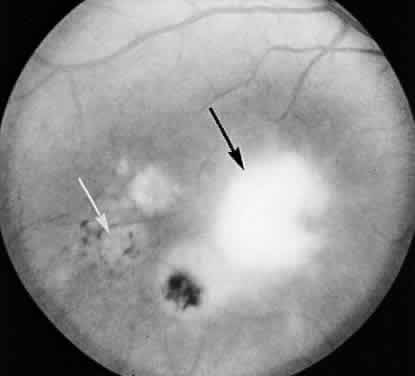
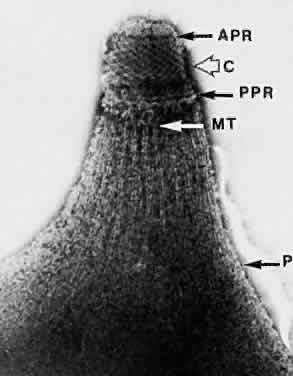
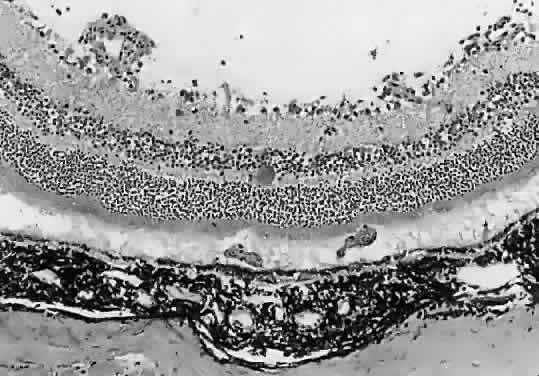
1. Jacobs L, Naquin H, Hoover R et al: A comparison of the toxoplasma skin tests, the Sabin-Feldman dye tests, and
the complement fixation tests for toxoplasmosis in various forms
of uveitis. Bull Johns Hopkins Hosp 99:1, 1956 2. Woods AC: Modern concepts of the etiology of uveitis. Am J Ophthalmol 50:1171, 1960 3. McCannel CA, Holland GN, Helm CJ et al: Causes of uveitis in the general practice of ophthalmology. Am J Ophthalmol 121:35, 1996 4. Tabarra KF. Toxoplasmosis: In Tasman W, Jaeger EA (eds): Duane's Foundations
of Clinical Ophthalmology, p 1. Vol 4. Hagerstown, MD, Harper & Row, 1996 5. Holland GN, O'Connor GR, Belfort R et al: Toxoplasmosis. In Pepose JS, Holland
GN, Wilhelmus KR (eds): Ocular Infection & Immunity, p 1183. St. Louis, Mosby-Year
Book, 1996 6. Weisz JM, Holland GN, Engstrom RE et al: The management of ocular toxoplasmosis. Surv
Ophthalmol (in press) 7. Feldman HA, Miller LT: Serological study of toxoplasmosis prevalence. Am J Hyg 64:320, 1956 8. Wallace GD: Serologic and epidemiologic observations on toxoplasmosis on three Pacific
atolls. Am J Epidemiol 90:103, 1969 9. Smith RE, Ganley JP: Ophthalmic survey of a community.1. Abnormalities of the ocular fundus. Am J Ophthalmol 74:1126, 1972 10. Glasner PD, Silveira C, Kruszon-Moran D et al: An unusually high prevalence of ocular toxoplasmosis in southern Brazil. Am J Ophthalmol 114:136, 1992 11. Desmonts G, Couvreur J: Congenital toxoplasmosis: A prospective study of 378 cases. N Engl J Med 290:1110, 1974 12. Wilson CB, Remington JS, Stagro S et al: Development of adverse sequelae in children born with subclinical congenital
toxoplasma infection. Paediatrics 66:767, 1980 13. Koppe JG, Rothova A: Congenital toxoplasmosis: A long term follow up of 20 years. Int Ophthalmol 13:387, 1989 14. Fair JR: Congenital toxoplasmosis, part IV. Case finding using the skin test and
Ophthalmoscope in state schools for mentally retarded children. Am J Ophthalmol 48:813, 1959 15. Perkins ES: Ocular toxoplasmosis. Br J Ophthalmol 57:1, 1973 16. Akstein RBV, Wilson LA, Teutsch SM: Acquired toxoplasmosis. Ophthalmology 89:1299, 1982 17. Bowie WR, King AS, Werker DH et al: Outbreak of toxoplasmosis associated with municipal drinking water. Lancet 350:173, 1997 18. Gilbert RE, Stanford MR, Jackson H et al: Incidence of acute symptomatic retinochoroiditis in South London according
to country of birth. Br Med J 310:1037, 1995 19. Friedmann CT, Knox DL: Variations in recurrent active toxoplasmic retinochoroiditis. Arch Ophthalmol 81:481, 1969 20. Hutchison WM, Dunachie JF, Siim JC et al: Coccidian-like nature of Toxoplasma gondii. Br Med J 1:142, 1970 21. Frenkel JK, Dubey JP, Miller NL: Toxoplasma gondii in cats: Fecal stages indentified as occidian cysts. Science 167:893, 1970 22. Shimada K, O'Connor GR, Yoneda C: Cyst formation by Toxoplasma gondii (RH strain) in vitro. The role of immunologic mechanisms. Arch Ophthalmol 92:496, 1974 23. Frenkel JK: Toxoplasmosis: Parasite life cycle, pathology, and immunology. In
Hammond DM, Long PL (eds): The Coccidia, p 343. Baltimore, University
Park Press, 1973 24. Fayer R: Coccidian taxonomy and nomenclature. J Protozool 28:266, 1981 25. Desmonts G, Couvreur J, Alison F et al: Etude epidemiologique sur la toxoplasmose: De l'influence de la cuisson
des boucherie sur la frequence de l'infection humaine. Rev Fr Clin Biol 10:952, 1965 26. Masur H, Lempert JA, Cherubini TD: An outbreak of toxoplasmosis in a family and ocumentation of acquired retinochoroiditis. Am J Med 64:396, 1978 27. Teutsch SM, Juranek DD, Sultzer A et al: Epidemic toxoplasmosis associated with cats. N Engl J Med 300:695, 1978 28. O'Connor GR: The mucosae as possible sites of entrance for Toxoplasma gondii. In O'Connor GR (ed): Immunology of the Mucous Membranes, p 71. New York, Masson, 1980 29. Appel BN, Mendelow H, Pasqual HN: Acquired toxoplasma lymphadenitis. Oral Surg 47:529, 1979 30. Räisänen S: Transmission of toxoplasmosis with trophozoites: An experimental study. Acta Univ Tamperensis 84: 1, 1977 31. Eichenwald H: Experimental toxoplasmosis Part I. Transmission of the infection in utero
and through the milk of lactating female mice. Am J Dis Child 76:307, 1948 32. Rieman HP, Meyer ME, Theis JH: Toxoplasmosis in an infant fed unpasteurized goat's milk. J Pediatr 87:573, 1975 33. Sacks J, Roberto RR, Brooks NF: Toxoplasmosis associated with raw goat's milk. JAMA 248:1728, 1982 34. Nichols BA, O'Connor GR: Penetration of mouse peritoneal macrophages by the protozoan Toxoplasma gondii.New evidence for active invasion and phagocytosis. Lab Invest 44:324, 1981 35. Amendoeira MMR, Coutino SG: Isolation of Toxoplasma gondii from the saliva and tonsils of a three-year-old child. J Infect Dis 145:587, 1982 36. Price JH: Toxoplasma infection in an urban community. Br Med J 4:141, 1969 37. Frenkel JK, Wallace GD: Transmission of toxoplasmosis by tachyzoites: Possibility and probability
of a hypothesis. Med Hypotheses 5:529, 1979 38. Vogel N, Krisits M, Michael E et al: Congenital toxoplasmosis transmitted from an immunologically competent
mother infected before conception. Clin Infect Dis 23:1055, 1996 39. Mets BM, Holfels E, Boyer KM et al: Eye manifestations of congenital toxoplasmosis. Am
J Ophthalmol 122:3 1996 40. Montoya JG, Reminton JS: Toxoplasmic chorioretinitis in the setting of acute acquired toxoplasmosis. Clin Infect Dis 23:277, 1996 41. Ronday MJ, Luyendijk L, Baarsma GS et al: Presumed acquired ocular toxoplasmosis. Arch Ophthalmol 113: 1524, 1995 42. Darrell RW, Kurkland LT, Jacobs L: Chorioretinopathy and toxoplasmosis: An epidemiologic study on a south
pacific island. Arch Ophthalmol 71:63, 1964 43. Silveira C, Belfort RJ, Burnier MJ et al: Acquired toxoplasmic infection as the cause of toxoplasmic retinochoroiditis
in families. Am J Ophthalmol 106:362, 1988 44. Hogan MJ: Ocular Toxoplasmosis. New York, Columbia University, 1951 45. Nozik RA, O'Connor GR: Experimental toxoplasmic retinochoroiditis. Arch Ophthalmol 79:485, 1968 46. O'Connor
GR,
Nozik
RA:
Studies on factors influencing the recurrence of ocular toxoplasmosis.
In Solanes MP (ed): Ophthalmology: Proceedings of the XXI International
Congress, Amsterdam, Excerpta Medica
222:1411,
1971 47. O'Connor GR: Experimental models of toxoplasmosis. In Tabbara KF, Cello
RM (eds): Animal Models of Ocular Diseases, p 97. Springfield, IL, Charles
C Thomas, 1984 48. Tabbara KF, Nozik RA, O'Connor GR: Clindamycin effects on experimental ocular toxoplasmosis in the rabbit. Arch Ophthalmol 92:244, 1974 49. Tabbara KF, Dy-Liacco J, Nozik RA et al: Clindamycin in chronic toxoplasmosis: Effect of periocular injections on
recoverability of organisms from healed lesions in the rabbit eye. Arch Ophthalmol 97:542, 1979 50. Culbertson WW, Tabbara KF, O'Connor GR: Experimental ocular toxoplasmosis in primates. Arch Ophthamol 100: 321, 1982 51. Newman PE, Ghosheh R, Tabbara KF et al: The role of hypersensitivity reactions to toxoplasma antigens in experimental
ocular toxoplasmosis in nonhuman primates. Am J Ophthalmol 94:159, 1982 52. Webb RM, Tabbara KF, O'Connor GR: Retinal vasculitis in ocular toxoplasmosis in nonhuman primates. Retina 4: 182, 1984 53. Holland GN, O'Connor GR, Diaz RF et al: Ocular toxoplasmosis in immunosuppressed nonhuman primates. Invest Ophthalmol Vis Sci 29:835, 1988 54. Pavesio CEN, Chiappino ML, Gormley P et al: Acquired retinochoroiditis in hamsters inoculated with ME 49 strain toxoplasma. Invest Ophthalmol Vis Sci 36:11, 1995 55. Lee WR, Hay J, Hutchison WM et al: A murine model of congenital toxoplasmic retinochoroiditis. Acta Ophthalmol 61:818, 1983 56. O'Connor GR: Uveitis and the immunologically compromised host. N Engl J Med 299:130, 1978 57. O'Connor GR: The roles of parasite invasion and of hypersensitivity in the pathogenesis
of toxoplasmic retinochoroiditis. Ocul Inflam Ther 1:37, 1983 58. Nichols BA, Chiappino ML, O'Connor GR: Secretion from the rhoptries of Toxoplasma gondii during host-cell invasion. J Ultrastruct Res 83:85, 1983 59. Russell DG, Sinden RE: The role of the cytoskeleton in the motility of coccidian sporozoites. J Cell Sci 50:345, 1981 60. de Duve C, Wattiaux R: Functions of lysosomes. Ann Rev Physiol 28:435, 1966 61. Holland GN, Engstrom RE, Glasgow BJ et al: Ocular toxoplasmosis in patients with the acquired immunodeficiency syndrome. Am J Ophthalmol 106:653, 1998 62. Kasper LH, Crabb JH, Pfefferkorn ER: Purification of a major membrane protein of Toxoplasma gondii by immunoabsorption with a monoclonal antibody. J Immunol 130:2407, 1983 63. Santoro F, Charif H, Capron A: The immunodominant epitope of the major membrane tachyzoite protein (P30) of Toxoplasma gondii. Parasite Immunol 8:631, 1986 64. Grinwood J, Smith JE: Toxoplasma gondii: The role of a 30-kDa surface protein in host cell invasion. Exp Parasitol 74:106, 1992 65. Kamiyama T, Hagiwara T: Augmented followed by suppressed levels of natural cell-mediated cytotoxicity
in mice infected with Toxoplasma. Infect Immun 36:628, 1982 66. Shirahata T, Shimizu K: Some physico-chemical characteristics of an immune lymphocyte product which
inhibits the multiplication of Toxoplasma within mouse macrophages. Microbiol Immunol 23:17, 1979 67. Beaman MH, Wong SY, Remington JS: Cytokines, Toxoplasma and intracellular parasitism. Immunol Rev 127: 97, 1992 68. Subauste CS, Remington JS: Immunity to Toxoplasma gondii. Curr Opin Immunol 5:532, 1993 69. Hunter CA, Suzuki Y, Subauste CS et al: Cells and cytokines in resistance to Toxoplasma gondii. Curr Top Microbiol Immunol 215:113, 1996 70. Langermans JA, Van der Hulst ME, Nibbering PH et al: IFN-γ-induced L-arginine-dependent toxoplamastatic activity in murine
peritoneal macrophages is mediated by endogenous tumor necrosis factor-α. J Immunol 148:568, 1992 71. Sibley LD, Adams LB, Fukutomi Y et al: Tumor necrosis factor-α triggers antitoxoplasmal activity of IFN-γ primed
macrophages. J Immunol 147:2340, 1991 72. Suzuki Y, Orellana MA, Schreiber RD et al: Intreferon-γ: The major mediator of resistance against Toxoplasma gondii. Science 240:516, 1988 73. Jones TC, Bienz KA, Erb P: In vitro cultivation of Toxoplasma gondii cysts in astrocytes in the presence of gamma interferon. Infect Immun 51:147, 1986 74. Gazzinelli RT, Brezin A, Li Q et al: Toxoplasma gondii: Acquired ocular toxoplasmosis in the murine model, protective role of
TNF-α and IFN-γ. Exp Parasitol 78:217, 1994 75. Ryning FW, Mills J: Pneumocystis carinii, Toxoplasma gondii, cytomegalovirus, and the compromised host. West J Med 130:18, 1979 76. Schuman JS: Ocular effects in the acquired immune deficiency syndrome. Mt Sinai J Med 50:443, 1983 77. Yeo JH, Jakobiec FA, Iwamoto T et al: Opportunistic toxoplasmic retinochoroiditis following chemotherapy for
systemic lymphoma: A light and electron microscopic study. Ophthalmology 90:885, 1983 78. Johnson MW, Greven CM, Jaffe GJ et al: Atypical, severe toxoplasmic retinochoroiditis in elderly patients. Ophthalmology 104:48, 1997 79. Suzuki Y, Wong WY, Grumet FC et al: Evidence of genetic regulation of susceptibilityto toxoplasmic encephalitis
in AIDS patients. J Infect Dis 173:265, 1996 80. Nussenblatt RB, Mittal KK, Fuhrman S et al: Lymphocyte proliferative responses of patients with ocular toxoplasmosis
to parasite and retinal antigens. Am J Ophthalmol 107: 632, 1989 81. Meenken C, Rothova A, de Waal LP et al: HLA typing in congenital toxoplasmosis. Br J Ophthalmol 79:494, 1995 82. Myrvik QN, Kohlweiss LA, Harpold D: Pathological potential of exaggerated
immunological reactions. In Schlessinger D (ed): Microbiology 1975, p 227. Washington
DC, American Society for Microbiology, 1975 83. Wong SY, Hajdu M, Ramirez R et al: Role of specific immunoglobulin E in diagnosis of acute toxoplasma infection
and toxoplasmosis. J Clin Microbiol 31:2952, 1993 84. Decoster A, Darcy F, Caron A et al: IgA antibodies against P30 as markers of congenital and acute toxoplasmosis. Lancet 2:1104, 1988 85. Sibalic D, Djurkovic-Djakovic O, Bobic B: Onset of ocular complications in congenital toxoplasmosis associated with
immunoglobulin M antibodies to Toxoplasma gondii. Eur J Clin Microbiol Infect Dis 9: 671, 1990 86. Holliman RE, Stevens PJ, Duffy KT et al: Serological investigation of ocular toxoplasmosis. Br J Ophthalmol 75: 353, 1991 87. Liesenfeld O, Press C, Montoya JG et al: False-positive results in immunoglobulin M (IgM) toxoplasma antibody tests
and importance of confirmatory testing: The platelia toxo IgM test. J Clin Microbiol 35:174, 1997 88. Danneman BR, Vaughan WC, Thulliez P et al: Differential agglutination test for diagnosis of recently acquired infection
with Toxoplasma gondii. J Clin Microbiol 28:1928, 1990 89. Desmonts G: Definitive serological diagnosis of ocular toxoplasmosis. Arch Ophthalmol 76:839, 1966 90. Rollins DF, Tabbara KF, O'Connor GR et al: Detection of toxoplasmal antigen and antibody in ocular fluids in experimental
ocular toxoplasmosis. Arch Ophthalmol 101: 455, 1983 91. Stur M, Grabner G, Huber-Spitzy V et al: A new quantitative fluorescence-immunoassay (FIAX) for
the detection of Toxoplasma IgG-antibodies in human aqueous humor. In Saari KM (ed): Uveitis Update, p 431. Amsterdam, Elsevier Biomedical, 1984 92. de Boer JH, Verhagen C, Bruinenberg M et al: Serologic and polymerase chain reaction analysis of intraocular fluids
in the diagnosis of infectious uveitis. Am J Ophthalmol 121:650, 1996 93. Davis JL, Feuer W, Culbertson WW et al: Interpretation of intraocular and serum antibody levels in necrotizing
retinitis. Retina 15:233, 1995 94. McHugh TD, Bathgate T, Mangan J et al: Recognition of tissue cyst-specific antigents in reactivating toxoplasmosis. J Med Microbiol 46:587, 1997 95. Rao NA, Font RL: Toxoplasmic retinochoroiditis: Electron-microscopic and immunofluorescence
studies of formalin-fixed tissue. Arch Ophthamol 95:273, 1977 96. Frenkel JK: Pathogenesis of toxoplasmosis with a considerationof cyst rupture in Besnoitia infection. Surv Ophthalmol 6:799, 1961 97. O'Connor GR: The influence of hypersensitivity on the pathogenesis of ocular toxoplasmosis. Trans Am Ophthalmol Soc 68:501, 1970 98. Wyler DJ, Blackman HJ, Lunde MN: Cellular hypersensitivity to toxoplasmal and retinal antigens in patients
with toxoplasmal retinochoroiditis. Am J Top Med Hyg 29: 1181, 1980 99. Nussenblatt RB, Gery I, Ballintine EJ: Cellular responsiveness of uveitis patients to retinal S-antigen. Am J Ophthalmol 89: 173, 1980 100. O'Connor GR: Factors related to the initiation and recurrence of uveitis: XL Edward
Jackson Memorial Lecture. Am J Ophthalmol 96:577, 1983 101. Nozik RA, O'Connor GR: Studies on experimental ocular toxoplasmosis in the rabbit. II. Attempts
to stimulate recurrence by local trauma, epinephrine, and corticosteroids. Arch Ophthalmol 84:788, 1970 102. Frenkel JK: Effect of cortisone, total body irradiation and nitrogen mustard on chronic
latent toxoplasmosis. Am J Pathol 33:618, 1957 103. Zimmerman LE: Ocular pathology of toxoplasmosis. Surv Ophthalmol 6:832, 1961 104. Kaufman HE: The effect of corticosteroid on experimental ocular toxoplasmosis. Am J Ophthalmol 50:919, 1960 105. Sibley LD, Boothroyd JC: Virulent strains of Toxoplasma gondii comprise a single clonal lineage. Nature 359:82, 1992 106. Nicholson DH, Wolchok EB: Ocular toxoplasmosis in an adult receiving long-term corticosteroid therapy. Arch Ophtahlmol 94:248, 1976 107. Engstrom RE, Holland GN, Nussenblatt RB et al: Current practices in the management of ocular toxoplasmosis. Am J Ophthalmol 111:601, 1991 108. Eyles DE, Coleman N: Synergistic effect of sulfadiazine and Daraprim against experimental toxoplasmosis
in the mouse. Antibiot Chemother 3:483, 1953 109. Sheffield HG, Melton ML: Effect of pyrimethamine and sulfadiazine on the fine structure and multiplication
of Toxoplasma gondii in cell cultures. J Parasitol 61:704, 1975 110. Thiermann E, Apt W, Atias A et al: A comparative study of some combined treatment regimens in acute toxoplasmosis
in mice. Am J Trop Med Hyg 27:747, 1978 111. Tabbara KF, O'Connor GR: Treatment of ocular toxoplasmosis with clindamycin and sulfadiazine. Ophthalmology 87:129, 1980 112. Tabbara KF, Sakuragi S, O'Connor GR: Minocycline in the chemotherapy of murine toxoplasmosis. Parasitology 84:297, 1982 113. Giles CL, Jacobs L, Melton ML: Chemotherapy of experimental toxoplasmosis: Evaluation of spiramycin alone
and in combination. Arch Ophthalmol 71:119, 1964 114. Chodos JP, Habegger-Chodos HE: The treatment of ocular toxoplasmosis with spiramycin. Arch Ophthalmol 65:401, 1961 115. Fajardo RV, Furgiule FP, Leopold IH: Treatment of toxoplasmosis uveitis. Arch Ophthalmol 67:712, 1962 116. Cassidy JV, Bahler JW, Hinkin MV: Spiramycin for toxoplasmosis. Am J Ophthalmol 57:227, 1964 117. Araujo FC, Huskinson J, Remington JS: Remarkable in vitro and in vivo activities of the hydroxynaphthoquinone 566C80 against
tachyzoites and tissue cysts of Toxoplasma gondii. Antimicrob Agents Chemother 35:293, 1991 118. Spalter HF, Campbell CJ, Noyori KS et al: Prophylactic photocoagulation of recurrent toxoplasmic retinochoroiditis. Arch Ophthalmol 75:21, 1966 119. Ghartey KN, Brockhurst RJ: Photocoagulation of active toxoplasmic retinochoroiditis. Am J Ophthalmol 89:85, 1980 120. Sabates R, Pruett RC, Brockhurst RJ: Fulminant ocular toxoplasmosis. Am J Ophthalmol 92:497, 1981 121. Wong SY, Remington JS: Toxoplasmosis in pregnancy. Clin Infect Dis 18:853, 1994 122. Alford CA, Stagno S, Reynolds DW: Congenital toxoplasmosis: Clinical, laboratory, and therapeutic considerations, with
special reference to subclinical disease. Bull NY Acad Med 50:160, 1974 123. McCauley JB, Boyer KM, Patel D et al: Early and longitudinal evaluations of treated infants and children and
untreated historical patients with congenital toxoplasmosis: The Chicago
Collaborative Treatment Trail. Clin Infect Dis 18:38, 1994 124. Work K: Resistance of Toxoplasma gondii encysted in pork. Acta Pathol Microbiol Immunol Scand 38:85, 1968 125. Darcy F, Maes P, Gras-Masse H et al: Protection of mice and nude rats against toxoplasmosis by a multiple antigenic
peptide construction derived from Toxoplasma gondii p30 antigen. J Immunol 149:3636, 1992 |