RETINITIS PIGMENTOSA (RP)
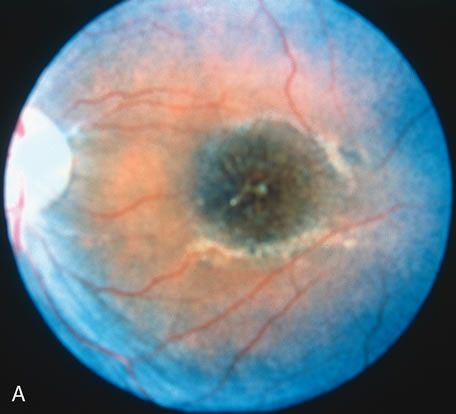
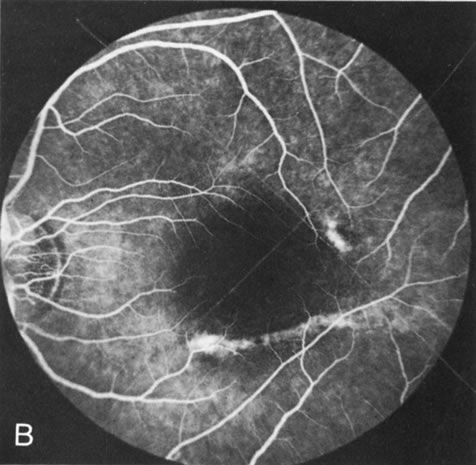
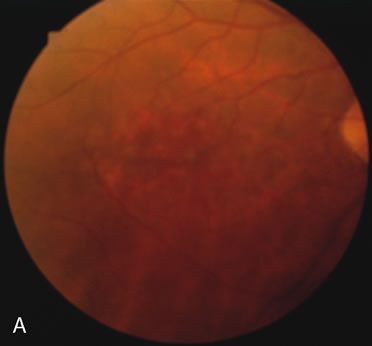
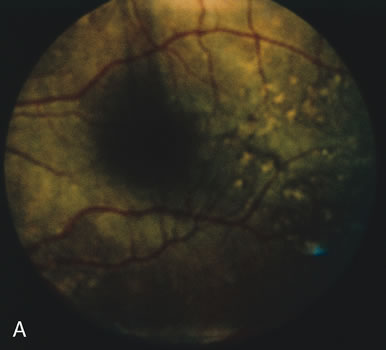
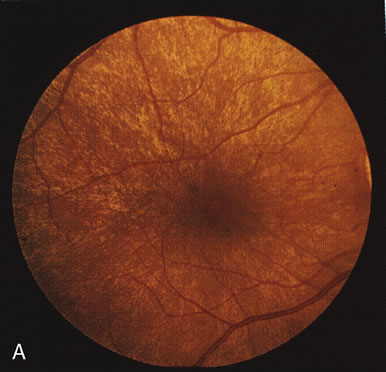
In retinitis pigmentosa (RP), the pigment abnormalities of atrophy, migration, and clumping are made apparent by transmitted hyperfluorescence and blocked hypofluorescence (Fig. 1A). Patients who have very minimal pigmentary alterations (pauci pigmentary RP) or no pigment abnormalities (RP sine pigmento) may show the abnormalities on fluorescien angiography (FA). It is uncommon to see choriocapillaris atrophy except in the late stages. This finding corresponds to the histopathology, which shows that the earliest abnormalities are in the photoreceptors and that the choroid is normal.1
|
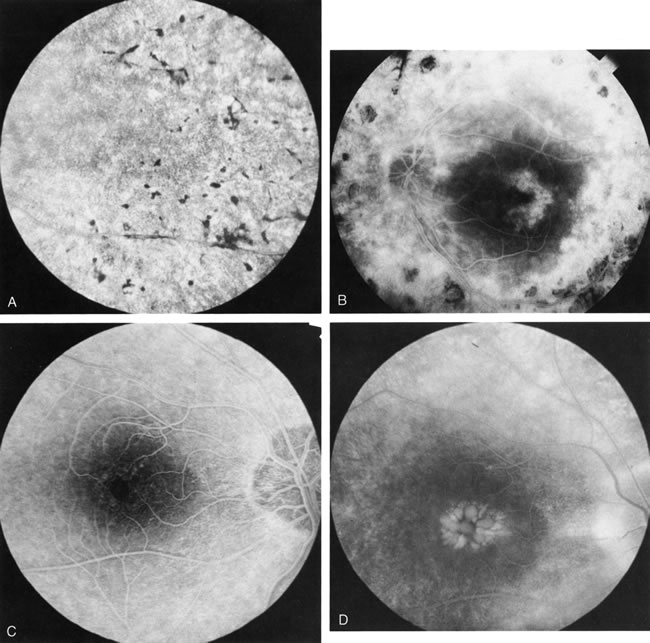
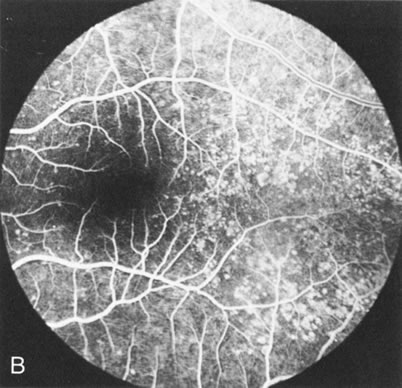
Dye leakage in RP may occur from the retinal vessels or at the level of the retinal pigment epithelium (Fig. 1B).2–4 The leakage may be seen in the macula and posterior pole, along the vascular arcades in the distribution of the radial peripapillary capillaries, and in the periphery (where an exudative vasculopathy resembling Coats' disease is suggested).
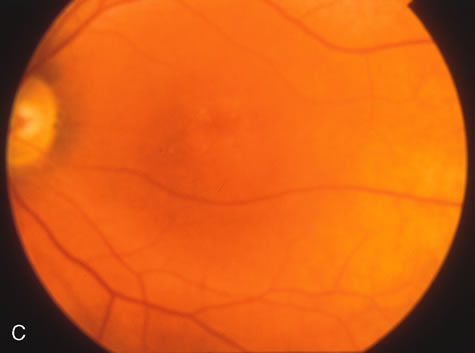
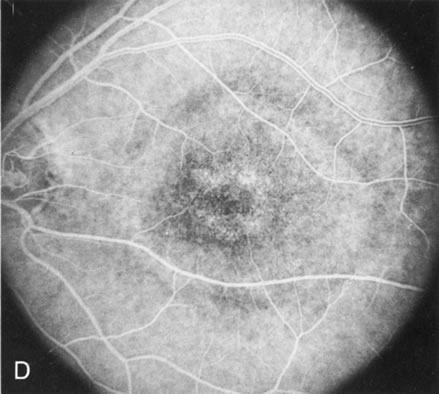
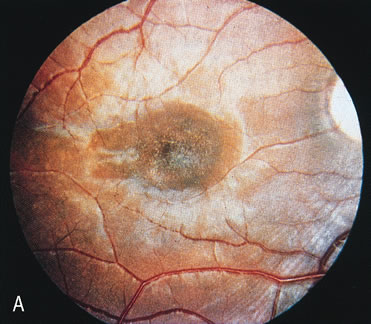
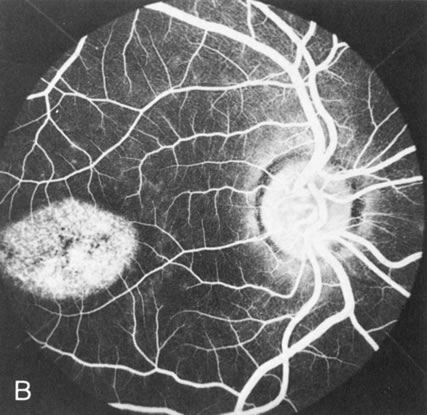
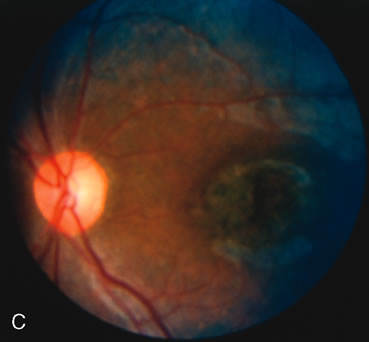
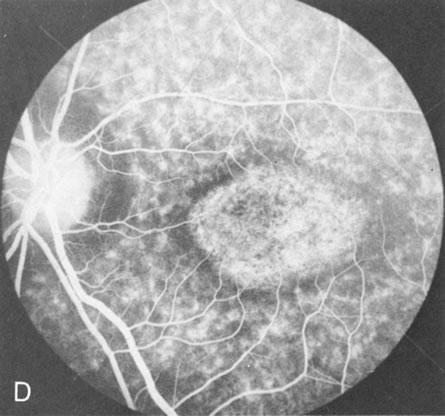
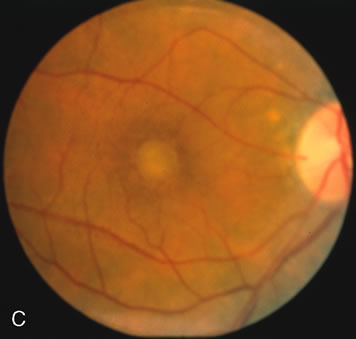
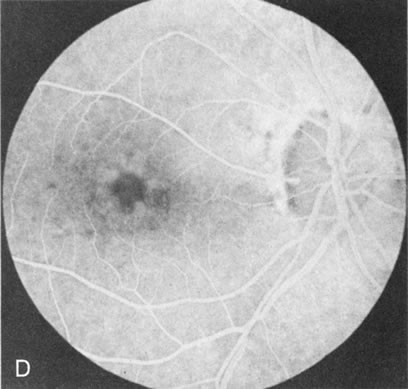
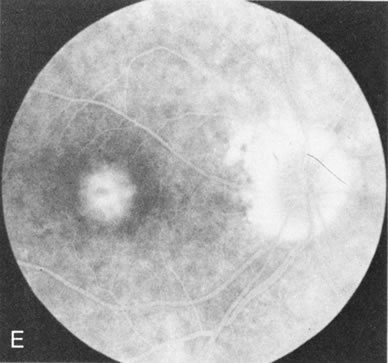
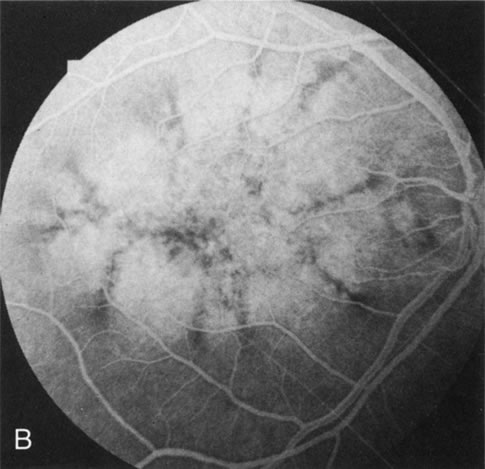
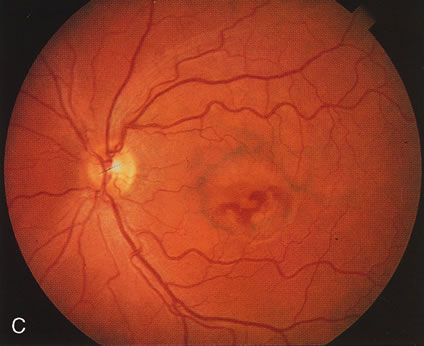
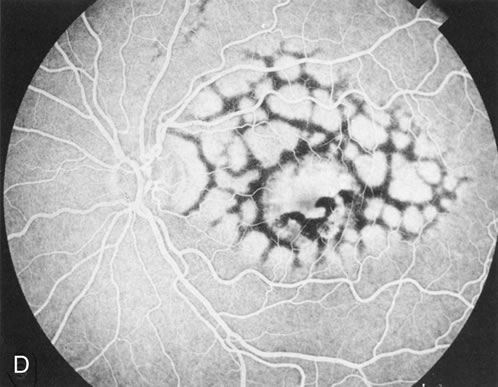
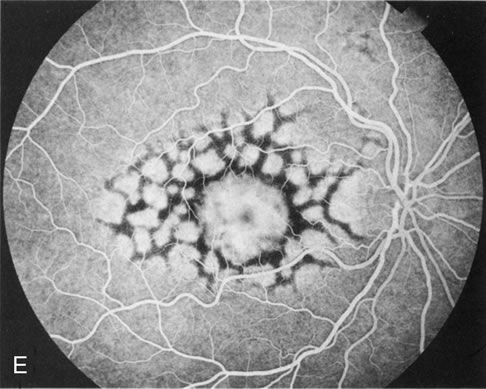
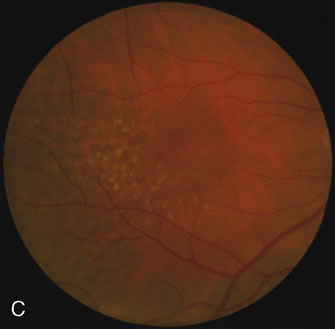
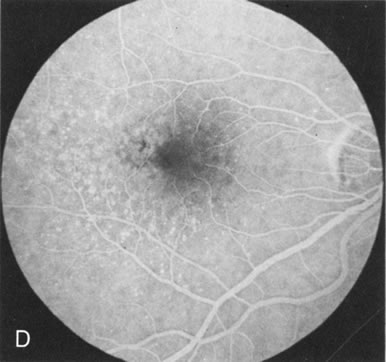
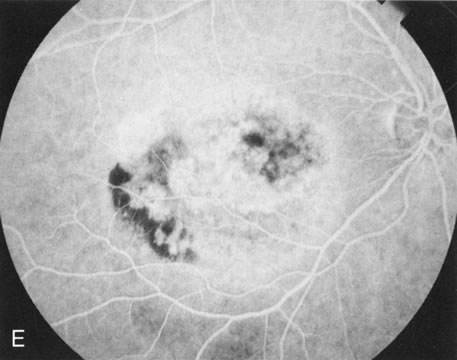
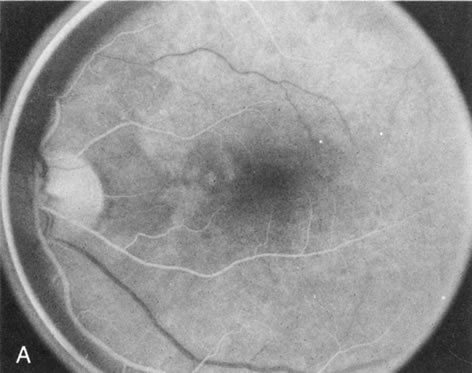
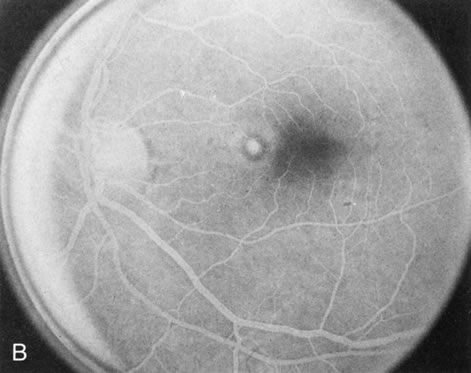
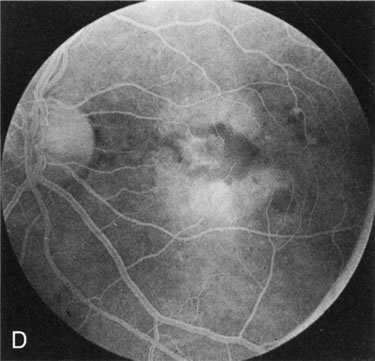
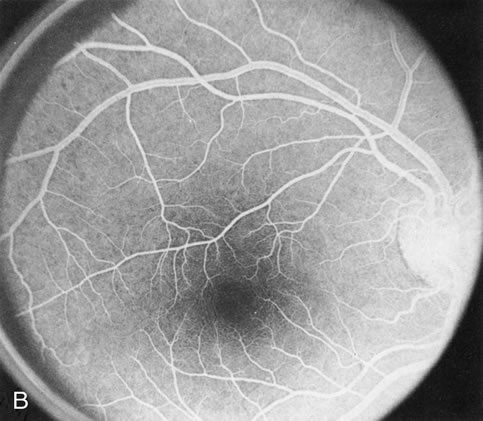
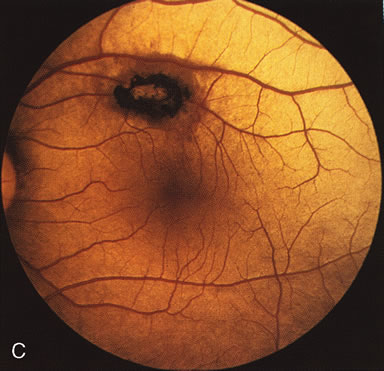
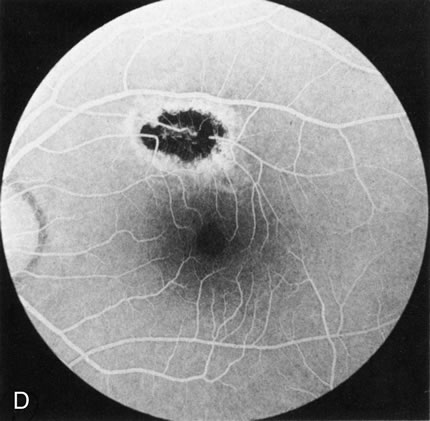
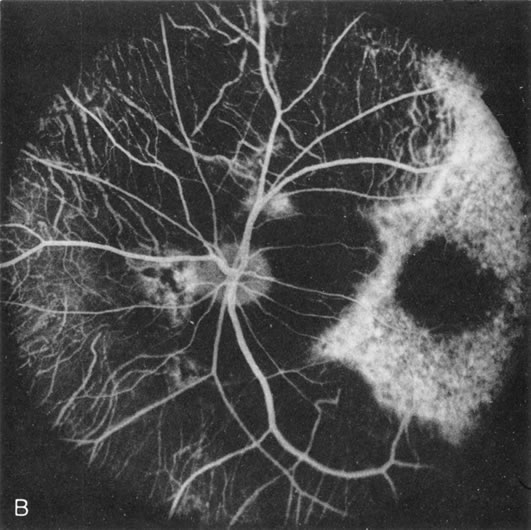
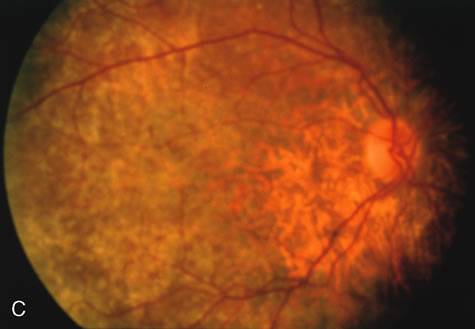
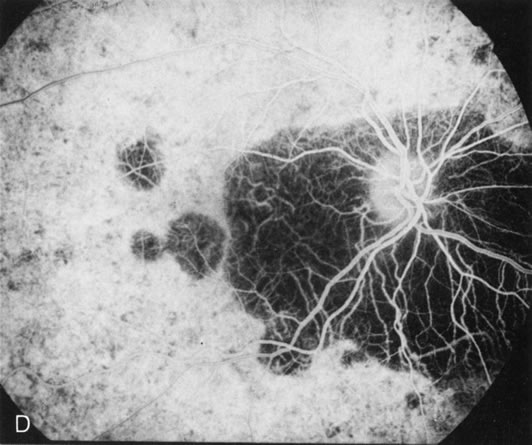
Of more clinical importance is the role of FA in the diagnosis and treatment of cystoid macular edema (CME) (Fig. 1C and D). Stereoscopic FA indicates that the leakage, which may be diffuse or have the typical petaloid stellate appearance of CME, can come from the perifoveal retinal capillaries, from the choroid through the RPE, or from a combination of both sources.4 With the recent suggestion that CME in RP may be successfully treated with acetazolamide,5, 6 FA is thus important to document the diagnosis of CME, establish the origin(s) of leakage, and follow patients during and after therapy.
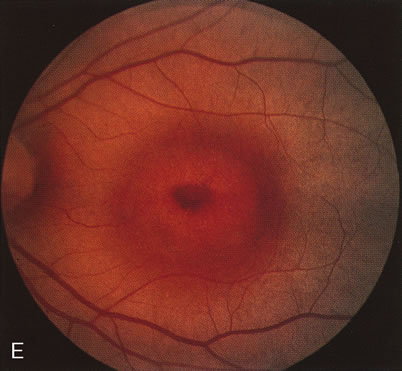
The carrier female with XLR RP, who has the golden tapetal sheen reflex, has normal FA findings. This suggests that the abnormal reflex is not due an abnormal pigment layer or deposition7 (Fig. 2A and B).
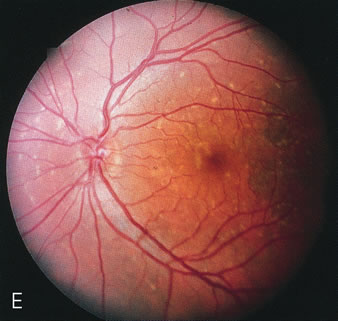
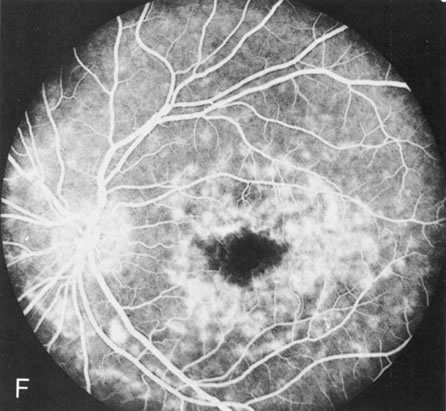
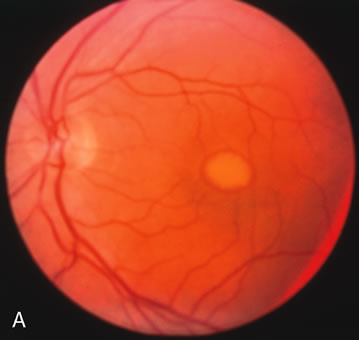
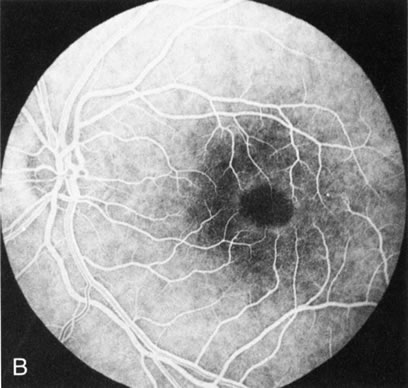
Fig. 2. Golden tapetal reflexes. There are three diseases in which there is an unusual golden reflex to the fundus: X-linked recessive RP in the carrier female, Oguchi's disease, and cone dystrophy. In all these diseases the angiogram is essentially normal, suggesting that this reflex is probably not related to pigment abnormalities. A, B. Carrier female of XLR retinitis pigmentosa. A golden scintillating reflex radiates from the macula. C, D. Oguchi's disease. A diffuse yellow metallic sheen is seen in the posterior pole (a pigmented chorioretinal lesion is an incidental finding). E, F. Progressive cone dystrophy. The typical bull's eye maculopathy is associated with a golden orange reflex. |
CONGENITAL STATIONARY NIGHT BLINDNESS
FA is normal in congenital stationary night blindness (CSNB) with a normal fundus. However, in the two types of CSNB with an abnormal fundus (Oguchi's disease, fundus albipunctatus) FA does provide some interesting information.
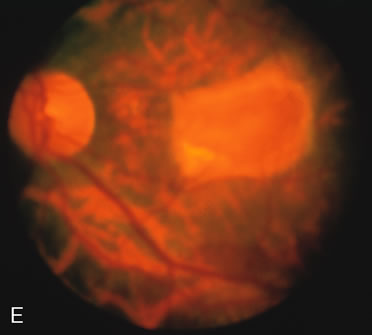
In Oguchi's disease the hallmark fundus finding is a yellow metallic sheen (Fig. 2C). A similar sheen has been seen in progressive cone dystrophies and juvenile macular degeneration.7 In all these diseases the abnormal reflex does not affect the normal transmission of fluorescein dye (Fig. 2D). The normal FA suggests that, like findings in the carrier female in XLR RP, the abnormal retinal reflex in this disease is unrelated to pigment concentration or distribution.
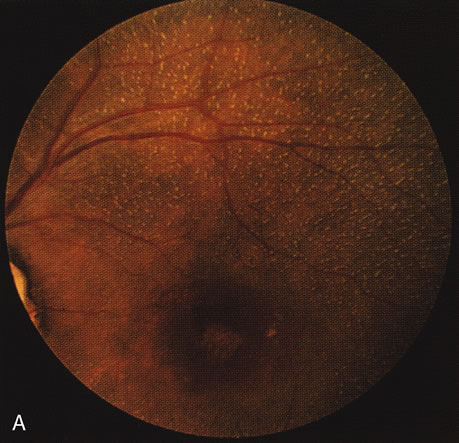
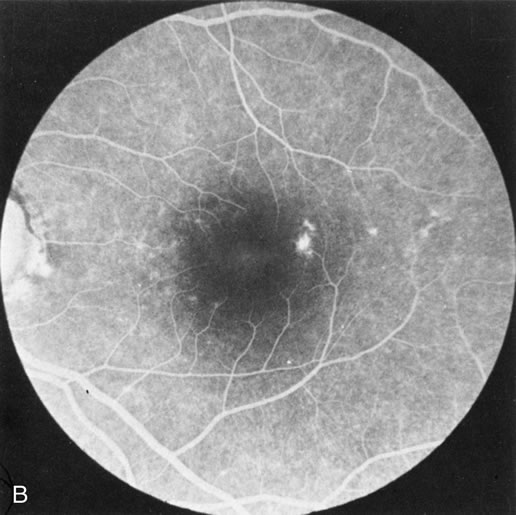
Fundus albipunctatus typically shows multiple, small white dots, which are deep in the retina, regular, and monotonous in their similar size, shape, and color and which involve the entire posterior pole into the equator while sparing the macula (Fig. 3A). These multiple dots are not apparent on FA. There may be a mottling of the background choroidal fluorescence and small areas of irregular transmission hyperfluorescence (especially surrounding the macula), but neither of these findings corresponds to the observed white dots8,9 (Fig. 3B).
|
|
CONE DYSTROPHIES
Fluorescein angiography highlights observable fundus findings. In patients with a golden reflex the FA is normal (Fig. 2E, F) or shows a mild transmission hyperfluorescence.7
Choroidal Dystrophies
The choroidal dystrophies may be generalized (choroideremia, gyrate atrophy, generalized choroidal dystrophy) or localized to the posterior pole (central areolar choroidal dystrophy, peripapillary or pericentral choroidal dystrophy, Bietti's crystalline retinopathy). The hallmark finding in all these disorders is an atrophy of the choriocapillaris early in the course of the disease. Subtle loss of the choriocapillaris is documented and confirmed by FA in which there is persistent visualization of the mid-sized choroidal vessels.10 In this regard, FA helps to differentiate disorders that affect the choroid early in the course of the disease from disorders that initially affect the retinal pigment epithelium (RPE). It is also an excellent way to determine if there is progression in the course of the disease.
Choroideremia
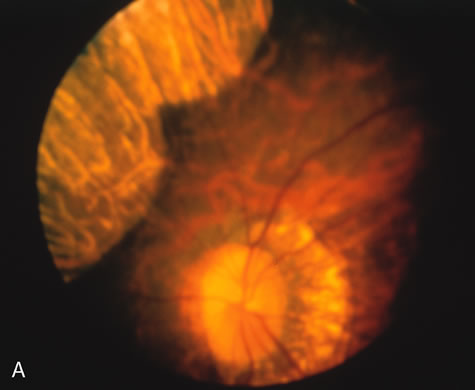
In the early stages of choroideremia, before choroidal atrophy is funduscopically obvious and when the picture resembles RP, FA indicates diffuse choroidal atrophy throughout the entire retina. Only the macular area remains preserved (Fig. 4A and B).
Fig. 4. Choroideremia and choroideremia carrier. A. The choroidal atrophy in this affected male is not apparent in the fundus. B. However, the angiogram shows diffuse atrophy of the choriocapillaris with persistent visualization of the larger choroidal vessels. C. This carrier female has peripapillary choroidal atrophy and diffuse pigment mottling. D. The patchy areas of focal choroidal atrophy that occasionally occurs in carriers is evident on angiography. |
The typical carrier female, with focal or diffuse pigment mottling, does not show choroidal atrophy. However, a few carrier females have a more severe form with focal areas of choroidal atrophy. The presence of these areas, and possible progression, can be documented by FA (Fig. 4C and D). These carriers exhibit a mosaicism, which is explained by the Lyon hypothesis of random X-chromosome inactivation.
Gyrate Atrophy
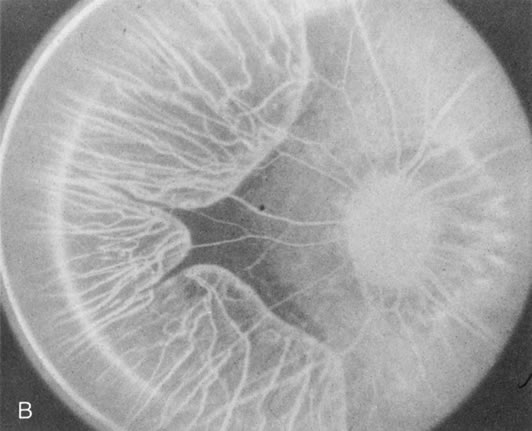
Unlike choroideremia, gyrate atrophy funduscopically has well-demarcated scalloped areas of choroidal atrophy. A hyperpigmented border separates the normal and abnormal tissue. These lesions begin as isolated areas in the midperiphery, which then merge to form a garland wreath, with progression peripherally and centrally, sparing only the macula.
FA demonstrates the sharp demarcation between normal and abnormal tissue, the former showing normal background fluorescence, the latter atrophy of the choriocapillaris (Fig. 5). Thus, the normal choriocapillaris background fluorescence in the early stages of gyrate atrophy is in contradistinction to the diffuse choriocapillaris atrophy in the early stages of choroideremia.
|
|
Generalized Choroidal Dystrophy
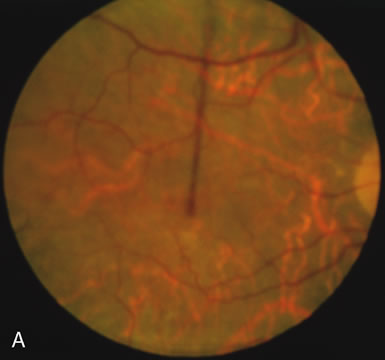
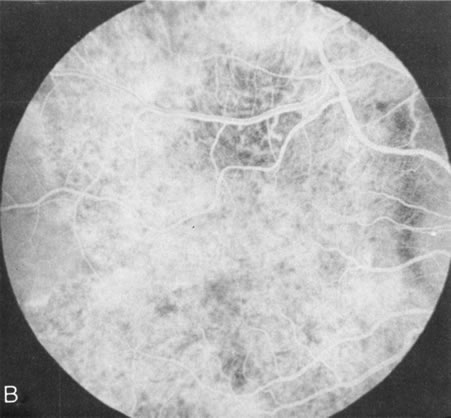
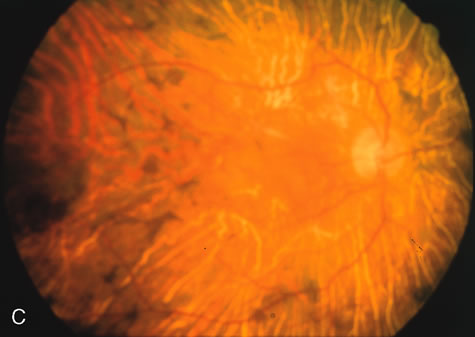
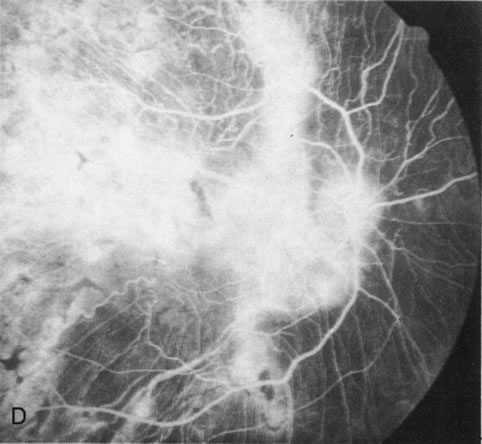
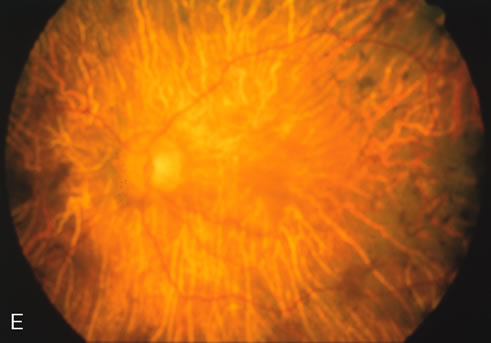
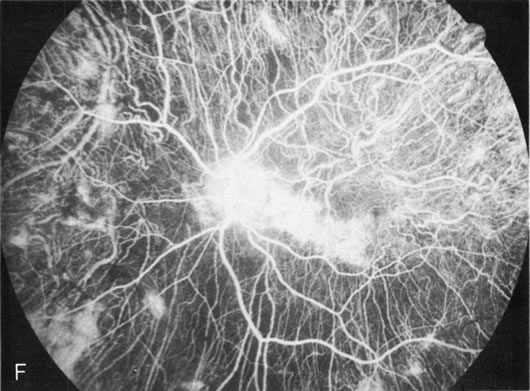
Generalized choroidal dystrophy is usually noted in middle-aged mildly symptomatic individuals who show a predominantly peripapillary or pericentral distribution of choroidal atrophy. Gradually, over the years these areas enlarge to eventually involve the entire retina. These changes are vividly seen on FA (Fig. 6).
Fig. 6. Generalized choroidal dystrophy. This 65-year-old woman gradually developed enlarging, progressive areas of choroidal atrophy over a 20-year period. When initially seen, the abnormalities were confined to the peripapillary and macular region (A,B). In a recent examination, the generalized choroidal atrophy is vividly demonstrated on angiography (C–F). |
Central Areolar Choroidal Dystrophy
Although central areolar choroidal dystrophy (CACD) and peripapillary (pericentral) choroidal dystrophy are not generalized, but rather localized disorders, they are discussed here as part of the choroidal dystrophies because the angiographic findings are similar.
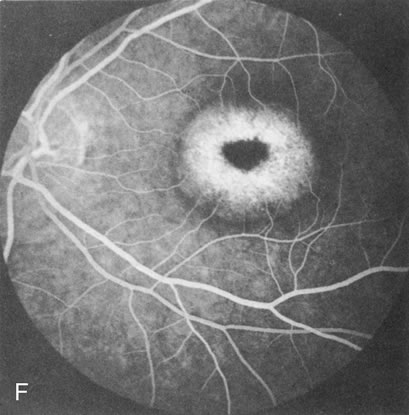
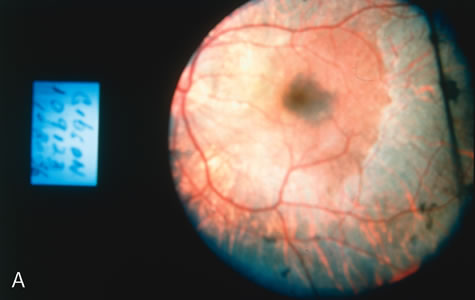
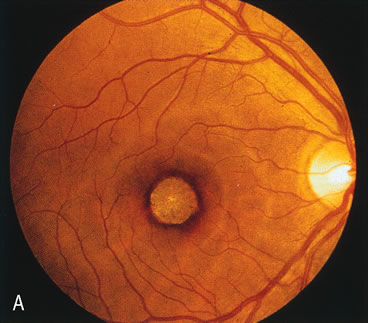
In CACD the bilateral macular lesions are solitary, circumscribed, and circular or ovoid in shape. They are unassociated with other findings such as drusen or flecks. FA will confirm the well-circumscribed area of choriocapillaris atrophy and further document that there are no associated findings that would lead to secondary choroidal atrophy in disorders such as age-related macular degeneration, Stargardt's fundus flavimaculatus, or dominant drusen of Bruch's membrane11 (Fig. 7 A and B).
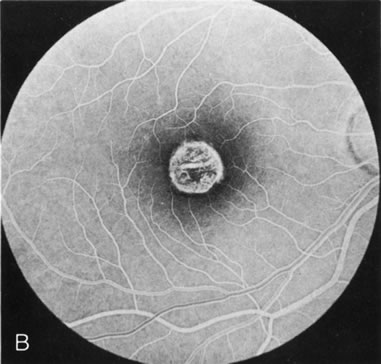
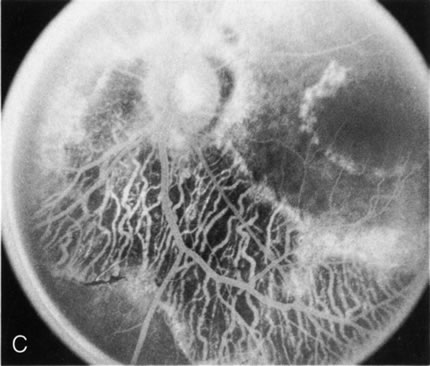
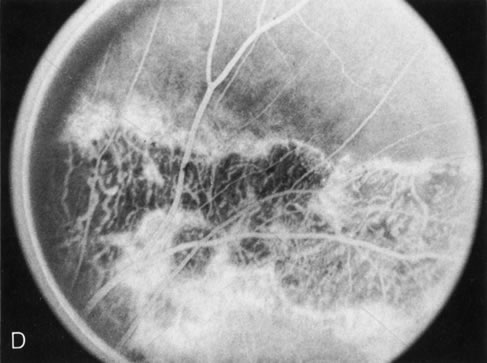
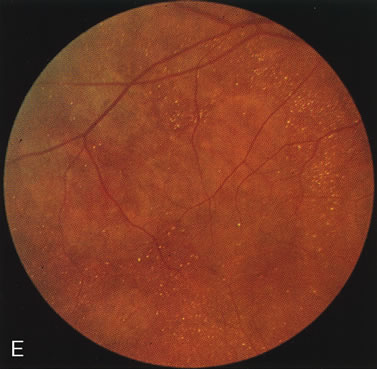
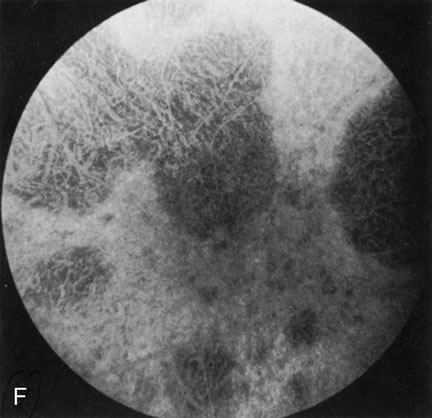
Fig. 7. Central areolar choroidal dystrophy. The presence of choroidal atrophy in this well-circumscribed macular lesion (A) is confirmed by persistent visualization of the larger choroidal vessels as seen on angiography (B). Peripapillary (pericentral) choroidal dystrophy. The areas of choroidal atrophy are well-demarcated and contrast with the areas of normal choroid (C, D). Crystalline retinopathy (of Bietti). The areas of choroidal atrophy correspond to areas of the retina where crystals are not present (E, F). |
Peripapillary (Pericentral) Choroidal Dystrophy
Peripapillary choroidal atrophy radiates from the optic nerve along the temporal vascular arcades. The macula is affected later in the course, and this is usually responsible for the onset of visual symptoms. FA shows the choroidal atrophy and the early macular changes (Fig. 7C and D).
Crystalline Retinopathy (of Bietti)
Crystalline retinopathy (of Bietti) is usually localized to the posterior pole but can be progressive. Glistening yellow crystals appear scattered throughout the retina (Fig. 7E). The angiogram has a typical and unique appearance. In the area of the crystals there is a transmitted hyperfluorescence; adjacent to these areas, where there are no crystals, choriocapillaris atrophy is evident (Fig. 7F). Progression of the disease is documented by FA,12 which shows enlarging areas of choriocapillaris atrophy.