain 119:1535, 1996 540. Vaina L: Functional segregation of color and motion processing in the human visual
cortex: clinical evidence. Cereb Cortex 5:555, 1994 541. Zeki S: Cerebral akinetopsia (visual motion blindness). A review. Brain 114:811, 1991 542. Clarke S, Miklossy J: Occipital cortex in man: organization of callosal connections, related
myelo- and cytoarchitecture, and putative boundaries of functional visual
areas. J Comp Neurol 298:188, 1990 543. Tootell R, Taylor J: Anatomical evidence for MT and additional cortical visual areas in humans. Cerebr Cortex 5:39, 1995 544. Watson J, Myers R, Frackowiak R, et al: Area V5 of the human brain: evidence from a combined study using positron
emission tomography and magnetic resonance imaging. Cereb Cortex 3:79, 1993 545. Barton J, Simpson T, Kiriakopoulos E, et al: Functional magnetic resonance imaging of lateral occipitotemporal cortex
during pursuit and motion perception. Ann Neurol 40:387, 1996 546. Plant G, Laxer K, Barbaro N, Schiffman J, Nakayama K: Impaired visual motion perception in the contralateral hemifield following
unilateral posterior cerebral lesions in humans. Brain 116:1303, 1993 547. Greenlee M, Lang H, Mergner T, Seeger W: Visual short-term memory of stimulus velocity in patients with unilateral
posterior brain damage. J Neurosci 15:2287, 1995 548. Barton J, Sharpe J, Raymond J: Retinotopic and directional defects in motion discrimination in humans
with cerebral lesions. Ann Neurol 37:665, 1995 549. Vaina L: Selective impairment of visual motion interpretation following lesions
of the right occipito-parietal area in humans. Biol Cybern 61:347, 1989 550. Regan D, Giaschi D, Sharpe J, Hong X: Visual processing of motion-defined form: selective failure in patients
with parietotemporal lesions. J Neurosci 12:2198, 1992 551. Vaina L, Cowey A, Kennedy D: Perception of first- and second-order motion: separable neurological mechanisms? Hum Brain Mapp 7:67, 1999 552. Braun D, Petersen D, Schonle P, Fahle M: Deficits and recovery of first- and second-order motion perception in patients
with unilateral cortical lesions. Eur J Neurosci 10:2117, 1998 553. Greenlee M, Smith A: Detection and discrimination of first- and second-order motion in patients
with unilateral brain damage. J Neurosci 17:804, 1997 554. Smith A, Greenlee M, Singh K, Kraemer F, Hennig J: The processing of first- and second-order motion in human visual cortex
assessed by functional magnetic resonance imaging. J Neurosci 18:3816, 1998 555. Batelli L, Cavanagh P, Intriligator J, et al: Unilateral right parietal brain damage leads to bilateral deficit for high-level
motion. Neuron 32:985, 2001 556. Batelli L, Cavanagh P, Martini P, Barton J: Bilateral deficits of transient visual attention in right parietal patients. Brain 126:2164, 2003 557. Freitag P, Greenlee M, Lacina T, Scheffler K, Radü E: Effect of eye movements on the magnitude of functional magnetic resonance
imaging responses in extrastriate cortex during visual motion perception. Exp Brain Res 119:409, 1998 558. Haarmeier T, Thier P, Repnow M, Petersen D: False perception of motion in a patient who cannot compensate for eye movements. Nature 389:849, 1997 559. Barton J, Sharpe J: Ocular tracking of step-ramp targets by patients with unilateral cerebral
lesions. Brain 121:1165, 1998 560. Yamasaki D, Wurtz R: Recovery of function after lesions in the superior temporal sulcus in the
monkey. J Neurophysiol 66:651, 1991 561. Rizzo M, Damasio H: Impairment of stereopsis with focal brain lesions. Ann Neurol 18:147, 1985 562. Patterson R, Fox R: The effect of testing method in stereoanomaly. Vision Res 24:403, 1984 563. Critchley M: Types of visual perseveration: ‘paliopsia’ and ‘illusory
visual spread.’rsquo; Brain 74:267, 1951 564. Bender M: Polyopia and monocular diplopia of cerebral origin. Arch Neurol Psychiatry 54:323, 1945 565. Michel EM, Troost BT: Palinopsia: cerebral localization with computed tomography. Neurology 30:887, 1980 566. Meadows J: Observations on a case of monocular diplopia of cerebral origin. J Neurol Sci 18:249, 1973 567. Lopez JR, Adornato BT, Hoyt WF: ‘Entomopia’: a remarkable case of cerebral polyopia. Neurology 43:2145, 1993 568. Kinsbourne M, Warrington E: A study of visual perseveration. J Neurol Neurosurg Psychiatr 26:468, 1963 569. Bender MB, Feldman M, Sobin AJ: Palinopsia. Brain 91:321, 1968 570. Cummings JL, Syndulko K, Goldberg Z, Treiman DM: Palinopsia reconsidered. Neurology 32:444, 1982 571. Meadows JC, Munro SS: Palinopsia. J Neurol Neurosurg Psychiatry 40:5, 1977 572. Blythe IM, Bromley JM, Ruddock KH, Kennard C: A study of systematic visual perseveration involving central mechanisms. Brain 106:661, 1986 573. Swash M: Visual perseveration in temporal lobe epilepsy. J Neurol Neurosurg Psychiatr 42:569, 1979 574. Young WB, Heros DO, Ehrenberg BL, Hedges TR 3rd: Metamorphopsia and palinopsia. Association with periodic lateralized epileptiform
discharges in a patient with malignant astrocytoma. Arch Neurol 46:820, 1989 575. Muller T, Buttner T, Kuhn W, Heinz A, Przuntek H: Palinopsia as sensory epileptic phenomenon. Acta Neurol Scand 91:433, 1995 576. Lefebre C, Kolmel HW: Palinopsia as an epileptic phenomenon. Eur Neurol 29:323, 1989 577. Joseph AB: Cotard's syndrome in a patient with coexistent Capgras' syndrome, syndrome
of subjective doubles, and palinopsia. J Clin Psychiatry 47:605, 1986 578. Kawasaki A, Purvin V: Persistent palinopsia following ingestion of lysergic acid diethylamide (LSD). Arch Ophthalmol 114:47, 1996 579. McGuire P, Cope H, Fahy T: Diversity of psychopathology associated with use of 3,4-methylenedioxymethamphetamine (‘Ecstasy’). Br J Psychiatry 165:391, 1994 580. Purvin V: Visual disturbances secondary to clomiphene citrate. Arch Ophthalmol 113:482, 1995 581. Friedman D, Hu E, Sadun A: Neuro-ophthalmic complications of interleukin 2 therapy. Arch Ophthalmol 109:1679, 1991 582. Hughes MS, Lessell S: Trazodone-induced palinopsia. Arch Ophthalmol 108:399, 1990 583. Faber RA, Benzick JM: Nafazodone-induced palinopsia. J Clin Psychopharmacol 20:275, 2000 584. Ihde-Scholl T, Jefferson JW: Mitrazapine-associated palinopsia. J Clin Psychiatry 62:373, 2001 585. Terao T: Palinopsia and paroxetine withdrawal. J Clin Psychiatry 63:368, 2002 586. Johnson SF, Loge RV: Palinopsia due to nonketotic hyperglycemia. West J Med 148:331, 1988 587. Marneros A, Korner J: Chronic palinopsia in schizophrenia. Psychopathology 26:236, 1993 588. Gates TJ, Stagno SJ, Gulledge AD: Palinopsia posing as a psychotic depression. Br J Psychiatry 153:391, 1988 589. Arnold RW, Janis B, Wellman S, Crouch E, Rosen C: Palinopsia with bacterial brain abscess and Noonan syndrome. Alaska Med 41:3, 1999 590. Werring DJ, Marsden CD: Visual hallucinations and palinopsia due to an occipital lobe tuberculoma. J Neurol Neurosurg Psychiatry 66:684, 1999 591. Hayashi R, Shimizu S, Watanabe R, Katsumata Y, Mimura M: Palinopsia and perilesional hyperperfusion following subcortical hemorrhage. Acta Neurol Scand 105:228, 2002 592. Purvin V, Bonnin J, Goodman J: Palinopsia as a presenting manifestation of Creutzfeldt-Jakob disease. J Clin Neuroophthalmol 9:242, 1989 593. Cleland P, Saunders M, Rosser R: An unusual case of visual perseveration. J Neurol Neurosurg Psychiatry 44:262, 1981 594. Lazaro RP: Palinopsia: rare but ominous symptom of cerebral dysfunction. Neurosurgery 13:310, 1983 595. Jacome DE: Palinopsia and bitemporal visual extinction on fixation. Ann Ophthalmol 17:251, 1985 596. Pomeranz HD, Lessell S: Palinopsia and polyopia in the absence of drugs or cerebral disease. Neurology 54:855, 2000 597. ffytche DH, Howard RJ: The perceptual consequences of visual loss: ‘positive’ pathologies
of vision. Brain 122:1247, 1999 598. Auzou P, Parain D, Ozsancak C, Weber J, Hannequin D: [EEG recordings during episodes of palinacousis and palinopsia]. Rev Neurol (Paris) 153:687, 1997 599. Silva JA, Tekell JL, Penny G, Bowden CL: Resolution of palinopsia with carbamazepine. J Clin Psychiatry 58:30, 1997 600. Jones MR, Waggoner R, Hoyt WF: Cerebral polyopia with extrastriate quadrantanopia: report of a case with
magnetic resonance documentation of V2/V3 cortical infarction. J Neuroophthalmol 19:1, 1999 601. Gottlieb D: The unidirectionality of cerebral polyopia. J Clin Neuroophthalmol 12:257, 1992 602. Kölmel H: Colored patterns in hemianopic fields. Brain 107:155, 1984 603. Plant G: A centrally generated colored phosphene. Clin Vision Sci 1:161, 1986 604. Anderson S, Rizzo M: Hallucinations following occipital lobe damage: the pathological activation
of visul representations. J Clin Exp Neurol 16:651, 1994 605. Manford M, Andermann F: Complex visual hallucinations. Clinical and neurobiological insights. Brain 121:1819, 1998 606. Weinberger L, Grant F: Visual hallucinations and their neuro-optical correlates. Arch Ophthalmol 23:166, 1940 607. Cogan D: Visual hallucinations as release phenomenon. Albrecht von Graefe's Arch klin exp Ophthal 188:139, 1973 608. Lance J: Simple formed hallucinations confined to the area of a specific visual
field defect. Brain 99:719, 1976 609. Lepore F: Spontaneous visual phenomena with visual loss: 104 patients with lesions
of retinal and neural afferent pathways. Neurology 40:444, 1990 610. Schultz G, Melzack R: The Charles Bonnet syndrome: ‘phantom visual images.’rsquo; Perception 20:809, 1991 611. Teunisse RJ, Cruysberg JR, Hoefnagels WH, Verbeek AL, Zitman FG: Visual hallucinations in psychologically normal people: Charles Bonnet's
syndrome. Lancet 347:794, 1996 612. Ames D, Wirshing W, Szuba M: Organic mental disorders associated with bupropion in three patients. J Clin Psychiatry 53:53, 1992 613. Rivas D, Chancellor M, Hill K, Freedman M: Neurological manifestations of baclofen withdrawal. J Urol 150:1903, 1993 614. Lera G, Vaamonde J, Rodriguez M, Obeso J: Cabergoline in Parkinson's disease: long-term follow-up. Neurology 43:2587, 1993 615. Zoldan J, Friedberg G, Livneh M, Melamed E: Psychosis in advanced Parkinson's disease: treatment with ondansetron, a 5-HT3 receptor
antagonist. Neurology 45:1305, 1995 616. Chen J, Brocavitch J, Lin A: Psychiatric disturbances associated with ganciclovir therapy. Ann Pharmacother 26:193, 1992 617. Gosh K, Sivakumaran M, Murphy P, Chapman C, Wood J: Visual hallucinations following treatment with vincristine. Clin Lab Hematol 16:355, 1994 618. Bourgeois JA, Thomas D, Johansen T, Walker DM: Visual hallucinations associated with fluoxetine and sertraline. J Clin Psychopharmacol 18:482, 1998 619. Platz W, Oberlaender F, Seidel M: The phenomenology of perceptual hallucinations in alcohol-induced delirium
tremens. Psychopathology 28:247, 1995 620. Gaillard MC, Borruat FX: Persisting visual hallucinations and illusions in previously drug- addicted
patients. Klin Monatsbl Augenheilkd 220:176, 2003 621. Oliveri M, Calvo G: Increased visual cortical excitability in ecstasy users: a transcranial
magnetic stimulation study. J Neurol Neurosurg Psychiatry 74:1136, 2003 622. Lerner A, Koss E, Patterson M, et al: Concomitants of visual hallucinations in Alzheimer's disease. Neurology 44:523, 1994 623. Chapman FM, Dickinson J, McKeith I, Ballard C: Association among visual hallucinations, visual acuity, and specific eye
pathologies in Alzheimer's disease: treatment implications. Am J Psychiatry 156:1983, 1999 624. Holroyd S, Shepherd ML, Downs JH, Downs JH 3rd: Occipital atrophy is associated with visual hallucinations in Alzheimer's
disease. J Neuropsychiatry Clin Neurosci 12:25, 2000 625. Paulsen J, Salmon D, Thal L, et al: Incidence of and risk factors for hallucinations and delusions in patients
with probable AD. Neurology 54:1965, 2000 626. Ballard C, McKeith I, Harrison R, et al: A detailed phenomenological comparison of complex visual hallucinations
in dementia with Lewy bodies and Alzheimer's disease. Int Psychogeriatr 9:381, 1997 627. Harding AJ, Broe GA, Halliday GM: Visual hallucinations in Lewy body disease relate to Lewy bodies in the
temporal lobe. Brain 125:391, 2002 628. Barnes J, David AS: Visual hallucinations in Parkinson's disease: a review and phenomenological
survey. J Neurol Neurosurg Psychiatry 70:727, 2001 629. Sanchez-Ramos J, Ortoll R, Paulson G: Visual hallucinations associated with Parkinson disease. Arch Neurol 53:1265, 1996 630. Holroyd S, Currie I, Wooten G: Prospective study of hallucinations and delusions in Parkinson's disease. J Neurol Neurosurg Psychiatry 70:734, 2001 631. Klein C, Kompf D, Pulkowski U, Moser A, Vieregge P: A study of visual hallucinations in patients with Parkinson's disease. J Neurol 244:371, 1997 632. Arnulf I, Bonnet A-M, Damier P, et al: Hallucinations, REM sleep, and Parkinson's disease. Neurology 55:281, 2000 633. Nomura T, Inoue Y, Mitani H, et al: Visual hallucinations as REM sleep behavior disorders in patients with
Parkinson's disease. Mov Disord 18:812, 2003 634. Barnes J, Boubert L, Harris J, Lee A, David AS: Reality monitoring and visual hallucinations in Parkinson's disease. Neuropsychologia 41:565, 2003 635. Lepore FE: Visual loss as a causative factor in visual hallucinations associated with
Parkinson disease. Arch Neurol 54:799, 1997 636. Bullock R, Cameron A: Rivastigmine for the treatment of dementia and visual hallucinations associated
with Parkinson's disease: a case series. Curr Med Res Opin 18:258, 2002 637. Menon GJ, Rahman I, Menon SJ, Dutton GN: Complex visual hallucinations in the visually impaired: the Charles Bonnet
Syndrome. Surv Ophthalmol 48:58, 2003 638. Scott IU, Schein OD, Feuer WJ, Folstein MF: Visual hallucinations in patients with retinal disease. Am J Ophthalmol 131:590, 2001 639. Kölmel H: Complex visual hallucinations in the hemianopic field. J Neurol Neurosurg Psychiatry 48:29, 1985 640. Teunisse RJ, Cruysberg JR, Verbeek A, Zitman FG: The Charles Bonnet syndrome: a large prospective study in The Netherlands. A
study of the prevalence of the Charles Bonnet syndrome and associated
factors in 500 patients attending the University Department of Ophthalmology
at Nijmegen [see comments]. Br J Psychiatry 166:254, 1995 641. Adair D, Keshaven M: The Charles Bonnet syndrome and grief reaction. Am J Psychiatry 145:895, 1988 642. Siatkowski RM, Zimmer B, Rosenberg PR: The Charles Bonnet syndrome. Visual perceptive dysfunction in sensory deprivation. J Clin Neuroophthalmol 10:215, 1990 643. Cohen SY, Bulik A, Tadayoni R, Quentel G: Visual hallucinations and Charles Bonnet syndrome after photodynamic therapy
for age related macular degeneration. Br J Ophthalmol 87:977, 2003 644. Nesher G, Nesher R, Rozenman Y, Sonnenblick M: Visual hallucinations in giant cell arteritis: association with visual
loss. J Rheumatol 28:2046, 2001 645. Cole MG: Charles Bonnet hallucinations: a case series. Can J Psychiatry 37:267, 1992 646. Heron W: The pathology of boredom. Sci Am 196:52, 1957 647. Schultz G, Melzack R: Visual hallucinations and mental state. A study of 14 Charles Bonnet syndrome
hallucinators. J Nerv Ment Dis 181:639, 1993 648. Shedlack K, McDonald W, Laskowitz D, Krishnan K: Geniculocalcarine hyperintensities on brain magnetic resonance imaging
associated with visual hallucinations in the elderly. Psychiatry Res 54:283, 1994 649. Schwartz TL, Vahgei L: Charles Bonnet syndrome in children. J Aapos 2:310, 1998 650. Mewasingh LD, Kornreich C, Christiaens F, Christophe C, Dan B: Pediatric phantom vision (Charles Bonnet) syndrome. Pediatr Neurol 26:143, 2002 651. Fernandes LH, Scassellati-Sforzolini B, Spaide RF: Estrogen and visual hallucinations in a patient with Charles Bonnet syndrome. Am J Ophthalmol 129:407, 2000 652. ffytche DH, Howard RJ, Brammer MJ, et al: The anatomy of conscious vision: an fMRI study of visual hallucinations. Nat Neurosci 1:738, 1998 653. Wunderlich G, Suchan B, Volkmann J, et al: Visual hallucinations in recovery from cortical blindness: imaging correlates. Arch Neurol 57:561, 2000 654. Adachi N, Watanabe T, Matsuda H, Onuma T: Hyperperfusion in the lateral temporal cortex, the striatum and the thalamus
during complex visual hallucinations: single photon emission computed
tomography findings in patients with Charles Bonnet syndrome. Psychiatry Clin Neurosci 54:157, 2000 655. Assadi M, Baseman S, Hyman D: Tc SPECT scan in a patient with occipital lobe infarction and complex visual
hallucinations. J Neurosci Nurs 35:175, 2003 656. Batra A, Bartels M, Wormstall H: Therapeutic options in Charles Bonnet syndrome. Acta Psychiatr Scand 96:129, 1997 657. Bhatia MS, Khastgir U, Malik SC: Charles Bonnet syndrome. Br J Psychiatry 161:409, 1992 658. Hori H, Terao T, Shiraishi Y, Nakamura J: Treatment of Charles Bonnet syndrome with valproate. Int Clin Psychopharmacol 15:117, 2000 659. Paulig M, Mentrup H: Charles Bonnet's syndrome: complete remission of complex visual hallucinations
treated by gabapentin. J Neurol Neurosurg Psychiatry 70:813, 2001 660. Maeda K, Shirayama Y, Nukina S, Yoshioka S, Kawahara R: Charles Bonnet syndrome with visual hallucinations of childhood experience: successful
treatment of 1 patient with risperidone. J Clin Psychiatry 64:1131, 2003 661. Panayiotopoulos CP: Elementary visual hallucinations, blindness, and headache in idiopathic
occipital epilepsy: differentiation from migraine. J Neurol Neurosurg Psychiatry 66:536, 1999 662. Bien C, Benninger F, Urbach H, et al: Localizing value of epileptic visual auras. Brain 123:244, 2000 663. Penfield W, Perot P: The brain's record of auditory and visual experience. Brain 86:595, 1963 664. Panayiotopoulos C: Elementary visual hallucinations in migraine and epilepsy. J Neurol Neurosurg Psychiatry 57:1371, 1994 665. Williamson P, Thadani V, Darcey T, et al: Occipital lobe epilepsy: clinical characteristics, seizure spread patterns, and
results of surgery. Ann Neurol 31:3, 1992 666. Lance J, Smee R: Partial seizures with visual disturbance treated by radiotherapy o cavernous
hemangioma. Ann Neurol 26:782, 1989 667. Sveinbjornsdottir S, Duncan J: Parietal and occipital lobe epilepsy: a review. Epilepsia 34:493, 1993 668. Rousseau M, Debrock D, Cabaret M, Steinling M: Visual hallucinations with written words in a case of left parietotemporal
lesion. J Neurol Neurosurg Psychiatry 57:1268, 1994 669. Gastaut H: A new type of epilepsy: benign partial epilepsy of childhood with occipital
spike-waves. Clin Electroencephalogr 13:13, 1982 670. Hupp S, Kline L, Corbett J: Visual disturbances of migraine. Surv Ophthalmol 33:221, 1989 671. Lance J, Anthony M: Some clinical aspects of migraine. A prospective survey of 500 patients. Arch Neurol 15:356, 1966 672. Plant G: The fortification spectra of migraine. BMJ 293:1613, 1986 673. Richards W: The fortification illusions of migraine. Sci Am 224:89, 1971 674. Grüsser O-J.: Migraine phosphenes and the retino-cortical magnification factor. Vision Res 35:1125, 1995 675. Walker M, Smith S, Sisodiya S, Shorvon S: Case of simple partial status epilepticus in occipital lobe epilepsy misdiagnosed
as migraine: clinical, electrophysiological, and magnetic resonance
imaging characteristics. Epilepsia 36:1233, 1995 676. Schulze-Bonhage A: [Differential diagnosis of visual aura in migraine and epilepsy]. Klin Monatsbl Augenheilkd 218:595, 2001 677. Sharma K, Wahi J, Phadke RV, Varma A, Jain VK: Migraine-like visual hallucinations in occipital lesions of cysticercosis. J Neuroophthalmol 22:82, 2002 678. Lhermitte J: Syndrome de la calotte du pedoncle cerebral: Les troubles psycho-sensoriels
dans les lesions du mescephale. Rev Neurol (Paris) 38:1359, 1922 679. van Bogaert L: L'hallucinose pedonculaire. Rev Neurol (Paris) 43:608, 1927 680. Geller T, Bellur S: Peduncular hallucinosis: magnetic resonance imaging confirmation of mesencephalic
infarction during life. Ann Neurol 21:602, 1987 681. Tsukamoto H, Matsushima T, Fujiwara S, Fukui M: Peduncular hallucinosis following microvascular decompression for trigeminal
neuralgia. Surg Neurol 40:31, 1993 682. McKee A, Levine D, Kowall N, Richardson E: Peduncular hallucinosis associated with isolated infarction of the substantia
nigra pars reticulata. Ann Neurol 27:500, 1990 683. de la Fuente Fernandez R, Lopez J, Rey del Corral P, de la Iglesia Martinez F: Peduncular hallucinosis and right hemi-parkinsonism caused by left mesencephalic
infarction. J Neurol Neurosurg Psychiatry 57:870, 1994 684. Nadvi S, van Dellen J: Transient peduncular hallucinosis secondary to brain stem compression by
a medulloblastoma. Surg Neurol 41:250, 1994 685. Noda S, Mizoguchi M, Yamamoto A: Thalamic experiential hallucinosis. J Neurol Neurosurg Psychiatry 56:1224, 1993 686. Rozanski J: Peduncular hallucinosis following vertebral angiography. Neurology 2:341, 1952 687. Feinberg W, Rapcsak S: ‘Peduncular hallucinosis’ following paramedian thalamic infarction. Neurology 39:1535, 1989 688. Cascino G, Adams R: Brainstem auditory hallucinosis. Neurology 36:1042, 1986 689. Serra Catafau J, Rubio F, Peres Serra J: Peduncular hallucinosis associated with posterio thalamic infarction. J Neurol 239:89, 1992 690. Caplan L: “Top of the basilar” syndromes. Neurology 30:72, 1980 691. Dunn D, Weisberg L, Nadell J: Peduncular hallucinations cause by brainstem compression. Neurology 33:1360, 1983 692. Fisher C: Visual hallucinations and racing thoughts on eye closure after minor surgery. Arch Neurol 48:1091, 1991 693. Fisher C: Visual hallucinations on eye closure associated with atropine toxicity: a
neurologic analysis and comparison with other visual hallucinations. Can J Neurol Sci 19:18, 1991 694. Heinemann E, Tulving E, Nachmias J: The effects of oculomotor adjustments on apparent size. Am J Psychol 72:32, 1959 695. Alexander K: On the nature of accommodative micropsia. Am J Optom Physiol Opt 52:79, 1975 696. Hollins M: Does accommodative micropsia exist? Am J Psychol 89:443, 1976 697. Frisen L, Frisen M: Micropsia and visual acuity in macular edema. A study of the neuro- retinal
basis of visual acuity. Albrecht Von Graefes Arch Klin Exp Ophthalmol 210:69, 1979 698. Sjostrand J, Anderson C: Micropsia and metamorphopsia in the re-attached macula following retinal
detachment. Acta Ophthalmol (Copenh) 64:425, 1986 699. Thiébaut F, Matavulj N: Hémi-micropsie relative homonyme driote en quadrant inférieur. Rev OtoNeuroOphthalmol 21:245, 1949 700. Cohen L, Gray F, Meyrignac C, Dehaene S, Degos J-D: Selective deficit of visual size perception: two cases of hemi-micropsia. J Neurol Neurosurg Psychiatry 57:73, 1994 701. Ebata S, Ogawa M, Tanaka Y, Mitzuno Y, Yoshida M: Apparent reduction in the size of one side of the face associated with
a small retrosplenial hemorrhage. J Neurol Neurosurg Psychiatry 54:68, 1991 702. Touge T, Takeuchi H, Yamada A, Miki H, Nishioka M: [A case of posterior cerebral artery territory infarction with micropsia
as the chief complaint]. Rinsho Shinkeigaku 30:894, 1990 703. Abe K, Oda N, Araki R, Igata M: Macropsia, micropsia, and episodic illusions in Japanese adolescents. J Am Acad Child Adolesc Psychiatry 28:493, 1989 704. Wilson S: Dysmetropsia and its pathogenesis. Trans Ophthalmol Soc UK 36:412, 1916 705. Iruela L, Ibanez-Rojo V, Baca E: Zolpidem-induced macropsia in an anorexic woman. Lancet 342:443, 1993 706. Brégeat P, Klein M, Thiébaut F, et al: Hémi-macropsia homonyme droite et tumeur occipitale gauche. Rev OtoNeuroOphthalmol 21:245, 1949 707. Ardila A, Botero M, Gomez J: Palinopsia and visual allesthesia. Int J Neurosci 32:775, 1987 708. Enoch JM, Schwartz A, Chang D, Hirose H: Aniseikonia, metamorphopsia and perceived entoptic pattern: some effects
of a macular epiretinal membrane, and the subsequent spontaneous separation
of the membrane. Ophthalmic Physiol Opt 15:339, 1995 709. Amemiya T, Iida Y, Yoshida H: Subjective and objective ocular disturbances in reattached retina after
surgery for retinal detachment, with special reference to visual acuity
and metamorphopsia. Ophthalmologica 186:25, 1983 710. Ida Y, Kotorii T, Nakazawa Y: A case of epilepsy with ictal metamorphopsia. Folia Psychiatr Neurol Jpn 34:395, 1980 711. Nass R, Sinha S, Solomon G: Epileptic facial metamorphopsia. Brain Dev 7:50, 1985 712. Imai N, Nohira O, Miyata K, Okabe T, Hamaguchi K: [A case of metamorphopsia caused by a very localized spotty infarct]. Rinsho Shinkeigaku 35:302, 1995 713. Brau RH, Lameiro J, Llaguno AV, Rifkinson N: Metamorphopsia and permanent cortical blindness after a posterior fossa
tumor. Neurosurgery 19:263, 1986 714. Krizek G: Metamorphopsias caused by pontine and peduncular lesions. Am J Psychiatry 142:999, 1985 715. Palca J: Insights from broken brains. Science 248:812, 1990 716. Weinberg J, Piasetsky E, Diller L, Gordon W: Treating perceptual organization deficits in non-neglecting RBD stroke
patients. J Clin Neuropsychol 4:59, 1982 717. Carlsson G, Svardsudd K, Welin L: Long-term effects of head injuries sustained during life in three male
populations. J Neurosurg 67:197, 1987 718. Feigenson J, McCarthy M, Greenberg S, et al: Factors influencing outcome and length of stay in a stroke rehabilitation
unit. Stroke 8:657, 1977 719. Jongbloed L: Prediction of function after stroke: a critical review. Stroke 17:765, 1986 720. Kerkhoff G: Neurovisual rehabilitation: recent developments and future directions. J Neurol Neurosurg Psychiatry 68:691, 2000 |