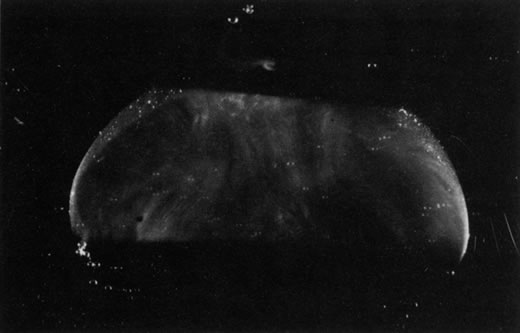
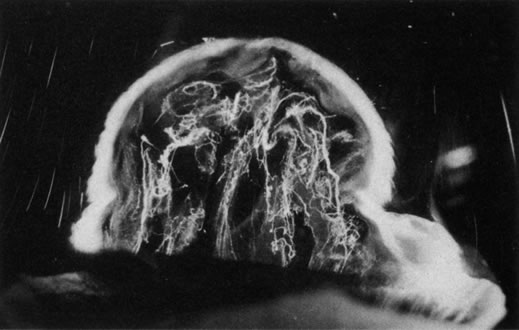
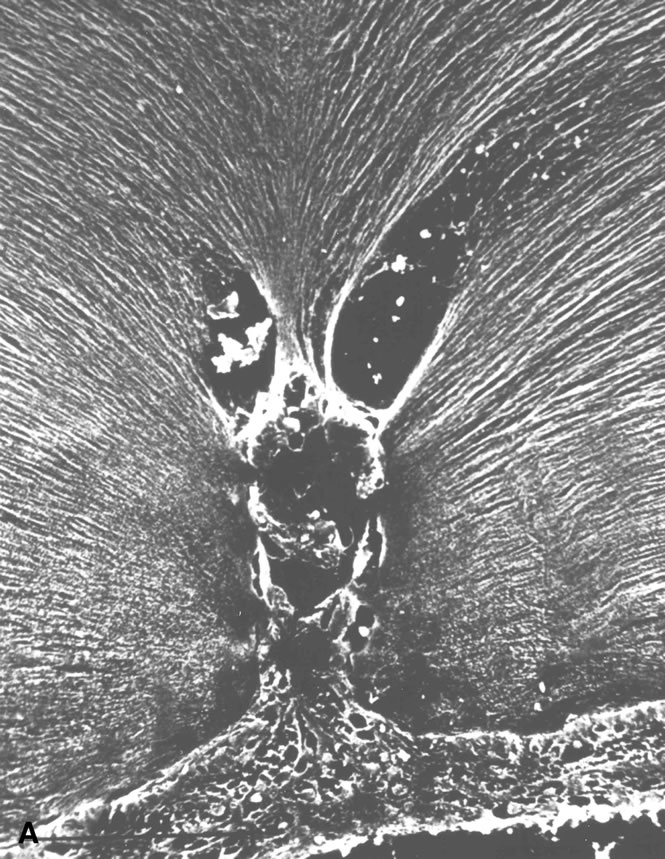
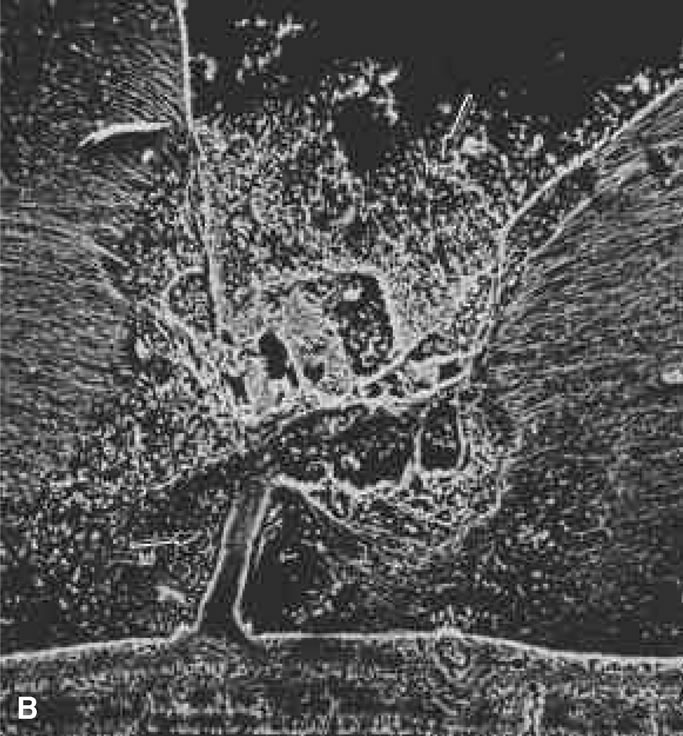
HYALOID VASCULAR SYSTEM
The embryonic vascular system of the vitreous (vasa hyaloidea propria) and lens (tunica vasculosa lentis) attains its maximum prominence during the ninth week of gestation or 40-mm stage.1 Atrophy of the vessels begins posteriorly with dropout of the vasa hyaloidea propria, followed by the tunica vasculosa lentis. Recent studies have detected the onset of apoptosis in the endothelial cells of the tunica vaculosa lentis as early as day 17.5 in the mouse embryo.2 At the 240-mm stage (seventh month) in the human, blood flow in the hyaloid artery ceases.3 Regression of the vessel itself begins with glycogen and lipid deposition in the endothelial cells and pericytes of the hyaloid vessels.3 Endothelial cell processes then fill the lumen, and macrophages form a plug that occludes the vessel. The cells in the vessel wall then undergo necrosis and are phagocytized by mononuclear phagocytes.4 Gloor5 claimed that macrophages are not involved in vessel regression within the embryonic vitreous but that autolytic vacuoles form in the cells of the vessel walls, perhaps in response to hyperoxia. Interestingly, the sequence of cell disappearance from the primary vitreous begins with endothelial and smooth muscle cells of the vessel walls, followed by adventitial fibroblasts and lastly phagocytes,6 consistent with a gradient of decreasing oxygen tension. Recent studies have suggested that the vasa hyaloidea propria and the tunica vasculosa lentis regress via apoptosis.7 These studies concluded that macrophages are important in this process. Subsequent studies by a different group confirmed the importance of macrophages in promoting regression of the fetal vitreous vasculature and further characterized these macrophages as hyalocytes.8
It is not known what stimulates regression of the hyaloid vascular system, but studies have identified a protein native to the vitreous that inhibits angiogenesis in various experimental models.9–11 Activation of this protein and its effect on the primary vitreous may be responsible for the regression of the embryonic hyaloid vascular system as well as the inhibition of pathologic neovascularization in the adult. Mitchell and colleagues12 point out that the first event in hyaloid vasculature regression is endothelial cell apoptosis and propose that lens development separates the fetal vasculature from vascular endothelial growth factor (VEGF)-producing cells, decreasing the levels of this survival factor for vascular endothelium, inducing apoptosis. Following endothelial cell apoptosis, there is loss of capillary integrity, leakage of erythrocytes into the vitreous, and phagocytosis of apoptotic endothelium by hyalocytes. Meeson and colleagues13 proposed that there are actually two forms of apoptosis that are important in regression of the fetal vitreous vasculature. The first (“initiating apoptosis”) results from macrophage induction of apoptosis in a single endothelial cell of an otherwise healthy capillary segment with normal blood flow. The isolated dying endothelial cells project into the capillary lumen and interfere with blood flow. This stimulates synchronous apoptosis of downstream endothelial cells (“secondary apoptosis”) and ultimately obliteration of the vasculature. Removal of the apoptotic vessels is achieved by hyalocytes.
Pathology
Regression of the hyaloid artery usually occurs completely and without complications. Persistence of the hyaloid vascular system occurs in 3% of full-term infants but in 95% of premature infants14 and can be associated with prepapillary hemorrhage.15 Anomalies involving incomplete regression of the embryonic hyaloid vascular system occur in more than 90% of infants born earlier than 36 weeks of gestation and in over 95% of infants weighing less than 5 lb at birth.16 There is a spectrum of disorders resulting from persistence of the fetal vasculature.17
Mittendorf's dot is a remnant of the anterior fetal vascular system located at the former site of anastomosis of the hyaloid artery and tunica vasculosa lentis. It is usually inferonasal to the posterior pole of the lens and is not associated with any known dysfunction.
Bergmeister's papilla is the occluded remnant of the posterior portion of the hyaloid artery with associated glial tissue. It appears as a gray, linear structure anterior to the optic disc and adjacent retina and does not cause any known functional disorders. Exaggerated forms can present as prepapillary veils.
Vitreous cysts are generally benign lesions that are found in eyes with abnormal regression of the anterior18 or posterior19 hyaloid vascular system; otherwise normal eyes20,21; and eyes with coexisting ocular disease, such as retinitis pigmentosa22 and uveitis.23 Some vitreous cysts contain remnants of the hyaloid vascular system,24 supporting the concept that the cysts result from abnormal regression of these embryonic vessels.25 However, one histologic analysis of aspirated material from a vitreous cyst purportedly revealed cells from the retinal pigment epithelium.26 Vitreous cysts are generally not symptomatic and thus do not require surgical intervention. However, argon laser photocoagulation has been employed, and a recent report27 described the use of neodymium:yttrium-aluminum-garnet (Nd:YAG) laser therapy to rupture a free-floating posterior vitreous cyst.
Persistent hyperplastic primary vitreous (PHPV) was first described by Reese28 as a congenital malformation of the anterior portion of the primary vitreous appearing as a plaque of retrolental fibrovascular connective tissue. This tissue is adherent to the posterior lens capsule and extends laterally to attach to the ciliary processes, which are elongated and displaced centrally. Although 90% of cases are unilateral, many of the fellow eyes have Mittendorf's dot or other anomaly of anterior vitreous development.29 A persistent hyaloid artery, often still perfused with blood, arises from the posterior aspect of the retrolental plaque in the affected eye. In severe forms, there is microphthalmos with anterior displacement of the lens-iris diaphragm, shallowing of the anterior chamber, and secondary glaucoma. PHPV is believed to arise from abnormal regression and hyperplasia of the primary vitreous.28 Experimental data suggest that the abnormality begins at the 17-mm stage of embryonic development.30 The hyperplastic features result from generalized hyperplasia of retinal astrocytes and a separate component of glial hyperplasia arising from the optic nerve head.31 The fibrous component of the PHPV membrane is presumably synthesized by these astrocytes and glial cells.32 A recent case report with clinicopathologic correlation found that collagen fibrils in this fibrous tissue had diameters of 40 to 50 nm with a cross-striation periodicity of 65 nm. The investigators concluded that the collagen fibrils differed from those of the primary vitreous and suggested that they arose either from a different population of cells or were the result of abnormal metabolism by the same cells that synthesize vitreous collagen.33
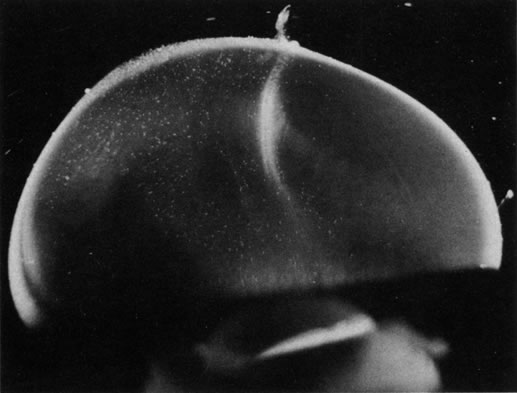
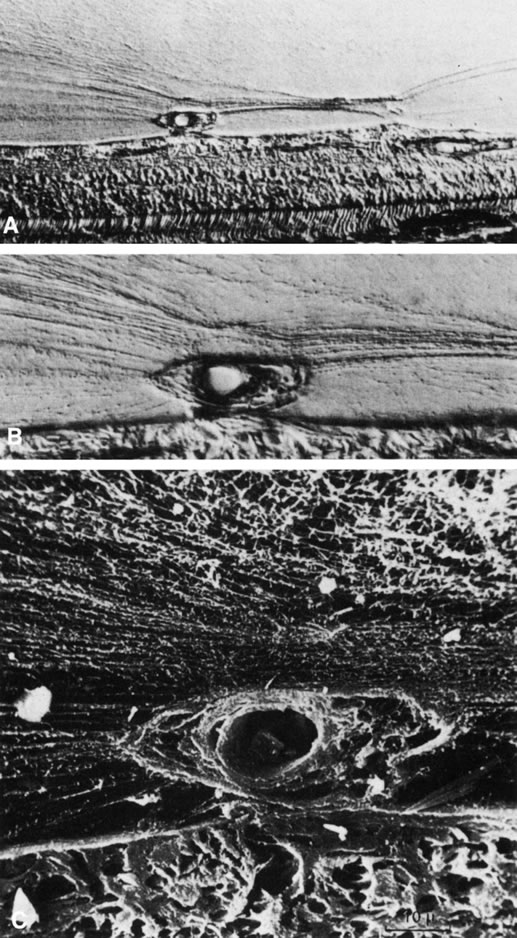
The retina is usually not involved in anterior PHPV. Indeed, previous studies have suggested that the anterior form is due to a primary defect in lens development and that vitreous changes are all secondary.34 This postulate has never been substantiated. There are rare instances of posterior PHPV in which opaque connective tissue arises from Bergmeister's papilla and persistent hyaloid vessels (Fig. 1).32,35 These can cause congenital falciform folds of the retina and, if severe, can cause tentlike retinal folds, leading on rare occasions to tractional and/or rhegmatogenous retinal detachment. Font and investigators36 demonstrated the presence of adipose tissue, smooth muscle, and cartilage within the retrolental plaque and suggested that PHPV arises from metaplasia of mesenchymal elements in the primary vitreous.
|
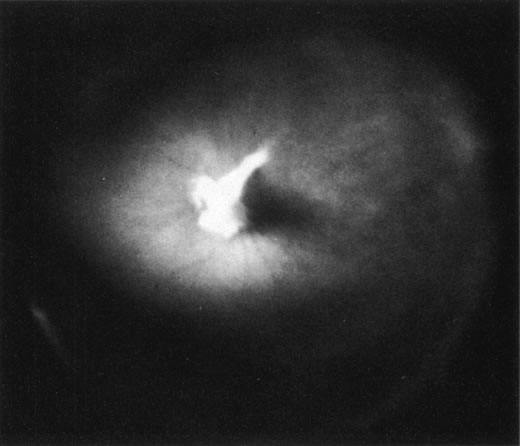
DOMINANT EXUDATIVE VITREORETINOPATHY
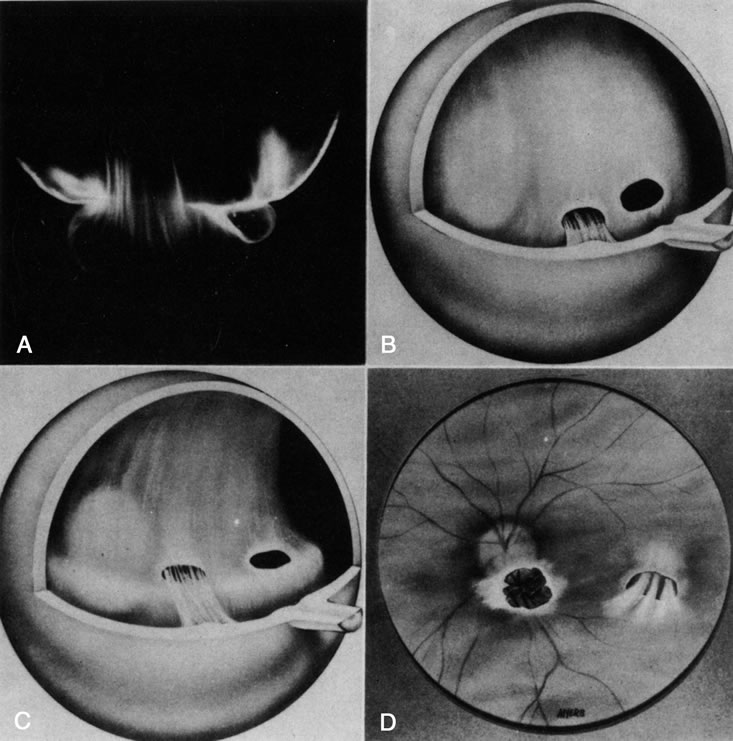
Dominant exudative vitreoretinopathy was first described in 1969 by Criswick and Schepens37 as a bilateral, slowly progressive abnormality of the vitreous and retina that resembles retinopathy of prematurity but with no history of prematurity or postnatal oxygen administration. Gow and Oliver38 identified this disorder as an autosomal dominant condition with complete penetrance. They characterized the course of this disease in stages ranging from posterior vitreous detachment with snowflake opacities (stage I), to thickened vitreous membranes and elevated fibrovascular scars (stage II), and vitreous fibrosis with subretinal and intraretinal exudates, ultimately developing retinal detachment due to fibrovascular proliferation arising from neovascularization in the temporal periphery (stage III). Plager and coworkers39 recently reported the same findings in four generations of three families, but found X-linked inheritance. Van Nouhuys40–42 studied 101 affected members in 16 Dutch pedigrees and five patients with sporadic manifestations. He found that the incidence of retinal detachment was 21%, all but one case occurring prior to the age of 30. These were all tractional or combined traction/rhegmatogenous detachments, and there were no cases of exudative retinal detachment. Van Nouhuys42 concluded that the etiology of dominant exudative vitreoretinopathy lies in premature arrest of development in the retinal vasculature, since the earliest findings in these patients were nonperfusion of the peripheral temporal retina with stretched retinal blood vessels and shunting with vascular leakage. Thus, Van Nouhuys considers dominant exudative vitreoretinopathy as a retinopathy with secondary vitreous involvement. However, Brockhurst and colleagues43 described that vitreous membrane formation begins just posterior to the ora serrata and that this precedes retinal vessel abnormalities, suggesting a vitreous origin to this disorder. Others suggested that there may be a combined etiology involving anomalies of the hyaloid vascular system and primary vitreous as well as retinovascular dysgenesis.44
RETINOPATHY OF PREMATURITY
Retinopathy of premature infants was first described in 1942 by Terry45 as “retrolental fibroplasia.” That term is no longer appropriate since it is actually a descriptive term of the pathology in advanced (stage V) cases of cicatricial retinopathy of prematurity (ROP). We now identify acute stages of the disease that are not retrolental and do not have much fibroplasia.46
The pathogenesis of ROP begins with birth, prior to complete maturation and development of the peripheral retina, followed by postnatal oxygen administration, triggering retinal vasoconstriction with endothelial cell necrosis and vaso-obliteration in response to hyperoxia.47 After the discontinuation of supplemental oxygen, arterial pO2 levels return to normal and the obliterated (or at best, highly constricted) vessels are not adequately reperfused, causing the peripheral retina they subserve to become ischemic and release neovascular growth factors. An alternative hypothesis of pathogenesis proposes that spindle cells in the immature peripheral retina are stimulated by excessive amounts of reactive oxygen species, whether related to oxygen therapy and subsequent relative hypoxia or other metabolic circumstances, to release angiogenic growth factors.48 In either case, the result is migration and proliferation of capillary endothelial cells that form new blood vessels at the posterior ridge of tissue between the vascularized and avascular retina. This results in neovascularization arising from the ridge that demarcates the developed posterior retina from the immature peripheral retina. The new vessels grow into the vitreous body, similar to neovascularization in diabetic retinopathy,49 although they grow farther anteriorly and more exuberantly. This is perhaps because of the participation of cells of the ocular fetal vasculature, whose apoptosis has been retarded or arrested by the presence of high levels of VEGF.50
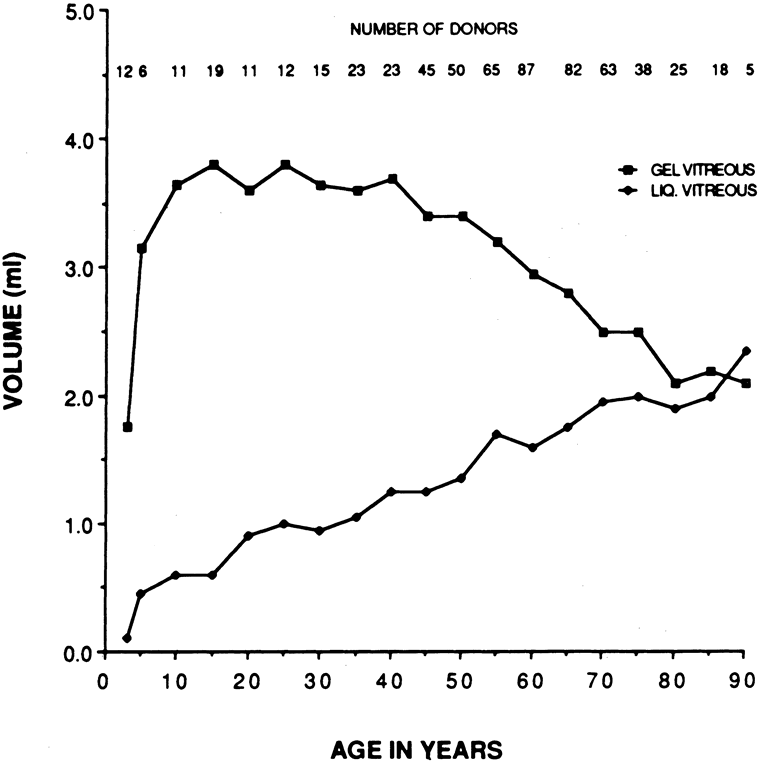
There are no clearly identified vitreous changes during stages I and II of acute ROP, although this may simply be due to our present inability to detect such abnormalities.51 Indeed, the abundance of reactive oxygen species in the retina and vitreous of premature infants could induce widespread vitreous liquefaction.52 There are also likely to be localized areas of liquid vitreous, particularly at the periphery. At surgery for stage IV-A ROP with retinal detachment, there is a “trough” in the periphery.53 This structure is most likely the consequence of underlying retinal immaturity in the periphery, with consequent lack of typical gel vitreous synthesis, normally a Müller cell function, overlying the immature retina. The liquid vitreous trough is probably present early in the natural history of disease but goes undetected by present vitreous imaging techniques.54 Such disruption of normal vitreous composition and structure probably alters a number of physiologic processes within the vitreous, including the ability of vitreous to inhibit cellular and vascular invasion.9–11 Furthermore, the interface between posterior gel vitreous and peripheral liquid vitreous at the ridge causes vitreous traction to be exerted at the retinal ridge.
In stage III ROP, new blood vessels extend from the inner retina into the vitreous cortex. The cortex, overlying the rear guard of differentiated capillary endothelial cells, becomes opaque and contains linear, fibrous structures adjacent to a large pocket of liquid vitreous.55 In advancing from stage III to stage IV, the neovascular tissue arising from the rear guard grows through the vitreous body toward Wiegert's ligament on the posterior lens capsule.56 This configuration of neovascularization is probably the result of cell migration and proliferation along the walls of the future Cloquet's canal or the tractus hyaloideus of Eisner. Cells of the primary vitreous likely contribute to the formation of the dense central vitreous stalk and retrolental membrane seen in the cicatricial stage, since these cells could also undergo migratory and proliferative responses to intraocular angiogenic stimuli.
VITREORETINAL DYSTROPHY OF GOLDMANN-FAVRE
The autosomal recessive condition vitreoretinal dystrophy of Goldmann-Favre was first described in 1957 by Goldmann57 as consisting of vitreous abnormalities with peripheral retinoschisis and chorioretinal atrophy. Francois58 characterized the vitreous changes as syneresis (collapse), fibrillar degeneration with white strand formation, and punctate deposits. The retinal changes are pigmentary degeneration (with a different appearance from the bone spicules of retinitis pigmentosa), attenuated blood vessels, and microcystic degeneration of the macula and retinal periphery. Night blindness is an important feature, and the electroretinogram is markedly abnormal. Fishman and collaborators59 used fluorescein angiography to demonstrate cystoid macular edema (CME) and vascular occlusion with leakage in the peripheral retina corresponding to the areas of schisis. Schepens60 described that there is vitreous organization attached to the areas of schisis. Histopathologic studies by Peyman and coworkers61 showed attenuation of the outer nuclear layer and absence of photoreceptors. Based on the presence of normal retinal pigment epithelium and choriocapillaris, these investigators suggested that the etiology was a primary retinal degeneration. In this regard, Müller cell dysfunction could lead to the vitreous abnormalities as well as peripheral retinoschisis. It follows that in these cases, CME results from peripheral and central vitreous changes that occur in the absence of posterior vitreous detachment, thus inducing traction on the macula, as has been noted in cases of peripheral anterior vitritis with CME62,63 and in some cases of aphakic macular edema.64–66
VITREOUS COLLAGEN DISORDERS
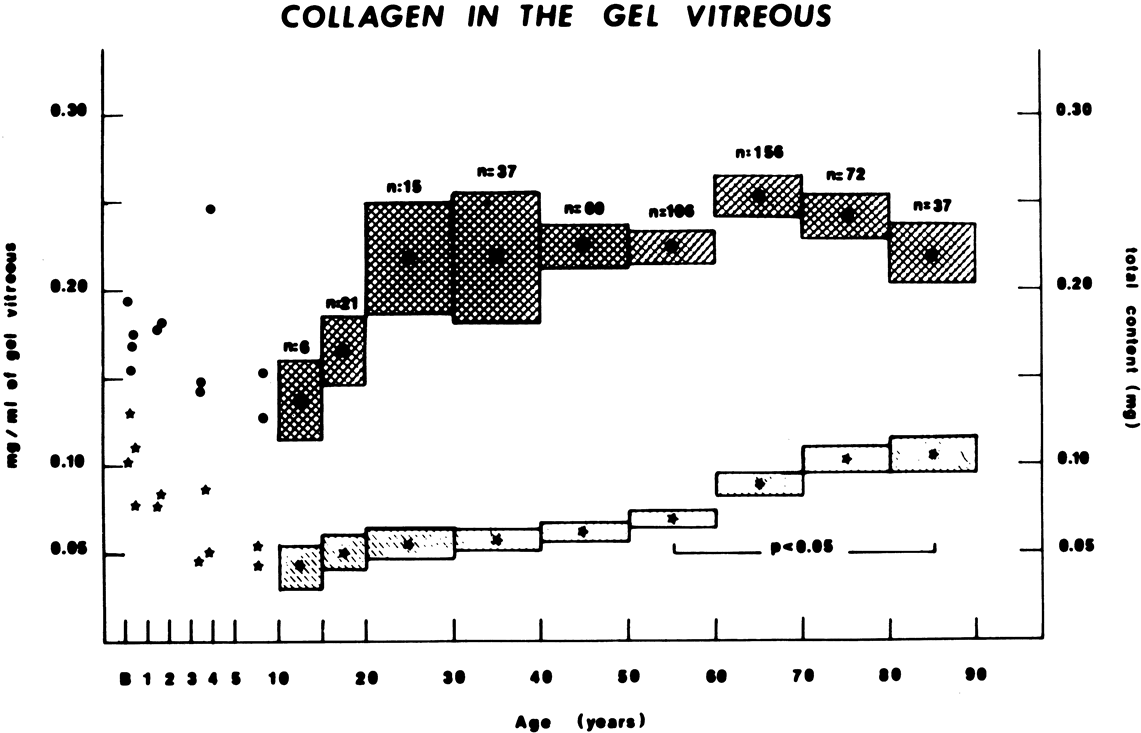
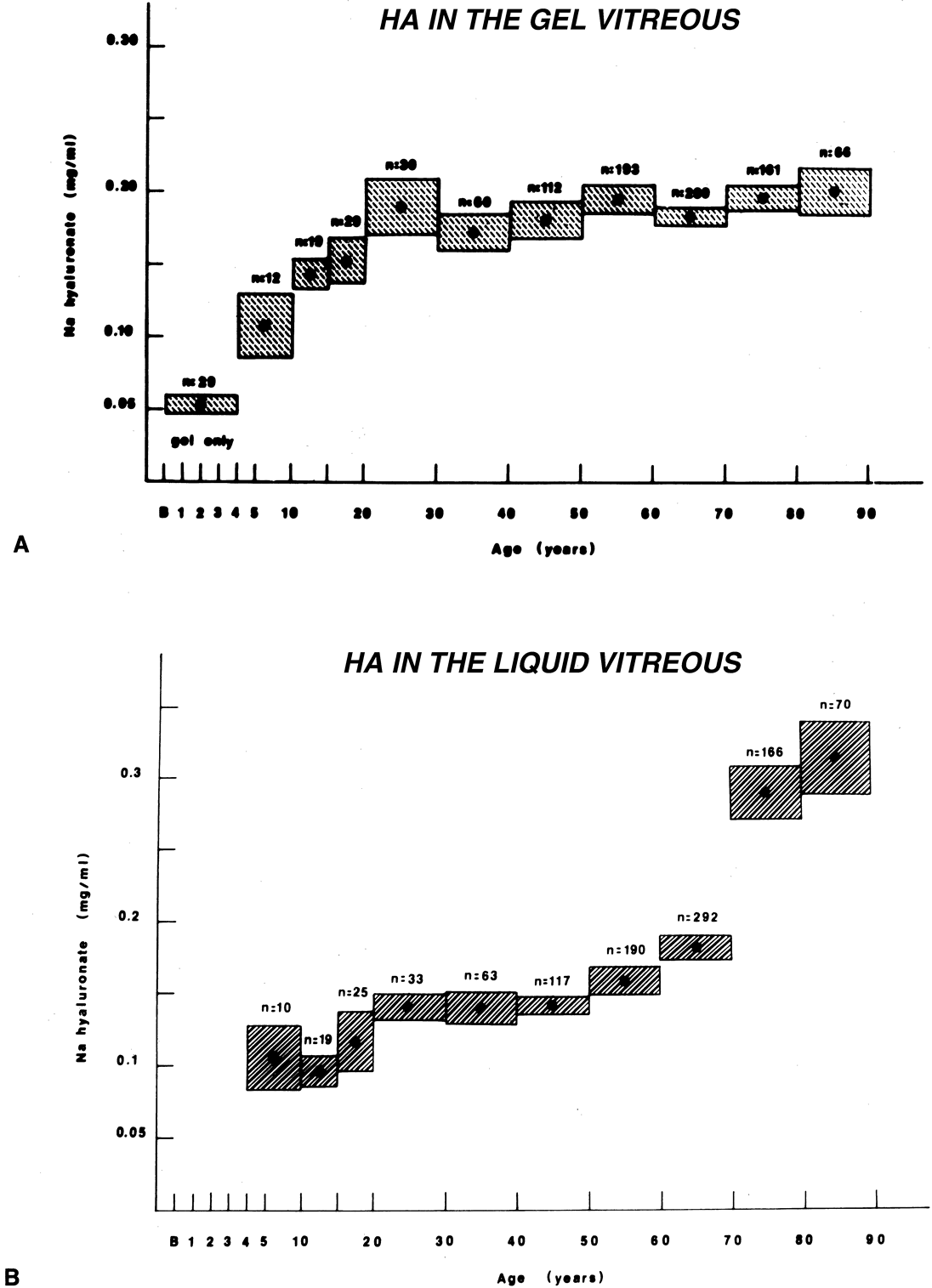
Vitreous is one of many connective tissues in the body. Collagen is one of the most important structural molecules in all connective tissues. Thus, it is of interest to consider parallel phenomena occurring in the vitreous and connective tissues elsewhere, especially as related to collagen. For example, Gartner67 pointed out the similarities between the intervertebral disk and the vitreous, in which age-related changes with herniation of the nucleus pulposus was associated with presenile vitreous degeneration in 40% of cases. He proposed that a generalized connective tissue disorder resulted in disk herniation and presenile vitreous degeneration in these cases. Based on these findings, Gartner likened herniation of the nucleus pulposus in the disk to prolapse of vitreous into the retrohyaloid space by way of the posterior vitreous cortex following posterior vitreous detachment (see Fig. 9).
|
Maumenee68 identified several different disorders with single-gene autosomal dominant inheritance in which dysplastic connective tissue primarily involves joint cartilage. In these conditions, there is associated vitreous liquefaction, collagen condensation, and vitreous syneresis (collapse). Since type II collagen is common to cartilage and vitreous, Maumenee suggested that the various arthro-ophthalmopathies may result from different mutations, perhaps of the same or neighboring genes, on the chromosome involved with type II collagen metabolism. In these disorders, probably including such conditions as Wagner's disease,69 the fundamental problem in the posterior segment of the eye is that the vitreous is liquefied and unstable, tending to syneresis at an early age. However, there is no dehiscence at the vitreoretinal interface in concert with the changes inside the vitreous body, perhaps owing to the fact that the internal limiting lamina of the retina is composed of type IV collagen. Thus, in these cases, abnormal type II collagen metabolism causes destabilization of the vitreous and results in traction on the retina that can lead to large posterior tears and difficult retinal detachments.
In Marfan's syndrome, an autosomal dominant disorder featuring poor musculature, lax joints, aortic aneurysms, and arachnodactyly, there is lens subluxation, thin sclera, peripheral fundus pigmentary changes, and vitreous liquefaction at an early age. The combination of myopia, vitreous syneresis, and abnormal vitreoretinal adhesions at the equator accounts for the frequency of rhegmatogenous retinal detachment due to equatorial or posterior horseshoe tears.60
Ehlers-Danlos syndrome has some similarities to Marfan's syndrome, most notably joint laxity, aortic aneurysms, and an autosomal dominant pattern of inheritance. However, there are as many as six types of Ehlers-Danlos patients, and the genetics of this condition are probably more heterogeneous than in Marfan's syndrome. A further distinction from Marfan's patients is the hyperelastic skin and poor wound healing of all connective tissues, including cornea and sclera, that is seen in Ehlers-Danlos patients. Ocular manifestations include lens subluxation, angioid streaks, thin sclera, and high myopia due to posterior staphyloma. Vitreous liquefaction and syneresis occur at a young age. Vitreous traction causes vitreous hemorrhage, perhaps also due to blood vessel wall fragility, and retinal tears with rolled edges, often causing bilateral retinal detachments.60
In 1965, Stickler and associates70 described a condition in five generations of a family that was found to be autosomal dominant with complete penetrance and variable expressivity. The features were a marfanoid skeletal habitus and orofacial and ocular abnormalities. Subsequent studies identified subgroups with short stature and a Weill-Marchesani habitus. The skeletal abnormalities now accepted as characteristic of Stickler's syndrome are radiographic evidence of flat epiphyses, broad metaphyses, and especially spondyloepiphyseal dysplasia.71 Ocular abnormalities are high myopia, greater than –10 diopters in 72% of cases,72 and vitreoretinal changes characterized by vitreous liquefaction, fibrillar collagen condensation, and a perivascular lattice-like degeneration in the peripheral retina believed to be the cause of a high incidence (greater than 50%) of retinal detachment.71 More recent studies correlated specific gene defects with particular phenotypes, thereby enabling the classification of Stickler syndrome patients into four subgroups.73 Patients with abnormalities in the genes coding for type II procollagen and type V/XI procollagen are the ones who have severe vitreous abnormalities.
Knobloch74 described a syndrome similar to Stickler's syndrome with hypotonia, relative muscular hypoplasia, and mild to moderate spondyloepiphysealdysplasia causing hyperextensible joints. The vitreoretinopathy is characterized by vitreous liquefaction, veils of vitreous collagen condensation, and perivascular lattice-like changes in the peripheral retina.
It is presently unknown whether myopia unrelated to the aforementioned arthro-ophthalmopathies should be considered a form of vitreous collagen disease as well. The extensive liquefaction of vitreous (myopic vitreopathy) and propensity for retinal detachment due to peripheral retinal traction and myopic peripheral retinal degeneration suggest that this postulate may deserve closer scrutiny.