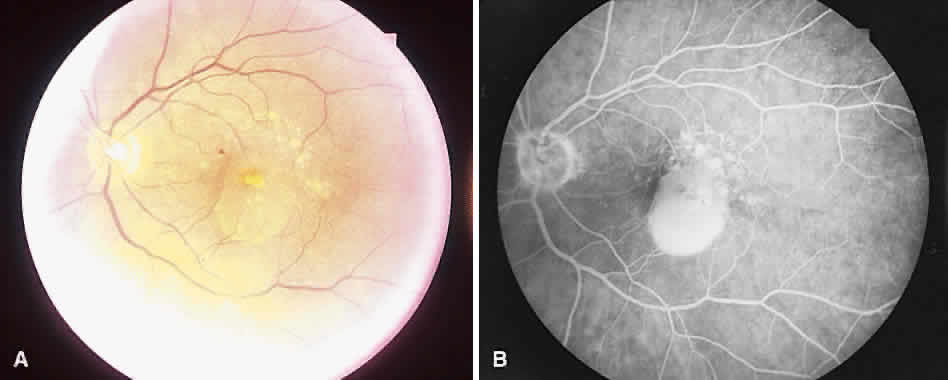
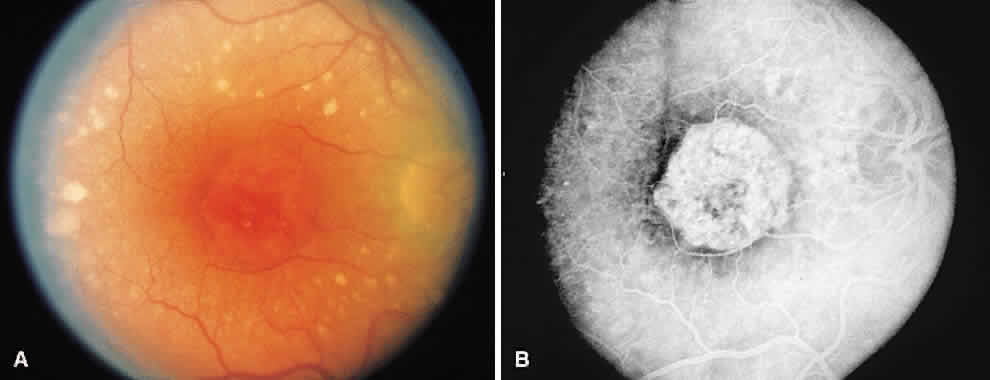
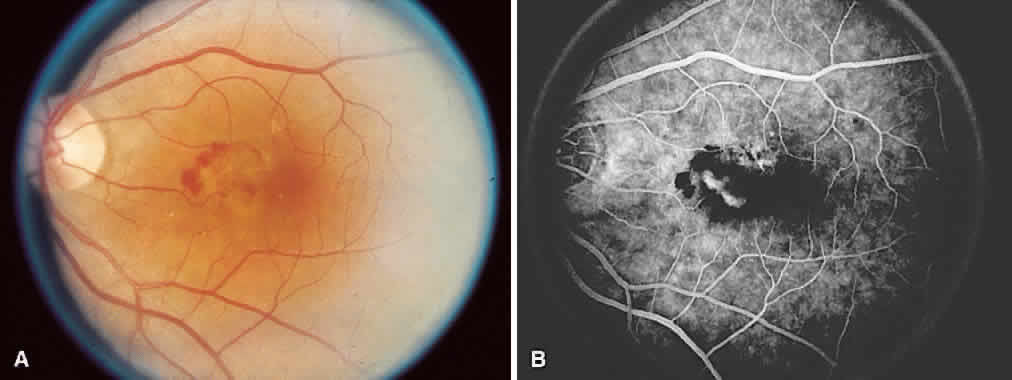
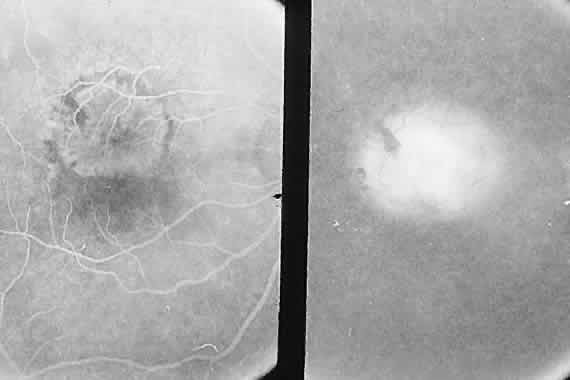
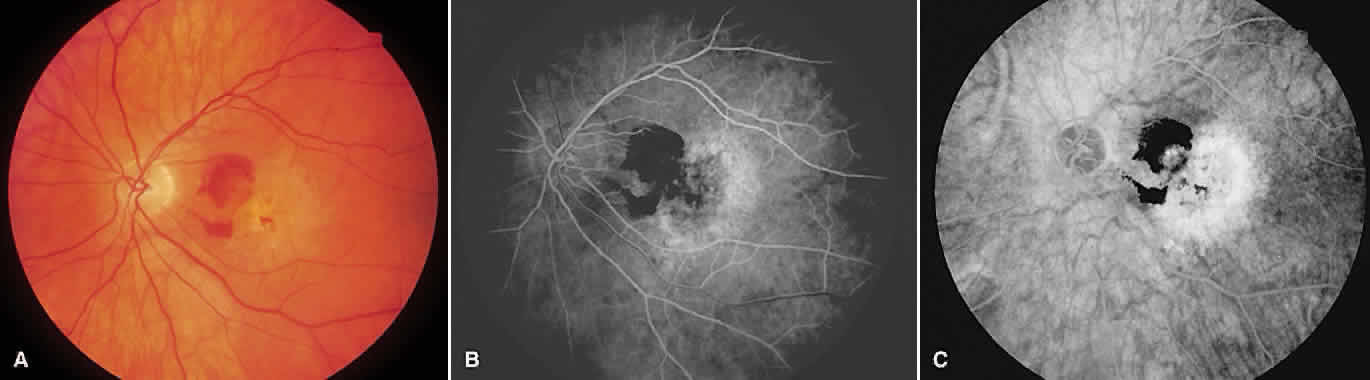
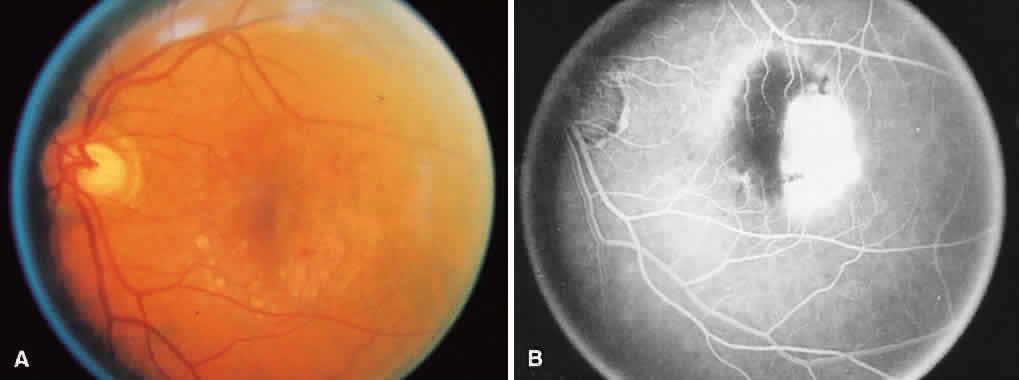
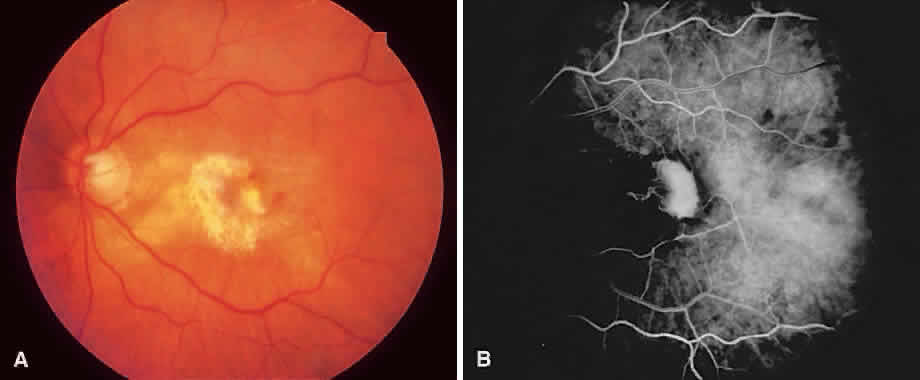
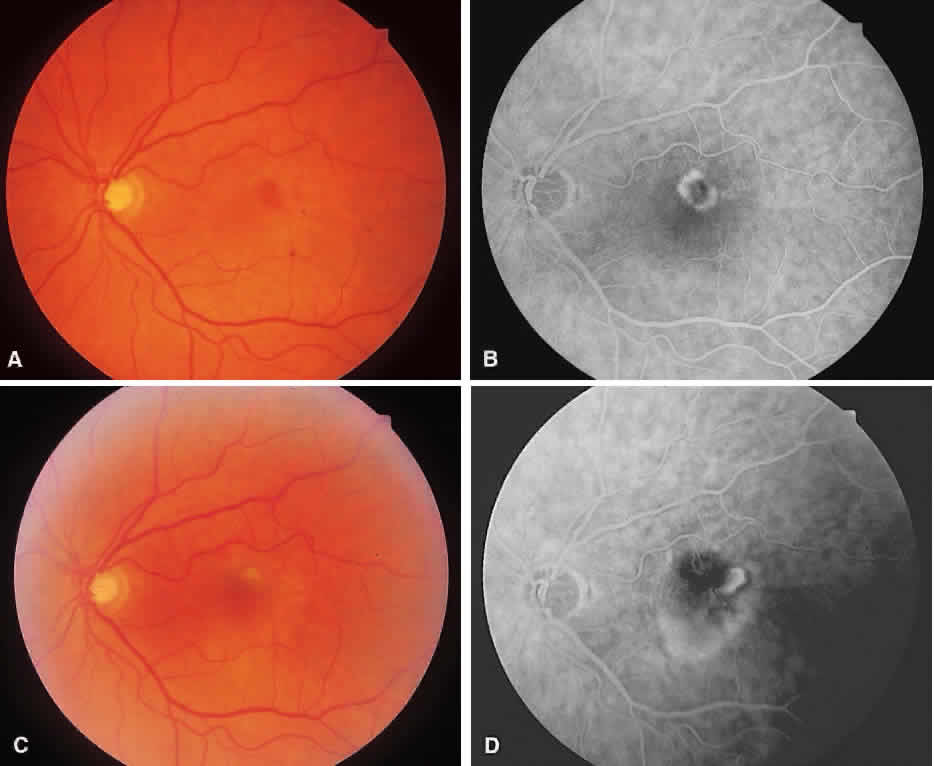
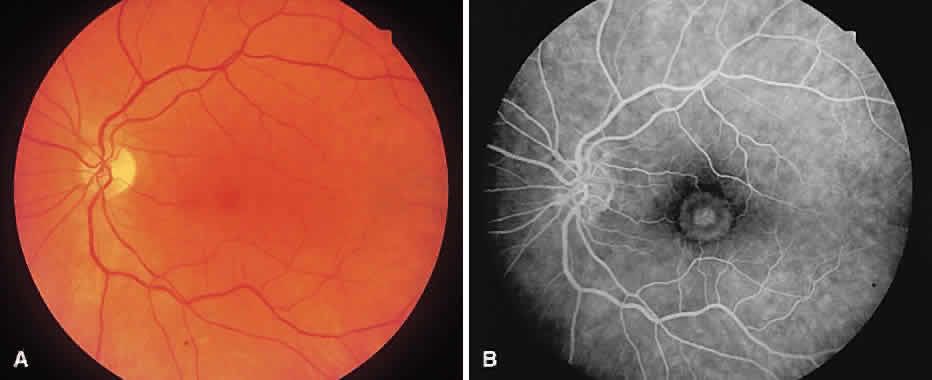
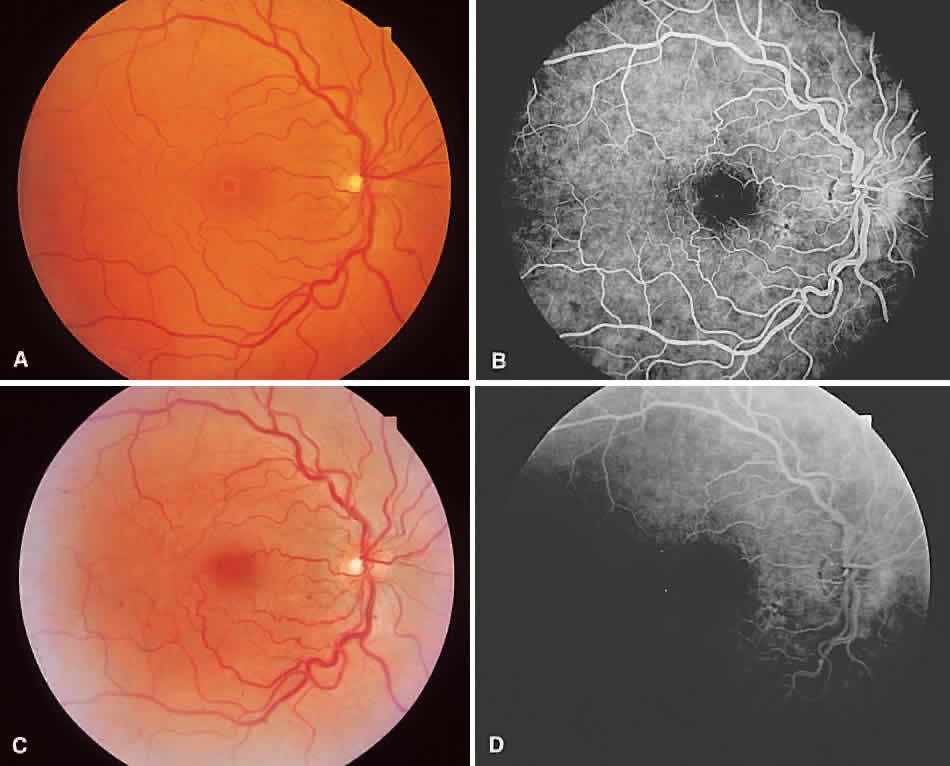
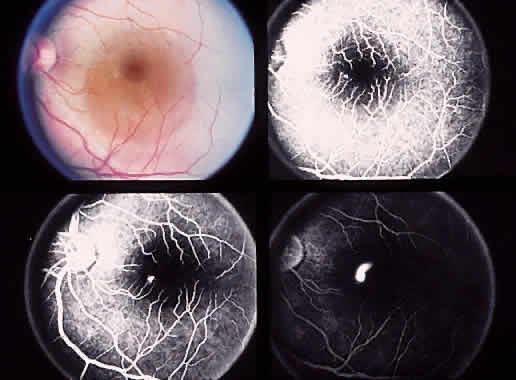
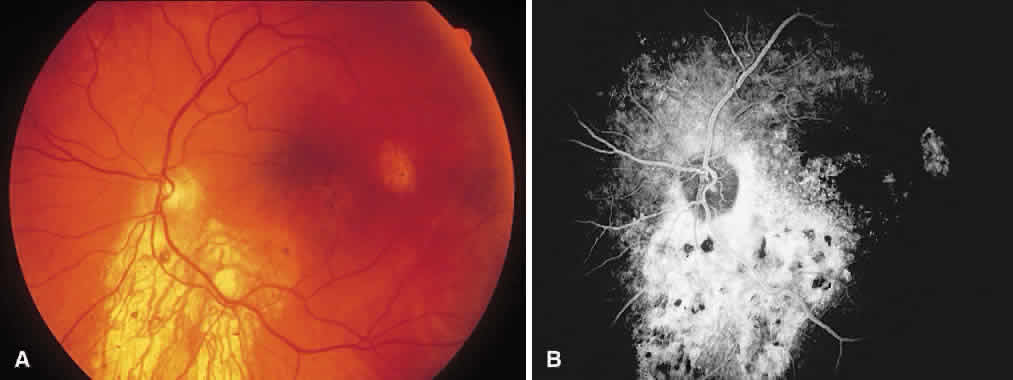
1. McDonnel JM: Ocular embryology and anatomy. In Ryan SJ (ed): Retina, pp 5–16. St
Louis: CV Mosby, 1989 2. Yanoff M: Macular pathology. In Yannuzzi LA, Gitter KA, Schatz H (eds): The
Macula: A Comprehensive Text and Atlas, pp 3–13. Baltimore: Williams & Wilkins, 1979 3. Guyer DR, Schachat AP, Green WR: The choroid: Structural considerations. In
Ryan SJ (ed): Retina, pp 17–36. St Louis: CV Mosby, 1989 4. Kahn HA, Moorehead HB: Statistics on Blindness in the Model Reporting Area, 1969-1970, Publication No. (NIH) 73–427. Washington DC: US
Government Printing Office, 1973 5. Leibowitz HM, Krueger DE, Maunder LR et al: The Framingham Eye Study monograph: An ophthalmological and epidemiological
study of cataract, glaucoma, diabetic retinopathy, macular degeneration, and
visual acuity in a general population of 2631 adults, 1973-1975. VI. Macular
degeneration. Surv Ophthalmol 24(Suppl):428–457, 1980 6. Klein BE, Klein R: Cataracts and macular degeneration in older Americans. Arch Ophthalmol 100:571–573, 1982 7. Sarks SH, Sarks JP: Age-related macular degeneration: Atrophic form. In
Ryan SJ (ed): Retina, pp 149–173. St Louis: CV Mosby, 1989 8. Ghafour IM, Allan D, Foulds WS: Common causes of blindness and visual handicap in the west of Scotland. Br J Ophthalmol 67:209–213, 1983 9. Ferris FL III: Senile macular degeneration: Review of epidemiologic features. Am J Epidemiol 118:132–151, 1983 10. Klein R, Rowland ML, Harris MI: Racial/ethnic differences in age-related maculopathy.Third National Health
and Nutrition Examination Survey.Ophthalmology 102:371–381, 1995 11. Klein R, Klein BEK, Linton KLP: Prevalence of age-related maculopathy. The Beaver Dam Eye Study. Ophthalmology 99:933–343, 1992 12. Bird AC, Bressler NM, Bressler SB et al: An international classification and grading system for age-related maculopathy
and age-related macular degeneration. Surv Ophthalmol 39:367–374, 1995 13. Young RW: Pathophysiology of age-related macular degeneration. Surv Ophthalmol 31:291–306, 1987 14. Eagle RC Jr: Mechanisms of maculopathy. Ophthalmology 91:613–625, 1984 15. Bressler NM, Bressler SB, Fine SL: Age-related macular degeneration. Surv Ophthalmol 32:375–413, 1988 16. Gass JDM: Stereoscopic Atlas of Macular Diseases: Diagnosis and Treatment, pp 46–50, 170–175. 2d ed. St Louis: CV Mosby, 1977 17. Schatz H: Essential Fluorescein Angiography: A Compendium of 100 Classic
Cases, pp 42–56. San Anselmo, CA: Pacific Medical Press, 1983 18. Kenyon KR, Maumenee AE, Ryan SJ et al: Diffuse drusen and associated complications. Am J Ophthalmol 100:119–228, 1985 19. Burns RP, Feeney-Burns L: Clinico-morphologic correlations of drusen of Bruch's membrane. Trans Am Ophthalmol Soc 78:206–225, 1980 20. El Baba F, Green WR, Fleischmann J et al: Clinicopathologic correlation of lipidization and detachment of the retinal
pigment epithelium. Am J Ophthalmol 101:576–583, 1986 21. Green WR, McDonnell PJ, Yeo JH: Pathologic features of senile macular degeneration. Ophthalmology 92:615–627, 1985 22. Sarks SH: Aging and degeneration in the macular region: A clinico-pathological study. Br J Ophthalmol 60:324–341, 1976 23. Sarks SH: Council lecture: Drusen and their relationship to senile macular degeneration. Aust J Ophthalmol 8:117–130, 1980 24. Bressler SB, Maguire MG, Bressler NM, Fine SL: The Macular Photocoagulation Study Group: Relationship of drusen and abnormalities
of the retinal pigment epithelium to the prognosis of neovascular
macular degeneration. Arch Ophthalmol 108:1442–1447, 1990 25. Klein R, Klein BEK, Jensen SC et al: The 5-year incidence and progression of age-related maculopathy: The Beaver
Dam Eye Study. Ophthalmology 104:7–21, 1997 26. Bastek JV, Siegel EB, Straatsma BR, Foos RY: Chorioretinal juncture: Pigmentary patterns of the peripheral fundus. Ophthalmology 89:1455–1463, 1982 27. Humphrey WT, Carlson RE, Valone JA Jr: Senile reticular pigmentary degeneration. Am J Ophthalmol 98:717–722, 1984 28. Bressler NM, Bressler SB, West SK et al: The grading and prevalence of macular degeneration in Chesapeake Bay watermen. Arch Ophthalmol 107:847–852, 1989 29. Bressler NM, Silva JC, Bressler, SB et al: Clinicopathologic correlation of drusen and retinal pigment epithelial
abnormalities in age-related macular degeneration. Retina 14: 130–142, 1994 30. Macular Photocoagulation Study Group: Risk factors for choroidal neovascularization
in the second eye of patients with juxtafoveal or subfoveal
choroidal neovascularization secondary to age-related macular degeneration. Arch
Ophthalmol 115:741–747, 1997 31. Elman MJ, Fine SL: Exudative age-related macular degeneration. In Ryan
SJ (ed): Retina, pp 175–200. St Louis: CV Mosby, 1989 32. Elman MJ, Fine SL, Murphy RP et al: The natural history of serous retinal pigment epithelium detachment in
patients with age-related macular degeneration. Ophthalmology 93:224–230, 1986 33. Meredith TA, Braley RE, Aaberg TM: Natural history of serous detachments of the retinal pigment epithelium. Am J Ophthalmol 88:643–651, 1979 34. Gass JDM, Norton EW, Justice J Jr: Serous detachment of the retinal pigment epithelium. Trans Am Acad Ophthalmol Otolaryngol 70:990–1015, 1966 35. Lewis ML: Idiopathic serous detachment of the retinal pigment epithelium. Arch Ophthalmol 96:620–624, 1978 36. Poliner LS, Olk RJ, Burgess D, Gordon ME: Natural history of retinal pigment epithelial detachments in age-related
macular degeneration. Ophthalmology 93:543–551, 1986 37. Moorfields Macular Study Group: Retinal pigment epithelial detachments
in the elderly: A controlled trial of argon laser photocoagulation. Br
J Ophthalmol 66:1–16, 1982 38. Blair CJ: Geographic atrophy of the retinal pigment epithelium: A manifestation of
senile macular degeneration. Arch Ophthalmol 93:19–25, 1975 39. Schatz H, McDonald HR: Atrophic macular degeneration: Rate of spread of geographic atrophy and
visual loss. Ophthalmology 96:1541–1551, 1989 40. Sarks SH: New vessel formation beneath the retinal pigment epithelium in senile eyes. Br J Ophthalmol 57:951–965, 1973 41. Green WR, Key SN III: Senile macular degeneration: A histopathologic study. Trans Am Ophthalmol Soc 75:180–254, 1977 42. Casswell AG, Kohen D, Bird AC: Retinal pigment epithelial detachments in the elderly: Classification and
outcome. Br J Ophthalmol 69:397–403, 1985 43. Gass JDM: Serous retinal pigment epithelial detachment with a notch: A sign of occult
choroidal neovascularization. Retina 4:205–220, 1984 44. Gass JDM: Pathogenesis of tears of the retinal pigment epithelium. Br J Ophthalmol 68:513–519, 1984 45. Bird AC: Pathogenesis of serous detachment of the retina and pigment epithelium. In
Ryan SJ (ed): Retina, pp 99–105. St Louis: CV Mosby, 1989 46. Gass JDM: Drusen and disciform macular detachment and degeneration. Arch Ophthalmol 90:206–217, 1973 47. Klein BA: Some aspects of classification and differential diagnosis of senile macular
degeneration. Am J Ophthalmol 58:927–939, 1964 48. Sarks SH: Senile choroidal sclerosis. Br J Ophthalmol 57:98–109, 1973 49. Burns RP, Feeney-Burns L: Clinico-morphologic correlations of drusen of Bruch's membrane. Trans Am Ophthalmol Soc 78:206–225, 1980 50. Bird AC, Marshall J: Retinal pigment epithelial detachments in the elderly. Trans Ophthalmol Soc UK 105(pt 6):674–682, 1986 51. Hyvarinen L, Maumenee AE, George T, Weinstein GW: Fluorescein angiography of the choriocapillaris. Am J Ophthalmol 67:653–666, 1969 52. Maguire P, Vine AK: Geographic atrophy of the retinal pigment epithelium. Am J Ophthalmol 102:621–625, 1986 53. Eye Disease Case-Control Study Group: Risk factors for neovascular age-related
macular degeneration. Arch Ophthalmol 110:1701–1708, 1991 54. Eye Disease Case-Control Study Group: Antioxidant status and neovascular
age-related macular degeneration. Arch Ophthalmol 111:104–109, 1993 55. Campochiaro PA, Glaser BM: A retina-derived stimulator(s) of retinal pigment epithelial cell and astrocyte
proliferation. Exp Eye Res 43:449–457, 1986 56. Gass JDM: Drusen and disciform macular detachment and degeneration. Trans Am Ophthalmol Soc 70:409–436, 1972 57. Ryan SJ: Subretinal neovascularization. In Ryan SJ (ed): Retina, pp 107–125. St
Louis: CV Mosby, 1989 58. Schatz H, Yannuzzi LA, Gitter KA: Subretinal neovascularization. In Yannuzzi
LA, Gitter KA, Schatz H (eds): The Macula: A Comprehensive Text
and Atlas, pp 180–201. Baltimore: Williams & Wilkins, 1979 59. Gass JDM: Stereoscopic Atlas of Macular Diseases: Diagnosis and Treatment. 4th
ed. St Louis: CV Mosby, 1997 60. Hyman LG, Lilienfeld AM, Ferris FL III, Fine SL: Senile macular degeneration: A case-control study. Am J Epidemiol 118:213–227, 1983 61. Bressler SB, Bressler NM, Fine SL et al: Natural course of choroidal neovascular membranes within the foveal avascular
zone in senile macular degeneration. Am J Ophthalmol 93:157–163, 1982 62. Cantrill HL, Ramsay RC, Knobloch WH: Rips in the pigment epithelium. Arch Ophthalmol 101:1074–1079, 1983 63. Decker WL, Sanborn GE, Ridley M et al: Retinal pigment epithelial tears. Ophthalmology 90:507–512, 1983 64. Macular Photocoagulation Study Group:
Subfoveal neovascular lesions in age-related macular degeneration: Guidelines
for evaluation and treatment in the Macular Photocoagulation Study. Arch
Ophthalmol 109:1242–1257, 1991 65. Shiraga F, Ojima Y, Matsuo T et al: Feeder vessel photocoagulation of subfoveal choroidal neovascularization
secondary to age-related macular degeneration. Ophthalmology 105:662–669, 1998 66. Staurenghi G, Orzalesi N, LaCarpia A, Aschero M: Laser treatment of feeder vessels in subfoveal choroidal neovascular membranes: A
revisitation using dynamic indocyanine green angiography. Ophthalmology 105:2297–2305, 1998 67. Heriot WJ, Henkind P, Bellhorn RW, Burns MS: Choroidal neovascularization can digest Bruch's membrane: A prior
break is not essential. Ophthalmology 91:1603–1608, 1984 68. Campochiaro PA, Jerdon JA, Glaser BM: The extracellular matrix of human retinal pigment epithelial cells in vivo and its synthesis in vitro. Invest Ophthalmol Vis Sci 27:1615–1621, 1986 69. Glaser BM, Campochiaro PA, Davis JL Jr, Jerdan JA: Retinal pigment epithelial cells release inhibitors of neovascularization. Ophthalmology 94:780–784, 1987 70. Berger JW, Maguire M, Fine SL: The Macular Photocoagulation Study. In Kertes
PJ, Conway MD (eds): Clinical Trials in Ophthalmology, pp 71–92. Baltimore: Williams & Wilkins, 1998 71. Macular Photocoagulation Study Group: Argon laser photocoagulation for
senile macular degeneration: Results of a randomized clinical trial. Arch
Ophthalmol 100:912–918, 1982 72. Macular Photocoagulation Study Group: Argon laser photocoagulation for
neovascular maculopathy: Three-year results from randomized clinical trials. Arch
Ophthalmol 104:694–701, 1986 73. Macular Photocoagulation Study Group: Krypton laser photocoagulation for
neovascular lesions of age-related macular degeneration. Results of
a randomized trial. Arch Ophthalmol 108:816–824, 1990 74. Macular Photocoagulation Study Group: Subfoveal neovascular lesions in
age-related macular degeneration. Guidelines for evaluation and treatment
in the Macular Photocoagulation Study. Arch Ophthalmol 109:1242–1257, 1991 75. Macular Photocoagulation Study Group: Visual outcome after laser photocoagulation
for subfoveal choroidal neovascularization secondary to age-related
macular degeneration. The influence of initial lesion size and
initial visual acuity. Arch Ophthalmol 112:480–488, 1994 76. Macular Photocoagulation Study Group: Persistent and recurrent neovascularization
after laser photocoagulation for subfoveal choroidal neovascularization
of age-related macular degeneration. Arch Ophthalmol 112:489–499, 1994 77. Yeo JH, Marcus S, Murphy RP: Retinal pigment epithelial tears: Patterns and prognosis. Ophthalmology 95:8–13, 1988 78. Young RW: Solar radiation and age-related macular degeneration. Surv Ophthalmol 32:252–269, 1988 79. Taylor HR, West S, Munoz B et al: The long-term effects of visible light on the eye. Arch Ophthalmol 110:99–104, 1992 80. The Eye Disease Case Control Study Group: Risk factors for neovascular
age-related macular degeneration. Arch Ophthalmol 110:1701–1708, 1992 81. Cruickshanks KJ, Klein R, Klein BEK: Sunlight and age-related macular degeneration: The Beaver Dam Eye Study. Arch Ophthalmol 111:514–518, 1993 82. Goldberg J, Flowerdew G, Smith E et al: Factors associated with AMD: Analysis of the first NHANES. Am J Epidemiol 128:700–710, 1988 83. Ho A, Javornik N, Maguire M et al: The Choroidal Neovascularization Prevention Trial (CNVPT): Assessment of
macular risk factors for CNV through 1 year. Invest Ophthalmol Vis Sci 38(Suppl):17, 1997 84. Olk RJ, Friberg TR, Stickney KL et al: Therapeutic benefits of infrared (810-nm) diode laser macular grid photocoagulation
in prophylactic treatment of nonexudative age-related macular
degeneration. Ophthalmology 106:2082–2090, 1999 85. Miller J, Walsh A, Kramer M et al: Photodynamic therapy of experimental choroidal neovascularization using
lipoprotein-delivered benzoporphyrin. Arch Ophthalmol 113:810–818, 1995 86. Husain D, Miller J, Michaud N et al: Intravenous infusion of liposomal benzoporphyrin derivative for photodynamic
therapy of experimental choroidal neovascularization. Arch Ophthalmol 114:978–985, 1996 87. Miller JW, Schmidt-Erfurth U, Sickenberg M et al: Photodynamic therapy with verteporfin for choroidal neovascularization
caused by age-related macular degeneration. Results of a single treatment
in a phase 1 and 2 study. Arch Ophthalmol 117:1161–1173, 1999 88. TAP Study Group: Photodynamic therapy of subfoveal choroidal neovascularization
in age-related macular degeneration with verteporfin. One-year
results of 2 randomized clinical trials—TAP report 1. Arch Ophthalmol 117:1329–1345, 1999 89. Schmidt-Erfurth U, Miller JW, Sickenberg M et al: Photodynamic therapy with verteporfin for choroidal neovascularization
caused by age-related macular degeneration. Results of retreatments in
a phase 1 and 2 study. Arch Ophthalmol 117:1177–1187,1999 90. Reichel E, Berrocal AM, Ip M et al: Transpupillary thermotherapy of occult subfoveal choroidal neovascularization
in patients with age-related macular degeneration. Ophthalmology, 106:1908–1914, 1999 91. Ciulla TA, Danis RP, Harris A: Age-related macular degeneration: A review of experimental treatments. Surv Ophthalmol 43:134–146, 1998 92. Spaide RF, Guyer DR, McCormick B et al: External beam radiation therapy for choroidal neovascularization. Ophthalmology 105:24–30, 1998 93. Spaide RF: Current status of radiotherapy for AMD. Presented at AAO Vitreoretinal
Update, New Orleans, 1998 94. Avery RL, Fekrat S, Hawkins BS, Bressler NM: Natural history of subfoveal subretinal hemorrhage in age-related macular
degeneration. Retina 16:183–189, 1996 95. Heriot WJ: Further experience in management of submacular hemorrhage with
intravitreal tPA. Presented at AAO Vitreoretinal Update, San Francisco, 1997 96. Johnson MW: Pneumatic displacement of submacular hemorrhage. Presented
at AAO Vitreoretinal Update, Orlando, 1999 97. McDonald HR: Submacular surgery: Where we stand today. Presented at AAO
Vitreoretinal Update, Orlando, 1999 98. Machemer R, Steinhors UH: Retinal separation, retinotomy and macular relocation, 1: Experimental
studies in rabbit eye. Graefes Arch Clin Exp Ophthalmol 231:629–634, 1993 99. Lewis H: Panel discussion: Complications of macular translocation. Presented
at AAO Vitreoretinal Update, Orlando, 1999 100. Holland GN: Endogenous fungal infections of the retina and choroid. In
Ryan SJ (ed): Retina, pp 625–636. St Louis: CV Mosby, 1989 101. Patz A, Fine SL: Subretinal neovascularization. In Yannuzzi LA, Gitter
KA, Schatz H (eds): The Macula: A Comprehensive Text and Atlas, pp 209–217. Baltimore: Williams & Wilkins, 1979 102. Klein ML, Fine SL, Knox DL, Patz A: Follow-up study in eyes with choroidal neovascularization caused by presumed
ocular histoplasmosis. Am J Ophthalmol 83:830–835, 1977 103. Lewis ML, Van Newkirk MR, Gass JDM: Follow-up study of presumed ocular histoplasmosis syndrome. Ophthalmology 87:390–399, 1980 104. Macular Photocoagulation Study Group: Five-year follow-up of fellow eyes
of individuals with ocular histoplasmosis and unilateral extrafoveal
or juxtafoveal neovascularization. Arch Ophthalmol 114:677–688, 1996 105. Ryan SJ Jr: De novo subretinal neovascularization in the histoplasmosis syndrome. Arch Ophthalmol 94:321–327, 1976 106. Macular Photocoagulation Study Group: Argon laser photocoagulation for
ocular histoplasmosis: Results of a randomized clinical trial. Arch Ophthalmol 101:1347–1357, 1983 107. Berger AS, Conway M, Del Priore LV et al: Submacular surgery for subfoveal choroidal neovascular membranes in patients
with presumed ocular histoplasmosis. Arch Ophthalmol 115:991–996, 1997 108. Akduman L, Del Priore LV, Desai VN et al: Perfusion of the subfoveal choriocapillaris affects visual recovery after
submacular surgery in presumed ocular histoplasmosis syndrome. Am J Ophthalmol 123:90–96, 1997 109. Melberg NS, Thomas MA, Burgess DB: The surgical removal of subfoveal choroidal neovascularization. Ingrowth
site as a predictor of visual outcome. Retina 16:190–195, 1996 110. Melberg NS, Thomas MA, Dickinson JD, Valluri S: Managing recurrent neovascularization after subfoveal surgery in presumed
ocular histoplasmosis syndrome. Ophthalmology 103:1064–1067, 1996 111. Cantrill HL, Folk JC: Multifocal choroiditis associated with progressive subretinal fibrosis. Am J Ophthalmol 101:170–180, 1986 112. Dreyer RF, Gass JDM: Multifocal choroiditis and panuveitis: A syndrome that mimics ocular histoplasmosis. Arch Ophthalmol 102:1176–1184, 1984 113. Morgan CM, Schatz H: Recurrent multifocal choroiditis. Ophthalmology 93:1138–1147, 1986 114. Watzke RC, Packer AJ, Folk JC et al: Punctate inner choroidopathy. Am J Ophthalmol 98:572–584, 1984 115. Sanborn G, Coscas G: Choroidal neovascular membrane in degenerative myopia. In
Ryan SJ (ed): Retina, pp 201–215. St Louis: CV Mosby, 1989 116. Curtin BJ: The Myopias: Basic Science and Clinical Management. Philadelphia: Harper & Row, 1985 117. Blach RK: Degenerative myopia. In Krill AE, Archer DB (eds): Hereditary
Retinal and Choroidal Diseases, Vol 2: Clinical Characteristics, pp 911–937. Hagerstown, MD: Harper & Row, 1977 118. Curtin BJ: Physiologic vs. pathologic myopia: Genetics vs. environment. Ophthalmology 86:681–691, 1979 119. Curtin BJ: The posterior staphyloma of pathologic myopia. Trans Am Ophthalmol Soc 75:67–86, 1977 120. Avila MP, Welter JJ, Jalkh AE et al: Natural history of choroidal neovascularization in regenerative myopia. Ophthalmology 91:1573–1581, 1984 121. Klein RM, Curtin BJ: Lacquer crack lesions in pathologic myopia. Am J Ophthalmol 79:386–392, 1975 122. Hotchkiss ML, Fine SL: Pathologic myopia and choroidal neovascularization. Am J Ophthalmol 91:177–183, 1981 123. Jalkh AE, Weiter JJ, Trempe CL et al: Choroidal neovascularization in degenerative myopia: Role of laser photocoagulation. Ophthalmic Surg 18:721–725, 1987 124. Yannuzzi LA, Shakin JL, Milch FA: Complications of laser photocoagulation
of subretinal neovascularization secondary to pathologic myopia. In
Gitter KA, Schatz H, Yannuzzi LA, McDonald HR (eds): Laser Photocoagulation
of Retinal Disease. San Anselmo, CA: Pacific Medical Press, 1988 125. Clarkson JG, Altman RD: Angioid streaks. Surv Ophthalmol 26:235–246, 1982 126. Yannuzzi LA, Shokin JL, Milch FA: Complications of laser photocoagulation
of subretinal neovascularization secondary to pathologic myopia. In
Gitter KA, Schatz H, Yannuzzi LA, McDonald HR (eds): Laser Photocoagulation
of Retinal Disease, pp 219–227. San Anselmo, CA: Pacific
Medical Press, 1988 127. Federman JL, Schields JA, Tomer TL: Angioid streaks. II. Fluorescein angiographic features. Arch Ophthalmol 93: 951–962, 1975 128. Gills JP Jr, Paton D: Mottled fundus oculi in pseudoxanthoma elasticum: A report on two siblings. Arch Ophthalmol 73: 792–795, 1965 129. Ferderman JL, Shields JA, Tomer TL, Arnesley WH: Angioid streaks. In Yannuzzi
LA, Gitter KA, Schatz H (eds): The Macula: A Comprehensive Text
and Atlas, pp 218–231. Baltimore: Williams & Wilkins, 1979 130. Condon PI, Serjeant GR: Ocular findings in homozygous sickle cell anemia in Jamaica. Am J Ophthalmol 73:533–543, 1972 131. Geeraets WJ, Guerry D III: Angioid streaks and sickle-cell disease. Am J Ophthalmol 49:450–470, 1960 132. Deutman AF: Macular dystrophies. In Ryan SJ (ed): Retina, pp 243–298. St
Louis: CV Mosby, 1989 133. McDonald HR, Schatz H, Aaberg TM: Reticular-like pigmentary patterns in pseudoxanthoma elasticum. Ophthalmology 95:306–311, 1988 134. Meislik J, Neldner K, Reeve EB, Ellis PP: Atypical drusen in pseudoxanthoma elasticum. Ann Ophthalmol 11:653–656, 1979 135. Gass JDM, Clarkson JG: Angioid streaks and disciform macular detachment in Paget's disease (osteitis
deformans). Am J Ophthalmol 75:576–586, 1973 136. Paton D: The Relation of Angioid Streaks to Systemic Disease. Springfield, IL: Charles
C Thomas, 1972 137. De Laey JJ: Choroidal neovascularization after traumatic choroidal rupture. In
Gitter KA, Schatz H, Yannuzzi LA, McDonald HR (eds): Laser Photocoagulation
of Retinal Disease, pp 227–233. San Anselmo, CA: Pacific
Medical Press, 1988 138. Hilton GF: Late serosanguineous detachment of the macula after traumatic choroidal
rupture. Am J Ophthalmol 79:997–1000, 1975 139. McDonald HR, Aaberg TM: Idiopathic epiretinal membranes. Semin Ophthalmol 1:189–195, 1986 140. McDonald HR, Schatz H: Introduction to epiretinal membranes. In Ryan SJ (ed) Retina, pp 789–795. St Louis: CV Mosby, 1989 141. Fine SL: Idiopathic preretinal macular fibrosis. Int Ophthalmol Clin 17:183–189, 1977 142. Lobes LA Jr, Burton TC: The incidence of macular pucker after retinal detachment surgery. Am J Ophthalmol 85:72–77, 1978 143. Machemer R, Laqua H: Pigment epithelium proliferation in retinal detachment (massive periretinal
proliferation). Am J Ophthalmol 80:1–23, 1975 144. Machemer R, Van Horn D, Aaberg TM: Pigment epithelial proliferation in human retinal detachment with massive
periretinal proliferation. Am J Ophthalmol 85:181–191, 1978 145. McDonald HR, Verre WP, Aaberg TM: Surgical management of idiopathic epiretinal membranes. Ophthalmology 93:978–983, 1986 146. Wilkins JR, Puliafito CA, Hee MR et al: Characterization of epiretinal membranes using optical coherence tomography. Ophthalmology 103:2142–2151, 1996 147. Bellhorn MB, Friedman AH, Wise GN, Henkind P: Ultrastructure and clinicopathologic correlation of idiopathic preretinal
macular fibrosis. Am J Ophthalmol 79:366–373, 1975 148. Foos RY: Vitreoretinal juncture: Epiretinal membranes and vitreous. Invest Ophthalmol Vis Sci 16:416–422, 1977 149. Green WR, Kenyon KR, Michels RG et al: Ultrastructure of epiretinal membranes causing macular pucker after retinal
reattachment surgery. Trans Ophthalmol Soc UK 99:65–77, 1979 150. Kampik A, Kenyon KR, Michels RG et al: Epiretinal and vitreous membranes: Comparative study of 56 cases. Arch Ophthalmol 99:1445–1454, 1981 151. Kampik A, Green WR, Michels RG, Nase PK: Ultrastructural features of progressive idiopathic epiretinal membrane
removed by vitreous surgery. Am J Ophthalmol 90:797–809, 1980 152. Smiddy WE, Maguire AM, Green WR et al: Idiopathic epiretinal membranes. Ultrastructural characteristics and clinicopathologic
correlation. Ophthalmology 96:811–820, 1989 153. Smiddy WE, Michels RG, Gilbert HD, Green WR: Clinicopathologic study of idiopathic macular pucker in children and young
adults. Retina 12:232–236, 1992 154. Margherio RR, Cox MS Jr, Trese MT et al: Removal of epimacular membranes. Ophthalmology 92:1075–1083, 1985 155. Trese MT, Chandler DB, Machemer R: Macular pucker. I. Prognostic criteria. Graefes Arch Clin Exp Ophthalmol 221:12–15, 1983 156. Ma SS, Barloon S, Maberley AL, Kestle J: Effect of macular edema on surgical visual outcome in eyes with idiopathic
epiretinal membrane. Can J Ophthalmol 31:183–186, 1996 157. Desai UR, Blinder KJ, Dennehy PJ: Vitrectomy and juvenile epiretinal membrane. Ophthalmic Surg Lasers 27:137–139, 1996 158. Michels RG: Vitrectomy for macular pucker. Ophthalmology 91:1384–1388, 1984 159. Rice TA, De Bustros S, Michels RG et al: Prognostic factor in vitrectomy for epiretinal membranes of the macula. Ophthalmology 93:602–610, 1986 160. Schepens CL: Fundus changes caused by alterations of vitreous body. Am J Ophthalmol 39:631–633, 1955 161. Maumenee AE: Further advances in the study of the macula. Arch Ophthalmol 78:151–165, 1967 162. Jaffe NS: Vitreous traction at the posterior pole of the fundus due to alterations
in the vitreous posterior. Trans Am Acad Ophthalmol Otolaryngol 71:642–652, 1967 163. Reese AB, Jones IS, Cooper WC: Macular changes secondary to vitreous traction. Am J Ophthalmol 64(Suppl):544–549, 1967 164. Reese AB, Jones IS, Cooper WC: Vitreomacular traction syndrome confirmed histologically. Am J Ophthalmol 69: 975–977, 1970 165. Boniuk M: Cystic macular edema secondary to vitreoretinal traction. Surv Ophthalmol 13:118–121, 1968 166. Smiddy WE, Michels RG, Glaser BM, De Bustros S: Vitrectomy for macular traction caused by incomplete vitreous separation. Arch Ophthalmol 106:624–628, 1988 167. Reese AB, Jones IS, Cooper WC: Vitreomacular traction syndrome confirmed histologically. Am J Ophthalmol 69: 975–977, 1970 168. Hikichi T, Yoshida A, Trempe CL: Course of vitreomacular traction syndrome. Am J Ophthalmol 119:55–61, 1995 169. Melberg NS, Williams DF, Balles MW et al: Vitrectomy for vitreomacular traction syndrome with macular detachment. Retina 15:192–197, 1995 170. Smiddy WE, Green WR, Michels RG, de la Cruz Z: Ultrastructural studies of vitreomacular traction syndrome. Am J Ophthalmol 107:177–185, 1989 171. McDonald HR, Johnson RN, Schatz H: Surgical results in the vitreomacular traction syndrome. Ophthalmology 101:1397–1402, 1994 172. Smiddy WE, Michels RG, Green WR: Morphology, pathology, and surgery of idiopathic vitreoretinal macular
disorders. A review. Retina 10:288–196, 1990 173. Ayyala RS, Cruz DA, Margo CE et al: Cystoid macular edema associated with latanoprost in aphakic and pseudophakic
eyes. Am J Ophthalmol 126:602–604, 1998 174. Irvine SR: A newly defined vitreous syndrome following cataract surgery, interpreted
according to recent concepts of structure of vitreous. Am J Ophthalmol 36:599–619, 1953 175. Gass JDM, Norton EW: Cystoid macular edema and papilledema following cataract extraction: A
fluorescein funduscopic and angiographic study. Arch Ophthalmol 76:646–661, 1966 176. Gass JDM, Norton EW: Follow-up study of cystoid macular edema following cataract extraction. Trans Am Acad Ophthalmol Otolaryngol 73:665–682, 1969 177. Gass JDM, Anderson DR, Davis EB: A clinical, fluorescein angiographic, and electron microscopic correlation
of cystoid macular edema. Am J Ophthalmol 100:82–86, 1985 178. Fine BS, Brucker AJ: Macular edema and cystoid macular edema. Am J Ophthalmol 92:466–481, 1981 179. Yanoff M, Fine BS, Brucker AJ, Eagle RC Jr: Pathology of human cystoid macular edema. Surv Ophthalmol 28(Suppl):505–511, 1984 180. Hitchings RA, Chisholm IH: Incidence of aphakic macular edema: A prospective study. Br J Ophthalmol 59:444–450, 1975 181. Allen A, Jaffe NS: Intraocular lenses: Cystoid macular edema—preliminary
study. Trans Am Acad Ophthalmol Otolaryngol 81:OP133–134, 1976 182. Fung WE, Custis PH: Aphakic and pseudophakic cystoid macular edema. In
Ryan SJ (ed): Retina, pp 1797–1810. St Louis: CV Mosby, 1994 183. Steinert RF, Puliafito CA, Kumar SR et al: Cystoid macular edema, retinal detachment, and glucoma after Nd:YAG laser
posterior capsulotomy. Am J Ophthalmol 112:373–380, 1991 184. Fung WE: Vitrectomy for chronic aphakic cystoid macular edema: Results of a national, collaborative, prospective, randomized investigation. Ophthalmology 92:1102–1111, 1985 185. Jaffe NS, Clayman HM, Jaffe MS: Cystoid macular edema after intracapsular and extracapsular cataract extraction
with and without an intraocular lens. Ophthalmology 89:25–29, 1982 186. Stark WJ Jr, Maumenee AE, Fagadau W et al: Cystoid macular edema in pseudophakia. Surv Ophthalmol 28(Suppl):442–451, 1984 187. Nussenblatt RB, Kaufman SC, Palestine AG et al: Macular thickening and visual acuity: Measurement in patients with cystoid
macular edema. Ophthalmology 94:1134–1139, 1987 188. Brown RM, Roberts CW: Preoperative and postoperative use of nonsteroidal antiinflammatory drugs
in cataract surgery. Insight 21:13–16, 1996 189. Thach AB, Dugel PU, Flindall RJ et al: A comparison of retrobulbar versus sub-Tenon's corticosteroid therapy
for cystoid macular edema refractory to topical medications. Ophthalmology 104:2003–2008, 1997 190. Marmor MF, Maack T: Enhancement of retinal adhesion and subretinal fluid absorption by acetazolamide. Invest Ophthalmol Vis Sci 23:121–124, 1982 191. Tripathi RC, Fekrat S, Tripathi BJ et al: A direct correlation of the resolution of pseudophakic cystoid macular
edema with acetazolamide therapy. Ann Ophthalmol 23:127–129, 1991 192. Grover S, Fishman GA, Fiscella RG, Adelman AE: Efficacy of dorzolamide hydrochloride in the management of chronic cystoid
macular edema in patients with retinitis pigmentosa. Retina 17:222–231, 1997 193. Whitcup SM, Csaky KG, Podgor MJ et al: A randomized, masked, cross-over trial of acetazolamide for cystoid macular
edema in patients with uveitis. Ophthalmology 103: 1054–1062, 1996 194. Fishman GA, Gilbert LD, Anderson RJ et al: Effect of methazolamide on chronic macular edema in patients with retinitis
pigmentosa. Ophthalmology 101:687–693, 1994 195. Suttorp-Schulten MS, Feron E, Postema F et al: Macular grid laser photocoagulation in uveitis. Br J Ophthalmol 79:821–824, 1995 196. Gass JD, Oyakawa RT: Idiopathic juxtafoveolar retinal telangiectasis. Arch Ophthalmol 100:769–780, 1982 197. Gass JDM, Blodi BA: Idiopathic juxtafoveolar retinal telangiectasis: Update of classification
and follow-up study. Ophthalmology 100:1536–1546, 1993 198. Casswell AG, Chaine G, Rush P, Bird AC: Paramacular telangiectasis. Trans Ophthalmol Soc UK 105:683–692, 1986 199. Patel B, Duvall J, Tullo AB: Lamellar macular hole associated with idiopathic juxtafoveolar telangiectasia. Br J Ophthalmol 72:550–551, 1988 200. Green WR, Quigley HA, De la Cruz Z et al: Parafoveal retinal telangiectasis. Light and electron microscopy studies. Trans Ophthalm Soc UK 100:162–170, 1980 201. Patel U, Gupta SC: Wyburn-Mason syndrome. Neuroradiology 31:544–546, 1990 202. Friedman SM, Mames RN, Stewart MW: Subretinal hemorrhage after grid laser photocoagulation for idiopathic
juxtafoveolar retinal telangiectasis. Ophthalm Surg 24:551–553, 1993 203. Hutton WL, Snyder WB, Fuller D et al: Focal parafoveal retinal telangiectasis. Arch Ophthalmol 96:1362–1367, 1978 204. Patel B, Duvall J, Tullo AB: Lamellar macular hole associated with idiopathic juxtafoveolar telangiectasia. Br J Ophthalmol 72:550–551, 1988 205. Moisseiev J, Lewis H, Bartov E et al: Superficial retinal refractile deposits in juxtafoveal telangiectasis. Am J Ophthalmol 109:604–605, 1990 206. Mansour AM, Schachat A: Foveal avascular zone in idiopathic juxtafoveolar telangiectasia. Ophthalmologica 207:9–12, 1993 207. Duke-EIder S, Dobree JH: System of Ophthalmology, Vol 10, Diseases of the
Retina, p 545. St Louis: CV Mosby, 1967 208. Lister W: Holes in the retina and their clinical significance. Br J Ophthalmol 8:1–20, 1924 209. Aaberg TM, Blair CJ, Gass JDM: Macular holes. Am J Ophthalmol 69:555–562, 1970 210. Avila MP, Jalkh AE, Murakami K et al: Biomicroscopic study of the vitreous in macular breaks. Ophthalmology 90:1277–1283, 1983 211. James M, Feman SS: Macular holes. Graefes Arch Clin Exp Ophthalmol 215:59–63, 1980 212. Johnson RN, Gass JDM: Idiopathic macular holes: Observations, stages of formation, and implications
for surgical intervention. Ophthalmology 95:917–924, 1988 213. McDonnell PJ, Fine SL, Hillis AI: Clinical features of idiopathic macular cysts and holes. Am J Ophthalmol 93:777–786, 1982 214. Morgan CM, Schatz H: Idiopathic macular holes. Am J Ophthalmol 99:437–444, 1985 215. Morgan CM, Schatz H: Involutional macular thinning: A pre-macular hole condition. Ophthalmology 93:153–161, 1986 216. Gass JDM: Macular dysfunction caused by vitreous and vitreoretinal interface
abnormalities: Vitreous traction maculopathies. In Gass JDM: Stereoscopic
Atlas of Macular Diseases: Diagnosis and Treatment, pp 676–693. 3d
ed. St Louis: CV Mosby, 1987 217. Evans JR, Schwartz SD, McHugh JD et al: Systemic risk factors for idiopathic macular holes: A case-control study. Eye 12:256–259, 1998 218. Eye Disease Case-Control Study Group: Risk factors for idiopathic macular
holes. Am J Ophthalmol 118:754–761, 1994 219. Ho AC, Guyer DR, Fine SL: Macular hole. Surv Ophthalmol 42:393–416, 1998 220. Worst JGF: Cisternal systems of the fully developed vitreous body in the young adult. Trans Ophthalmol Soc UK 97:550–554, 1977 221. Gass JD: Reappraisal of biomicroscopic classification of stages of development of
a macular hole. Am J Ophthalmol 119:752–759, 1995 222. Gass JDM: Idiopathic senile macular hole: Its early stages and pathogenesis. Arch Ophthalmol 106:629–639, 1988 223. Ezra E, Munro PM, Charteris DG et al: Macular hole opercula. Ultrastructural features and clinicopathological
correlation. Arch Ophthalmol 115:1381–1387, 1997 224. Yooh H, Brooks HL Jr, Capone A Jr et al: Ultrastructural features of tissue removed during idiopathic macular hole
surgery. Am J Ophthalmol 122:67–75, 1996 225. Martinez J, Smiddy WE, Kim J, Gass JD: Differentiating macular holes from macular pseudoholes. Am J Ophthalmol 117:762–767, 1994 226. Tsujikawa M, Ohji M, Fujikado T et al: Differentiating full-thickness macular holes from impending macular holes
and macular pseudoholes. Br J Ophthalmol 81:117–122, 1997 227. Frangieh GT, Green WR, Engel HM: A histopathologic study of macular cysts and holes. Retina 1:311–336, 1981 228. Blain P, Paques M, Massin P et al: Epiretinal membranes surrounding idiopathic macular holes. Retina 18:316–321, 1998 229. Madreperla SA, Geiger GL, Funata M et al: Clinicopathologic correlation of a macular hole treated by cortical vitreous
peeling and gas tamponade. Ophthalmology 101:682–686, 1994 230. Rosa RH Jr, Glaser BM, de la Cruz Z et al: Clinicopathologic correlation of an untreated macular hole and a macular
hole treated by vitrectomy, transforming growth factor-beta-2, and gas
tamponade. Am J Ophthalmol 122:853–863, 1996 231. de Bustros S: Vitrectomy for prevention of macular holes. Results of a randomized multicenter
clinical trial. Vitrectomy for Prevention of Macular Hole Study
Group. Ophthalmology 101:1055–1059, 1994 232. Kokame GT, de Bustros S: Visual acuity as a prognostic indicator in stage I macular holes. Vitrectomy
for Prevention of Macular Hole Study Group. Am J Ophthalmol 120:112–114, 1995 233. Kim JW, Freeman WR, Azen SP et al: Prospective randomized trial of vitrectomy or observation for stage 2 macular
holes. Vitrectomy for Macular Hole Study Group. Am J Ophthalmol 121:605–614, 1996 234. Kim JW, Freeman WR, el-Haig W et al: Baseline characteristics, natural history, and risk factors to progression
in eyes with stage 2 macular holes. Results from a prospective randomized
clinical trial. Vitrectomy for Macular Hole Study Group. Ophthalmology 102:1818–1828, 1995 235. Akiba J, Kakehashi A, Arzabe CW et al: Fellow eyes in idiopathic macular hole cases. Ophthalmic Surg 23:594–597, 1992 236. Ezra E, Wells JA, Gray RH et al: Incidence of idiopathic full-thickness macular holes in fellow eyes. A 5-year
prospective natural history study. Ophthalmology 105:353–359, 1998 237. Chew EY, Sperduto RD, Hiller R et al: Clinical course of macular holes: The Eye Disease Case-Control Study. Arch Ophthalmol 117:242–246, 1999 238. Kelly NE, Wendel RT: Vitreous surgery for idiopathic macular holes: Results of a pilot study. Arch Ophthalmol 109:654–659, 1991 239. Wendel RT, Patel AC, Kelly NE et al: Vitreous surgery for macular holes. Ophthalmology 100:1671–1676, 1993 240. Ryan EH Jr, Gilbert HD: Results of surgical treatment of recent-onset full-thickness idiopathic
macular holes. Arch Ophthalmol 112:1545–1553, 1994 241. Willis AW, Garcia-Cosio JF: Macular hole surgery. Comparison of longstanding versus recent macular
holes. Ophthalmology 103:1811–1814, 1996 242. Olsen TW, Sternberg P Jr, Capone A Jr et al: Macular hole surgery using thrombin-activated fibrinogen and selective
removal of the internal limiting membrane. Retina 18:322–329, 1998 243. Park DW, Lee JH, Min WK: The use of internal limiting membrane maculorrhexis in treatment of idiopathic
macular holes. Korean J Ophthalmol 12:92–97, 1998 244. Gaudric A, Massin P, Paques M et al: Autologous platelet concentrate for the treatment of full-thickness macular
holes. Graefes Arch Clin Exp Ophthalmol 233:549–554, 1995 245. Glaser BM, Michels RG, Kupperman BD et al: Transforming growth factor beta 2 for the treatment of full-thickness macular
holes: A prospective randomized study. Ophthalmology 99:1162–1172, 1992 246. Thompson JT, Sjaarda RN, Glaser BM et al: Increased intraocular pressure after macular hole surgery. Am J Ophthalmol 121:615–622, 1996 247. Thompson JT, Smiddy WE, Williams GA et al: Comparison of recombinant transforming growth factor-beta-2 and placebo
as an adjunctive agent for macular hole surgery. Ophthalmology 105:700–706, 1998 248. Smiddy WE, Pimentel S, Williams GA: Macular hole surgery without using adjunctive additives. Ophthalmic Surg Lasers 28:713–717, 1997 249. Goldbaum MH, McCuen BW 2nd, Hanneken AM et al: Silicone oil tamponade to seal macular holes without position restrictions. Ophthalmology 105:2140–2148, 1998 250. Freeman WR, Azen SP, Kim JW et al: Vitrectomy for the treatment of full-thickness stage 3 or 4 macular holes: Results
of a multicentered randomized clinical trial. Vitrectomy for
Treatment of Macular Hole Study Group. Arch Ophthalmol 115:11–21, 1997 251. Leonard RE II, Smiddy WE, Flynn HW Jr, Feuer W: Long-term visual outcomes in patients with successful macular hole surgery. Ophthalmology 104:1648–1652, 1997 252. Chen CJ: Glaucoma after macular hole surgery. Ophthalmology 105:94–100, 1998 253. Banker AS, Freeman WR, Kim JW et al: Vision-threatening complications of surgery for full-thickness macular
holes. Vitrectomy for Macular Hole Study Group. Ophthalmology 104:1442–1452, 1997 254. Sjaarda RN, Glaser BM, Thompson JT et al: Distribution of iatrogenic retinal breaks in macular hole surgery. Ophthalmology 102:1387–1392, 1995 255. Park SS, Marcus DM, Duker JS et al: Posterior segment complications after vitrectomy for macular hole. Ophthalmology 102:1071–1076, 1995 256. Heier JS, Topping TM, Frederick AR Jr et al: Visual and surgical outcomes of retinal detachment following macular hole
repair. Retina 19:110–115, 1999 257. Hutton WL, Fuller DG, Snyder WB et al: Visual field defects after macular hole surgery. A new finding. Ophthalmology 103:2152–2158, 1996 258. Boldt HC, Munden PM, Folk JC et al: Visual field defects after macular hole surgery. Am J Ophthalmol 122:371–381, 1996 259. Pendergast SD, McCuen BW 2d: Visual field loss after macular hole surgery. Ophthalmology 103:1069–1077, 1996 260. Paques M, Massin P, Santiago PY et al: Visual field loss after vitrectomy for full-thickness macular holes. Am J Ophthalmol 124:88–94, 1997 261. Yan H, Dhurjon L, Chow DR et al: Visual field defect after pars plana vitrectomy. Ophthalmology 105:1612–1616, 1998 262. Poliner LS, Tornambe PE: Retinal pigment epitheliopathy after macular hole surgery. Ophthalmology 99:1671–1677, 1992 263. Duker JS, Wendel R, Patel AC et al: Late re-opening of macular holes after initially successful treatment with
vitreous surgery. Ophthalmology 101:1373–1378, 1994 264. Johnson RN, McDonald HR, Schatz H, Ai E: Outpatient postoperative fluid-gas exchange after early failed vitrectomy
surgery for macular hole. Ophthalmology 104:2009–2013, 1997 265. Ohana E, Blumenkranz MS: Treatment of reopened macular hole after vitrectomy by laser and outpatient
fluid-gas exchange. Ophthalmology 105:1398–1403, 1998 266. Annesley WH Jr: Augsburger JJ, Shakin JL: Ten-year follow-up of photocoagulated central serous choroidopathy. Trans Am Ophthalmol Soc 79:335–346, 1981 267. Burton TC: Central serous retinopathy. In Blodi FC (ed): Current Concepts
in Ophthalmology, Vol 3, pp 1–28. St Louis: CV Mosby, 1972 268. De Lacy JJ: Central serous chorioretinopathy: To treat or not to treat. Doc Ophthalmol 61:367–372, 1986 269. Lyons DE: Conservative management of central serous retinopathy. Trans Ophthalmol Soc UK 97:214–216, 1977 270. Straatsma BR, Allen RA, Pettit TH: Central serous retinopathy. Transactions of the Pacific Coast Ophthalmologic Society Annual Meeting 47:107–127, 1966 271. Spitznas M, Huke J: Number, shape, and topography of leakage points in acute type I central
serous retinopathy. Graefes Arch Clin Exp Ophthalmol 225:437–440, 1987 272. Bedrossian RH: Central serous retinopathy and pregnancy [letter]. Am J Ophthalmol 78:152, 1974 273. Eastenberg DM, Ober RR: Central serous choroidopathy in pregnancy. Arch Ophthalmol 101:1055–1058, 1983 274. Chumbley LC, Frank RN: Central serous retinopathy and pregnancy. Am J Ophthalmol 7:158–160, 1974 275. Cruysberg JR, Deutmann AF: Visual disturbances during pregnancy caused by central serous choroidopathy. Br J Ophthalmol 66:240–241, 1982 276. Yannuzzi LA, Gitter KA, Schatz H (eds): The Macula: A Comprehensive Text
and Atlas, pp 145–165. Baltimore: Williams & Wilkins, 1979 277. Klein ML, Van Buskirk M, Friedman E et al: Experience with nontreatment of central serous choroidopathy. Arch Ophthalmol 91:247–250, 1974 278. Gelber GS, Schatz H: Loss of vision due to central serous chorioretinopathy following psychological
stress. Am J Psychiatry 144:46–50, 1987 279. Yannuzzi LA, Shakin JL, Fisher YL, Altomonte MA: Peripheral retinal detachments and retinal pigment epithelial atrophic
tracts secondary to central serous pigment epitheliopathy. Ophthalmology 91:1554–1572, 1984 280. Benson WE, Shields JA, Annesley WH Jr: Tasman W: Idiopathic central serous chorioretinopathy with bullous retinal detachment. Ann Ophthalmol 12:920–924, 1980 281. Gass JDM: An unusual manifestation of idiopathic central serous choroidopathy. Am J Ophthalmol 75:810–821, 1973 282. Mazzuca DE, Benson WE: Central serous retinopathy: Variants. Surv Ophthalmol 31:170–174, 1986 283. Sharma T, Badrinath SS, Gopal L et al: Subretinal fibrosis and nonrhegmatogenous retinal detachment associated
with multifocal central serous chorioretinopathy. Retina 18:23–29, 1998 284. Shimizu K, Tobari I: Central serous retinopathy dynamics of subretinal fluid. Mod Probl Ophthalmol 9:152–157, 1971 285. Nanjiani M: Long-term follow-up of central serous retinopathy. Trans Ophthalmol Soc UK 97:656–661, 1977 286. Dellaporta A: Central serous retinopathy. Trans Am Ophthalmol Soc 74:144–153, 1977 287. Robertson DM, Ilstrup D: Direct, indirect, and sham laser photocoagulation in the management of
central serous chorioretinopathy. Am J Ophthalmol 95:457–466, 1983 288. Robertson DM: Argon laser photocoagulation treatment in central serous chorioretinopathy. Ophthalmology 93: 972–974, 1986 289. Yannuzzi LA: Type A behavior and central serous chorioretinopathy. Retina 7:111–131, 1987 290. Gass JD, Little H: Bilateral bullous exudative retinal detachment complicating idiopathic
central serous chorioretinopathy during systemic corticosteroid therapy. Ophthalmology 102:737–747, 1995 291. Haimovici R, Gragoudas ES, Duker JS et al: Central serous chorioretinopathy associated with inhaled or intranasal
corticosteroids. Ophthalmology 104:1653–1660, 1997 292. Zamir E: Central serous retinopathy associated with adrenocorticotrophic hormone
therapy. A case report and a hypothesis. Graefes Arch Clin Exp Ophthalmol 235:339–344, 1997 293. Marmor MF: New hypothesis on the pathogenesis and treatment of serous retinal detachment. Graefes Arch Clin Exp Ophthalmol 226:548–552, 1988 294. Spitznas M: Pathogenesis of central serous retinopathy: A new working hypothesis. Graefes Arch Clin Exp Ophthalmol 224:321–324, 1986 295. Ficker L, Vafidis G, While A, Leaver P: Long-term follow-up of a prospective trial of argon laser photocoagulation
in the treatment of central serous retinopathy. Br J Ophthalmol 72:829–834, 1988 296. Gass JDM: Photocoagulation treatment of idiopathic central serous choroidopathy. Trans Am Acad Ophthalmol Otolaryngol 83(pt 1):456–467, 1977 297. Landers MB III, Shaw HE Jr: Anderson WB Jr: Sinyai AJ: Argon laser treatment of central serous chorioretinopathy. Ann Ophthalmol 9:1567–1572, 1977 298. Leaver P, Williams C: Argon laser photocoagulation in the treatment of central serous retinopathy. Br J Ophthalmol 63:674–677, 1979 299. Watzke RC, Burton TC, Leaverton PE: Ruby laser photocoagulation therapy of central serous retinopathy. I. A
controlled clinical study. II. Factors affecting prognosis. Trans Am Acad Ophthalmol Otolaryngol 78:205–211, 1974 300. Watzke RC, Burton TC, Woolson RF: Direct and indirect laser photocoagulation of central serous choroidopathy. Am J Ophthalmol 88:914–918, 1979 301. Khosla PK, Rana SS, Tewari HK et al: Evaluation of visual function following argon laser photocoagulation in
central serous retinopathy. Ophthalmic Surg Lasers 28:693–697, 1997 302. Novak MA, Singerman LJ, Rice TA: Krypton and argon laser photocoagulation for central serous chorioretinopathy. Retina 7:162–169, 1987 303. Slusher MM: Krypton red laser photocoagulation in selected cases of central serous
chorioretinopathy. Retina 6:81–84, 1986 304. Theodossiadis G, Tongos D: Treatment of central serous retinopathy: A comparative study with and without
light coagulation. Ophthalmologica 169:416–431, 1974 305. Wakakura M, Ishikawa S: Central serous chorioretinopathy complicating systemic corticosteroid treatment. Br J Ophthalmol 68:329–331, 1984 306. Burumcek E, Mudun A, Karacorlu S, Arslan MO: Laser photocoagulation for persistent central serous retinopathy: Results
of long-term follow-up. Ophthalmology 104:616–622, 1997 307. Yannuzzi LA, Sorenson J, Spaide RF, Lipson B: Idiopathic polypoidal choroidal vasculopathy. Retina 10:1–8, 1990 308. Spaide RF, Yannuzzi LA, Slakter JS et al: Indocyanine green videoangiography of idiopathic polypoidal choroidal vasculopathy. Retina 15:100–110, 1995 309. Yannuzzi LA, Ciardella A, Spaide RF et al: The expanding clinical spectrum of idiopathic polypoidal choroidal vasculopathy. Arch Ophthalmol 115:478–485, 1997 310. Moorthy RS, Lyon AT, Rabb MF et al: Idiopathic polypoidal choroidal vasculopathy of the macula. Ophthalmology 105:1380–1385, 1998 311. Yannuzzi LA, Wong DWK, Sforzolini BS et al: Polypoidal choroidal vasculopathy and neovascularized age-related macular
degeneration. Arch Ophthalmol 117:1503–1510, 1999 |