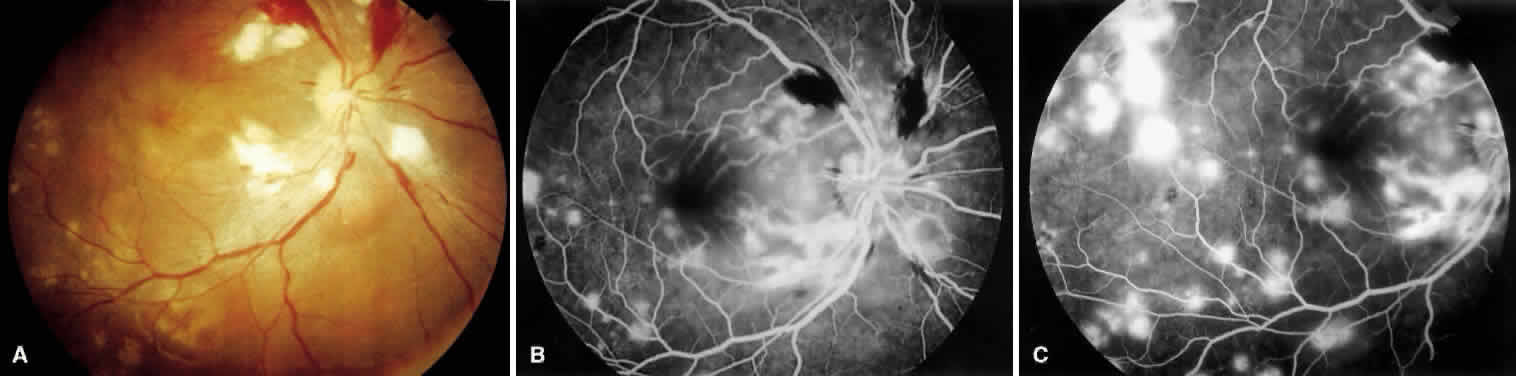
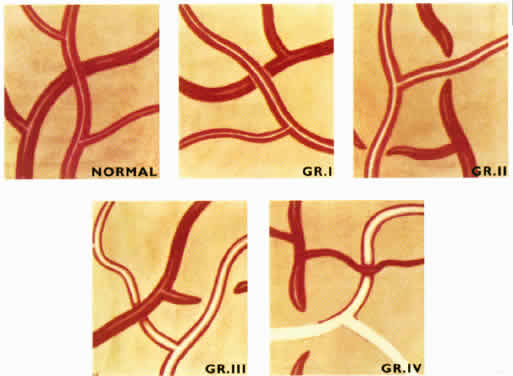
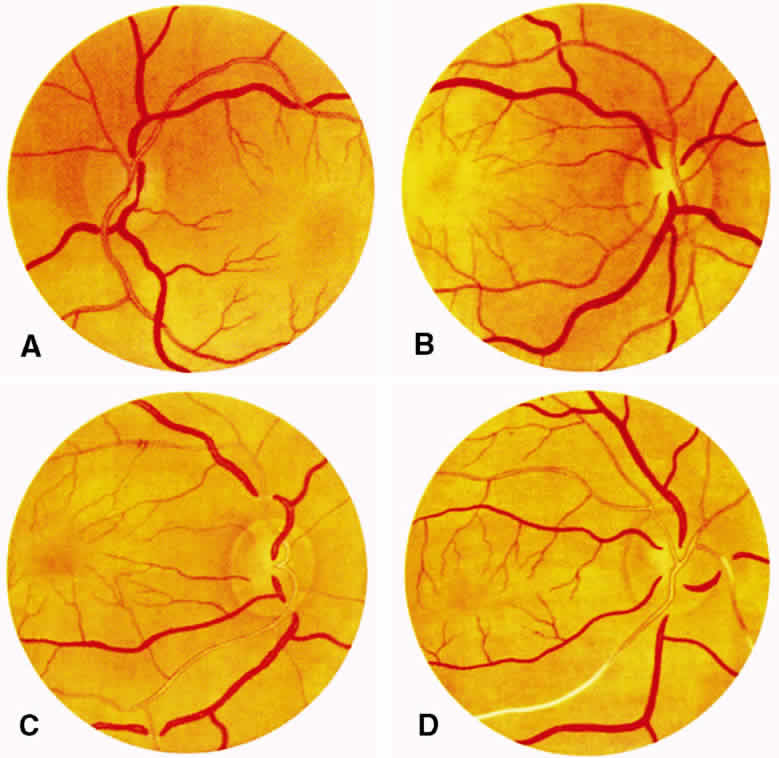
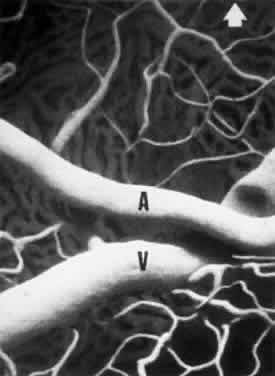
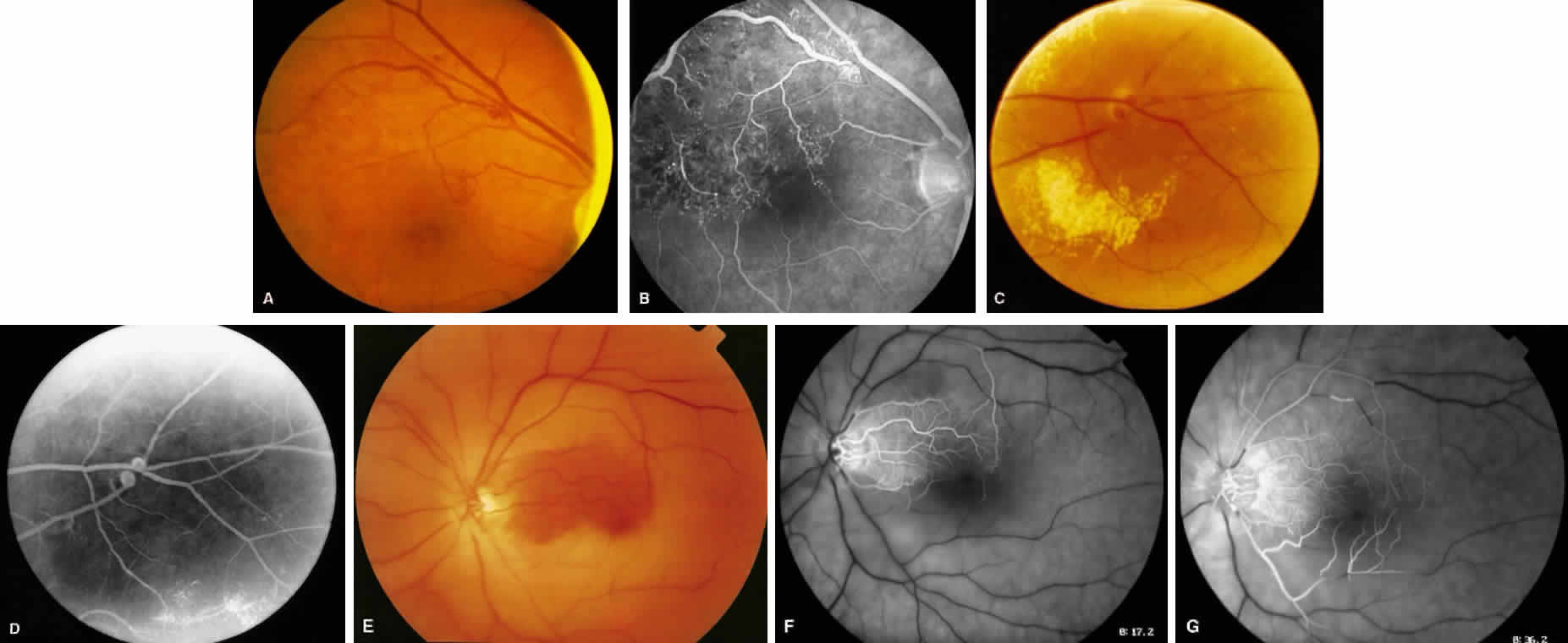
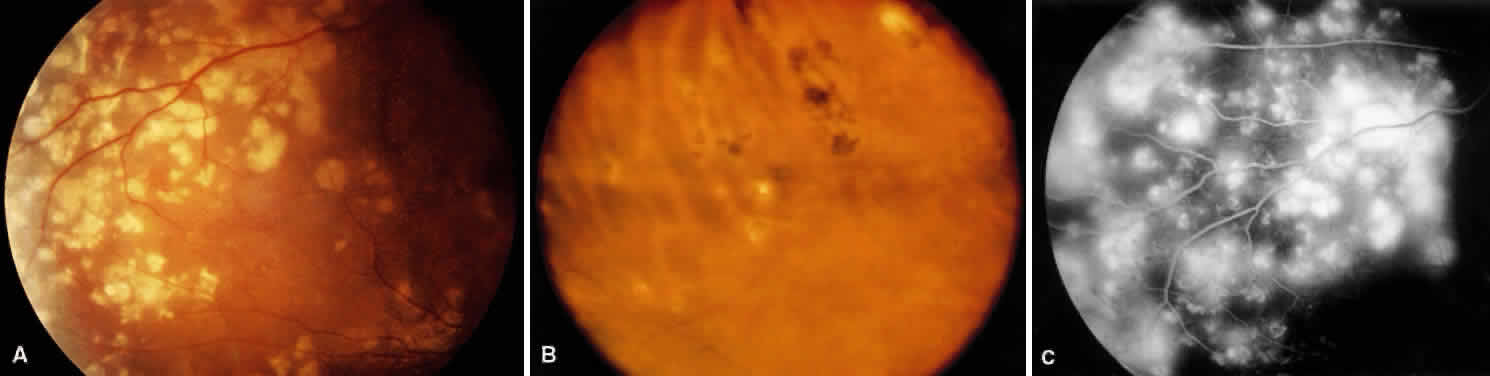
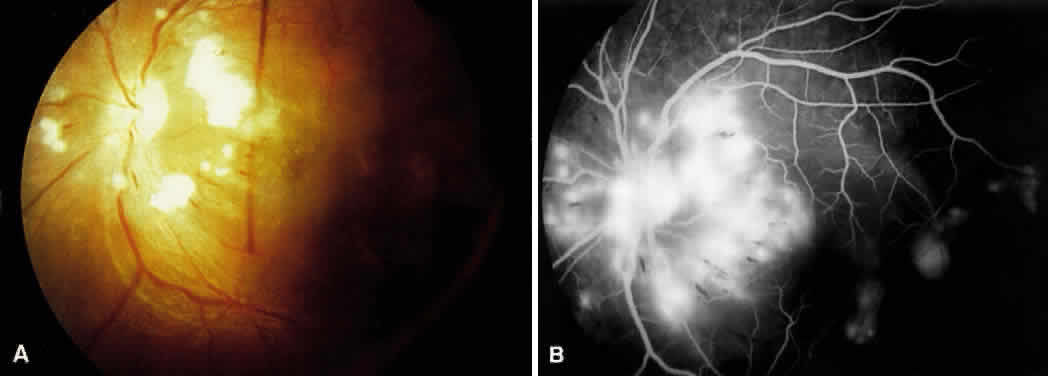
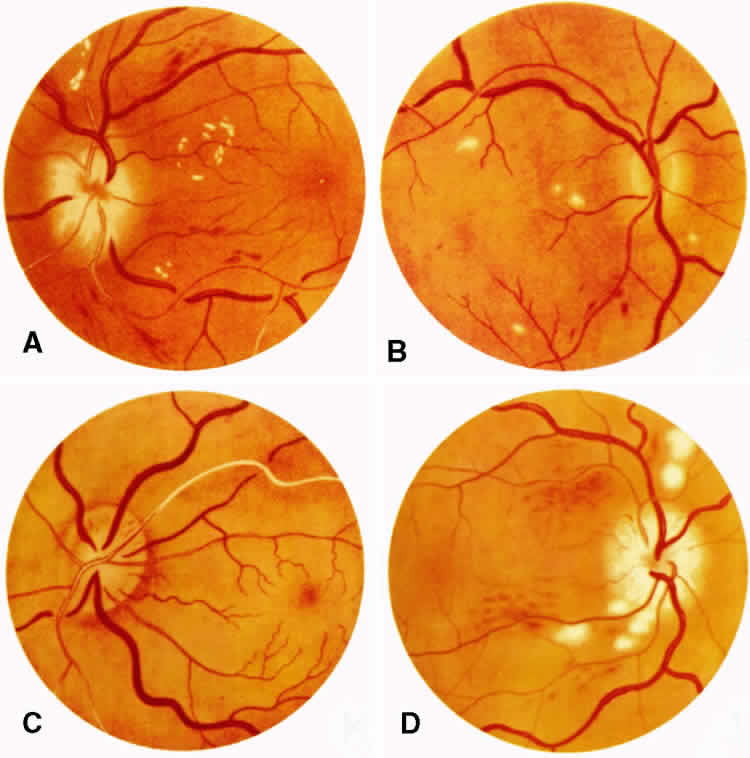
1. Burt VL, Whelton R, Roccella EJ: Prevalence of hypertension in the U.S. adult population: Results from the
Third National Health and Nutrition Examination Survey, 1988-1991. Hypertension 2S:305, 1995 2. The Joint National Committee on Detection, Evaluation and Treatment of
High Blood Pressure: The fifth report of the Joint National Committee
on detection, evaluation and treatment of high blood pressure. Arch Intern
Med 153:154, 1993 3. Garner A, Ashton N, Tripathi R et al: Pathogenesis of hypertensive retinopathy: An experimental study in the
monkey. Br J Ophthalmol 59:3, 1975 4. Hayreh SS, Servais G, Virdi PS: Fundus lesions in malignant hypertension: III. Arterial blood pressure, biochemical, and
fundus changes. Ophthalmology 93:45, 1986 5. Kishi S, Tso MOM, Hayreh SS: Fundus lesions in malignant hypertension: I. A pathologic study of experimental
hypertensive choroidopathy. Arch Ophthalmol 103:1189, 1985 6. Kishi S, Tso MOM, Hayreh SS: Fundus lesions in malignant hypertension: II. A pathologic study of experimental
hypertensive optic neuropathy. Arch Ophthalmol 103:1198, 1985 7. Leishman R: The eye in general vascular disease: Hypertension and arteriosclerosis. Br J Ophthalmol 41:641, 1957 8. Tso MOM, Jampol LM: Pathophysiology of hypertensive retinopathy. Ophthalmology 89:1132, 1982 9. Tso MOM, Abrams GW, Jampol LM: Hypertensive retinopathy, choroidopathy, and
optic neuropathy: A clinical and pathophysiological approach to classification. In
Singerman LJ, Jampol LM (eds): Retinal and Choroidal
Manifestations of Systemic Disease, pp 79–127. Baltimore: Williams & Wilkins, 1991 10. Hayreh SS, Servais G, Virdi PS: Retinal arteriolar changes in malignant arterial hypertension. Ophthalmologica 198:178, 1989 11. Hayreh SS: Duke-Elder Lecture: Systemic arterial blood pressure and the eye. Eye 10:5, 1996 12. Hayreh SS, Servais G, Virdi PS: Fundus lesions in malignant hypertension. IV. Focal intraretinal periarteriolar
transudates. Ophthalmology 93:60, 1986 13. Hayreh SS, Servais G: Retinal hemorrhages in malignant arterial hypertension. Int Ophthalmol 12:137, 1988 14. Wise GN, Dollery CT, Henkind P: The Retinal Circulation. New York: Harper
and Row, 1971 15. Ashton N: The eye in malignant hypertension. Trans Am Acad Ophthalmol Otolaryngol 76:17, 1972 16. Hayreh SS, Servais G, Virdi PS: Macular lesions in malignant arterial hypertension. Ophthalmologica 198:230, 1989 17. Hayreh SS, Servais G, Virdi PS: Fundus lesions in malignant hypertension: VI. Hypertensive choroidopathy. Ophthalmology 93:1383, 1986 18. Hayreh SS, Servais G, Virdi PS. Cotton-wool spots (inner retinal ischemic
spots) in malignant arterial hypertension. Ophthalmologica 198:197, 1989 19. Garner A: Vascular diseases. In: Garner A, Klintworth GK (eds). Pathobiology
of Ocular Disease: A Dynamic Approach, pp 1625–1710. New York: Marcel
Dekker Inc, 1994 20. Cohen SM, Gass JDM: Macular hole following severe hypertensive retinopathy. Arch Ophthalmol 112:878, 1994 21. Talks SJ, Good P, Clough CG et al: The acute and long-term ocular effects of accelerated hypertension: A clinical
and electrophysiological study. Eye 10:321, 1996 22. Klein R, Klein BEK, Moss SE et al: Hypertension and retinopathy, arteriolar narrowing, and arteriovenous nicking
in a population. Arch Ophthalmol 112:92, 1994 23. Leibowitz HM, Krueger DE, Maunder LR et al: The Framingham Eye Study monograph. Surv Ophthalmol 24:335, 1980 24. McDonough JR, Garrison GE, Harnes CG: Blood pressure and hypertensive disease among Negroes and whites: A study
in Evans County, Georgia. Ann Intern Med 61:208, 1964 25. Elschnig A: Die diagnostische und prognostische Bedeutung der Netzhauterkrankungen
bei Nephritis. Wien Med Wochenschrift 54:446, 1904 26. deVenecia G, Wallow I, Houser D et al: The eye in accelerated hypertension: I. Elschnig's spots in non human
primates. Arch Ophthalmol 98:913, 1980 27. Klien BA: Ischemic infarcts of the choroid (Elschnig spots): A cause of retinal separation
in hypertensive disease with renal insufficiency: A clinical
and histopathologic study. Am J Ophthalmol 66:1069, 1968 28. deVenecia G, Jampol LM: The eye in accelerated hypertension: II. Localized serous detachments of
the retina in patients. Arch Ophthalmol 102:68, 1984 29. Morse PH: Elschnig's spots and hypertensive choroidopathy. Am J Ophthalmol 66:844, 1968 30. Friedman E, Smith TR, Kuwabara T et al: Choroidal vascular patterns in hypertension. Arch Ophthalmol 71:842, 1964 31. MacCumber MW, Flower RW, Langham ME: Ischemic hypertensive choroidopathy: Fluorescein angiography, indocyanine
green videoangiography, and measurement of pulsatile blood flow. Arch Ophthalmol 111:704, 1993 32. Hayreh SS, Servais G, Virdi PS: Fundus lesions in malignant hypertension: V. Hypertensive optic neuropathy. Ophthalmology 93:74, 1986 33. Sossi N, Anderson DR: Blockage of axonal transport in optic nerve induced by elevation of intraocular
pressure; Effect of arterial hypertension induced by angiotensin
I. Arch Ophthalmol 101:94, 1983 34. Gunn M: On ophthalmoscopic evidence of general arterial disease. Trans Ophthalmol Soc UK 18:356, 1898 35. Keith NM, Wagener HP, Barker NW: Some different types of essential hypertension: Their course and prognosis. Am J Med Sci 197:332, 1939 36. Wagener HP, Clay GE, Gipner JF: Classification of retinal lesions in the presence of vascular hypertension. Trans Am Ophthalmol Soc 45:57, 1947 37. Scheie HG: Evaluation of ophthalmoscopic changes of hypertension and arteriolar sclerosis. Arch Ophthalmol 49:117, 1953 38. Hayreh SS: Structure and blood supply of the optic nerve. In Heilmann K, Richardson
KT (eds): Glaucoma: Conceptions of a Disease, Pathogenesis, Diagnosis, Therapy, pp 78–96. Philadelphia: WB Saunders, 1978 |  210
210