| Color changes in the fundus indicative of possible disease include the
following: (1) white, (2) gray opacification, (3) yellow, (4) black, and (5) red. The
region of the retina involved affects the color and the
shape of the hemorrhage. Fresh hemorrhage on the retinal surface appears bright red, whereas fresh hemorrhage
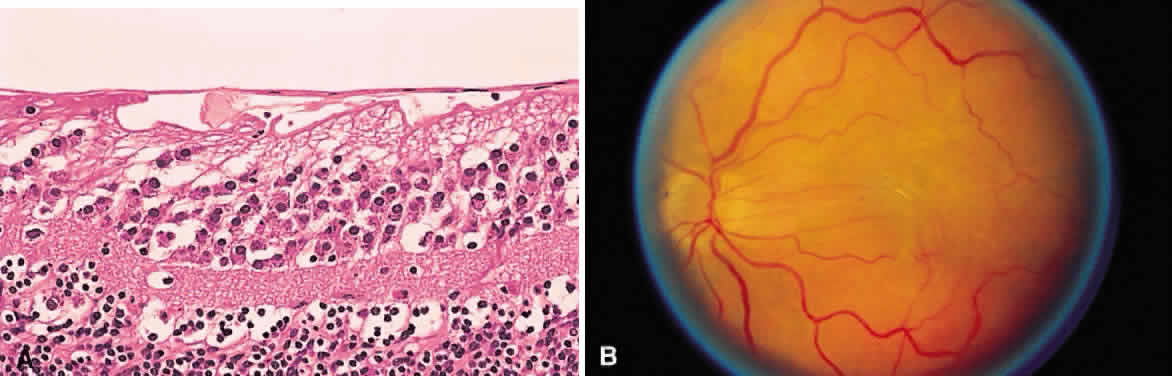
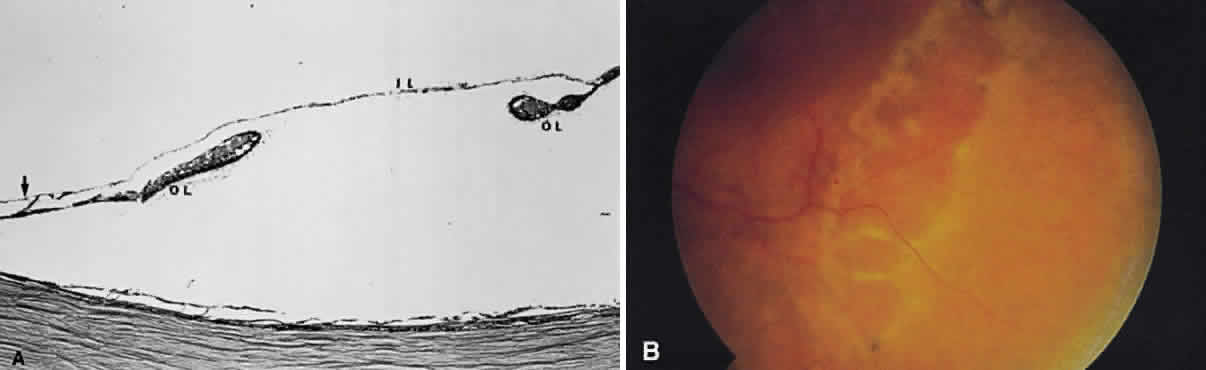
beneath the retinal pigment epithelium seems much darker. A hemorrhage in the nerve fiber layer (Fig. 11A) dissects along the plane of the layer parallel to the orientation of
the internal limiting membrane (see Fig. 11B). A hemorrhage located between the retinal pigment epithelium and Bruch's
membrane also spreads in a plane parallel to the orientation
of the membrane (Fig. 12). However, its extent is limited by the adhesion of the pigment epithelium
to Bruch's membrane, in contrast to a nerve fiber layer hemorrhage, where
no such delineating structure is present. Therefore, a fresh
nerve fiber layer hemorrhage appears bright red and has feathery
borders, whereas a subpigment epithelial hemorrhage appears brown-black
and has sharp borders (Fig. 13). 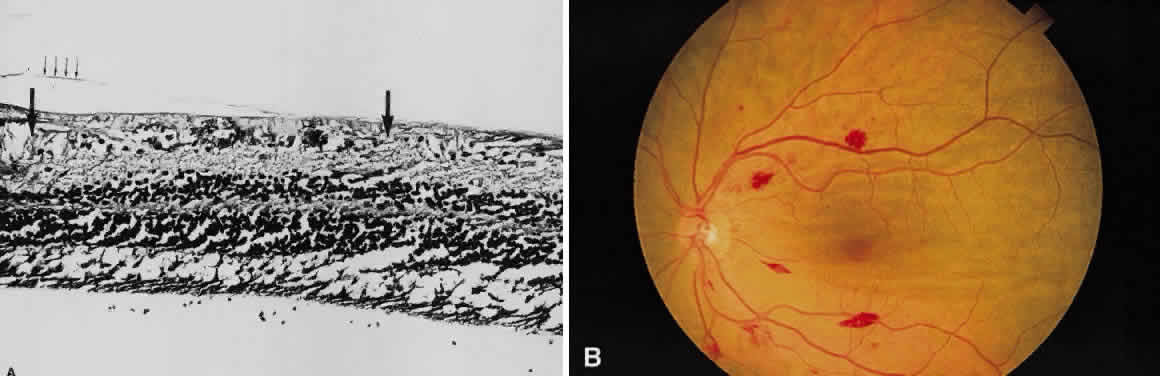 Fig. 11. A. Section of retina with hemorrhage in the nerve fiber layer (between the two large arrows). Notice that the limits of the hemorrhage are not clearly defined, since
scattered red blood cells can be seen to the right of the right-hand
large arrow. This histologic picture corresponds to a clinically observed
fame-shaped hemorrhage with an indistinct border. The detached
posterior hyaloid is marked by four small arrows. B. Fundus photograph of nerve fiber layer hemorrhage. They are oriented parallel
to the plane of the internal limiting membrane. Because of their
dispersal within the ganglion cell layer, the borders are “feathery” (flame
shaped). Fig. 11. A. Section of retina with hemorrhage in the nerve fiber layer (between the two large arrows). Notice that the limits of the hemorrhage are not clearly defined, since
scattered red blood cells can be seen to the right of the right-hand
large arrow. This histologic picture corresponds to a clinically observed
fame-shaped hemorrhage with an indistinct border. The detached
posterior hyaloid is marked by four small arrows. B. Fundus photograph of nerve fiber layer hemorrhage. They are oriented parallel
to the plane of the internal limiting membrane. Because of their
dispersal within the ganglion cell layer, the borders are “feathery” (flame
shaped).
|
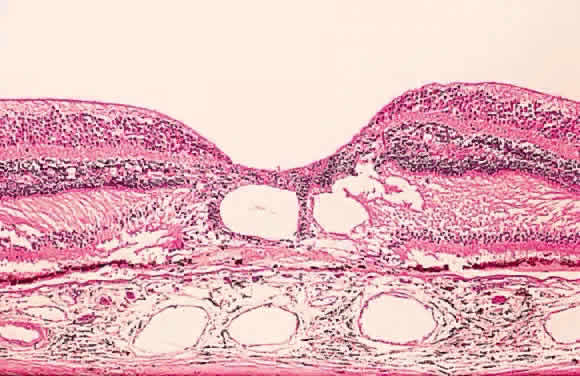
 Fig. 12. Section of the eye showing subretinal hemorrhage. (Courtesy of Ralph C. Eagle Jr, MD, Philadelphia, PA) Fig. 12. Section of the eye showing subretinal hemorrhage. (Courtesy of Ralph C. Eagle Jr, MD, Philadelphia, PA)
|
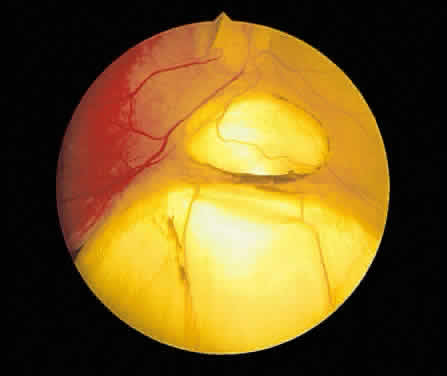
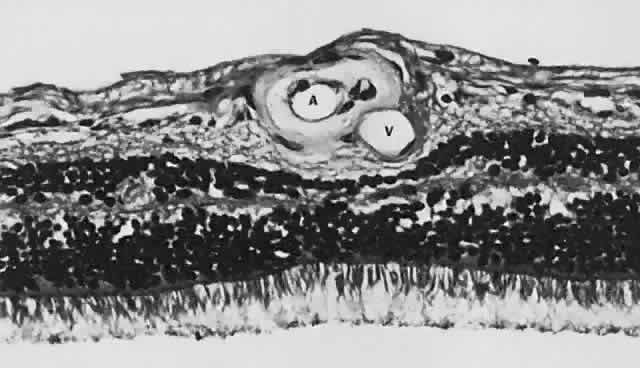
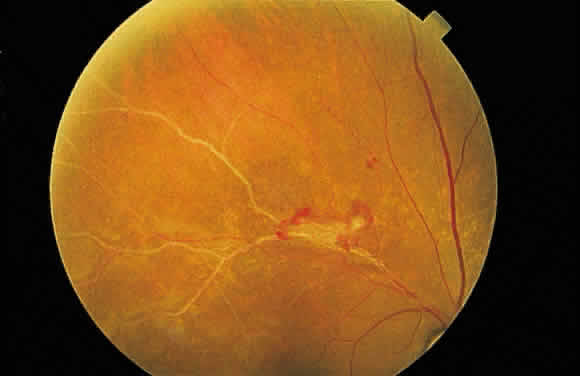
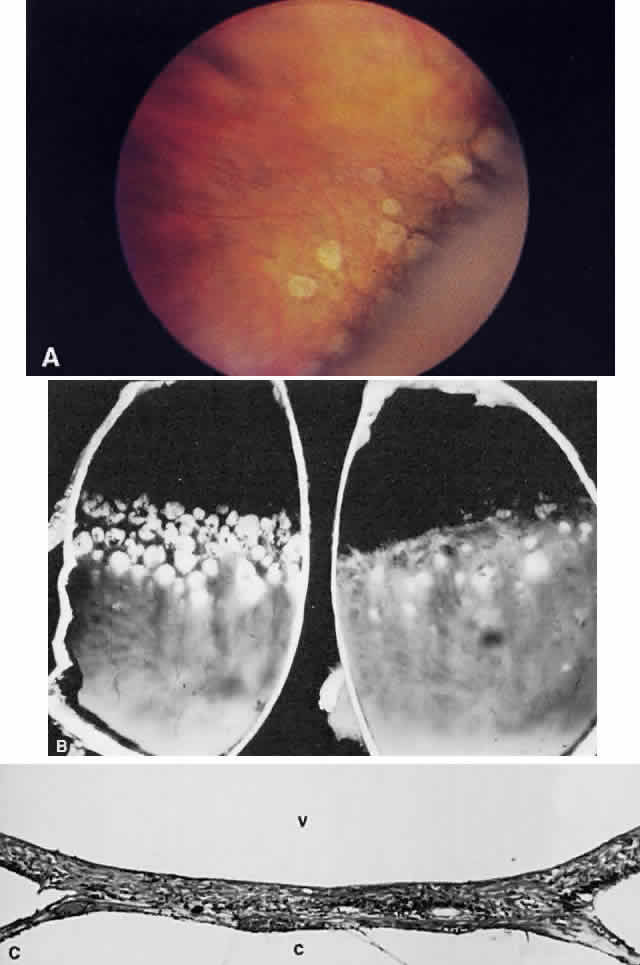
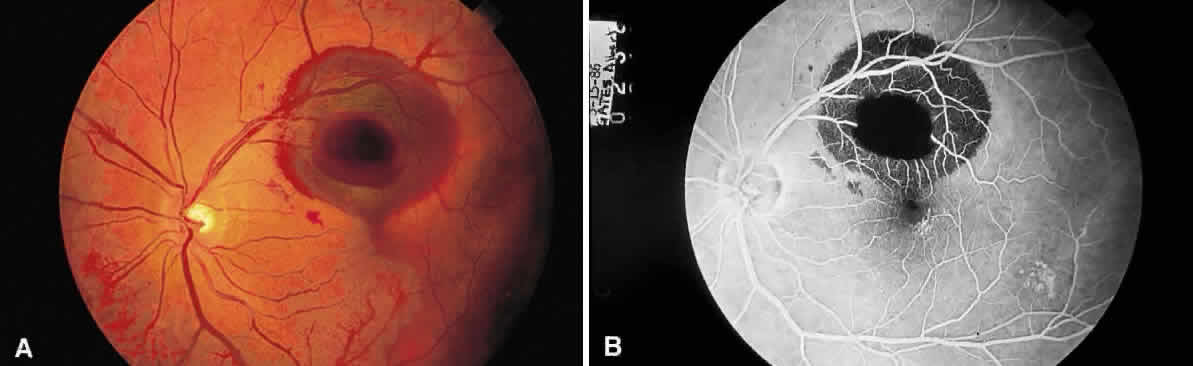 Fig. 13. A. Fundus photograph of a subpigment epithelium hemorrhage superotemporal
to the disc secondary to a macroaneurysm. Notice its dark color and sharp
border. The central portion of the hemorrhage has extended through
the sensory retinal to the subinternal limiting membrane area. B. Fluorescein angiography showing the retinal vessels overlying the deep
hemorrhage but obscured by the central extension anteriorly. (B, courtesy of William Tasman, MD, Philadelphia, PA) Fig. 13. A. Fundus photograph of a subpigment epithelium hemorrhage superotemporal
to the disc secondary to a macroaneurysm. Notice its dark color and sharp
border. The central portion of the hemorrhage has extended through
the sensory retinal to the subinternal limiting membrane area. B. Fluorescein angiography showing the retinal vessels overlying the deep
hemorrhage but obscured by the central extension anteriorly. (B, courtesy of William Tasman, MD, Philadelphia, PA)
|
The color of emboli in the retinal vasculature facilitates the identification
of their composition and their origin. White emboli are of composed
of calcium, and their source may be either a calcified cardiac valve
or calcified atheromatous plaque. Cholesterol and lipid emboli, most
likely from noncalcified atheromatous plaques in the carotids, appear
yellow (Hollenhorst plaque) (Fig. 14). The more evanescent platelet emboli are gray-white. 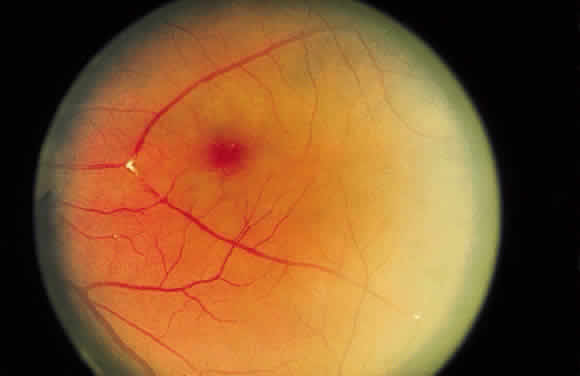 Fig. 14. Hollenhorst plaque lodged at the bifurcation of a cilioretinal artery. Fig. 14. Hollenhorst plaque lodged at the bifurcation of a cilioretinal artery.
|
WHITE CHANGES IN THE FUNDUS Fibrous connective tissue appears white because of its collagen content. It
is not difficult to remember this relation, since the sclera is white. The
white appearance of a retinal choroidal coloboma, as an example
of collagen accounting for a white fundus appearance, already has
been cited. The temporal disc crescent seen in myopia is another example
of the ophthalmoscopist viewing the sclera directly (Fig. 15). In some cases of myopia, the pigment epithelium and choroid are stretched
to the point that they do not approximate the temporal margin of
the disc. The normally transparent retina does not contribute to the
color of this crescent. The black rim of the crescent is the contribution
of the retinal pigment epithelium. 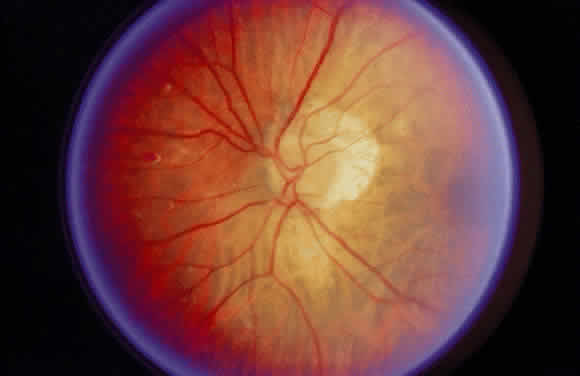 Fig. 15. “Myopic crescent” in a patient with 9 diopters of myopia. Fig. 15. “Myopic crescent” in a patient with 9 diopters of myopia.
|
Fibrous tissue may originate from the choroid and proliferate through breaks
in Bruch's membrane into the subpigment epithelial or subretinal
spaces,4 hence the white appearance of disciform macular degeneration (Fig. 16). Fibrous tissue also may originate from fibroblasts located in the adventitia
of retinal vessels and may contribute to the white appearance
of vascularized membranes (Fig. 17A), such as those seen in proliferative diabetic retinopathy (see Fig. 17B) or arterioles after vascular occlusion (see Fig. 6). The accretion of collagen in the wall of the vessel in arteriolosclerosis
may thicken the vessel wall (see Fig. 5) and alter the color of the blood column to a copper or silver color. Injury
to the pigment epithelium results in scar formation (Fig. 18). Collagen deposition from pigment epithelium metaplasia may be identified
by the presence of pigment within a scar (Fig. 19). Injured nonpigmented epithelium may undergo similar fibrous metaplasia
and may contribute to the formation of membranes such as cyclitic membranes
in the region of the ciliary body. 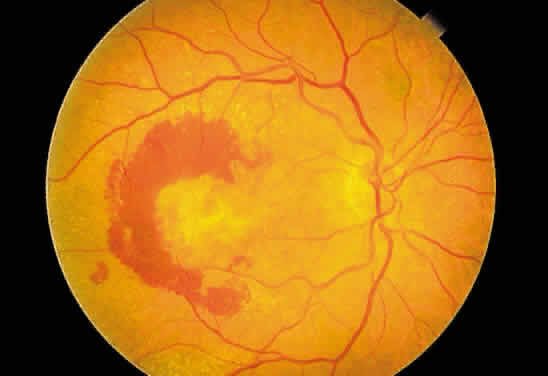 Fig. 16. Fundus photograph of a disciform macular scar, white because of fibrosis
and atrophy of the pigment epithelium. A broad, C-shaped, fresh hemorrhage
within the sensory retina surrounds it. Fig. 16. Fundus photograph of a disciform macular scar, white because of fibrosis
and atrophy of the pigment epithelium. A broad, C-shaped, fresh hemorrhage
within the sensory retina surrounds it.
|
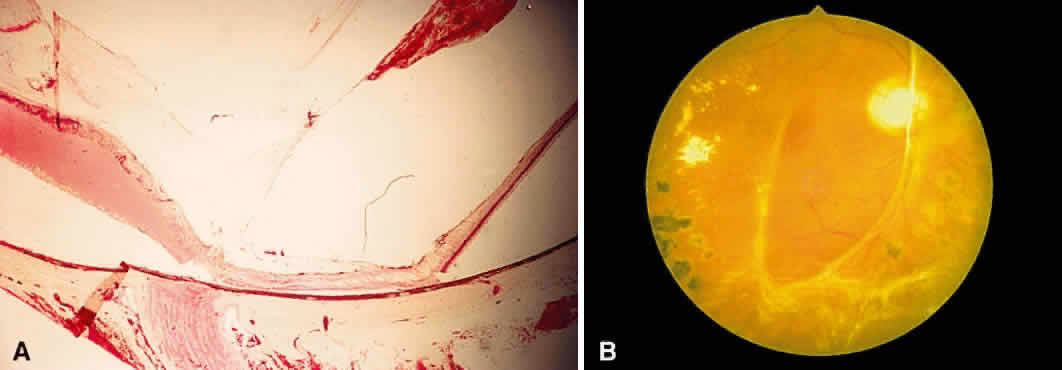 Fig. 17. A. A histologic section stained with H & E demonstrating vascularized
membranes on the disc and retina. Tractional retinal detachment is present. Notice
the subretinal fluid (amorphous eosinophilic material). B. Fundus photograph of fibrous tissue emanating from the disc in patient
with proliferative diabetic retinopathy. The disc is pale; the macula
is edematous with exudate. Pigmented laser spots are visible temporally. Fig. 17. A. A histologic section stained with H & E demonstrating vascularized
membranes on the disc and retina. Tractional retinal detachment is present. Notice
the subretinal fluid (amorphous eosinophilic material). B. Fundus photograph of fibrous tissue emanating from the disc in patient
with proliferative diabetic retinopathy. The disc is pale; the macula
is edematous with exudate. Pigmented laser spots are visible temporally.
|
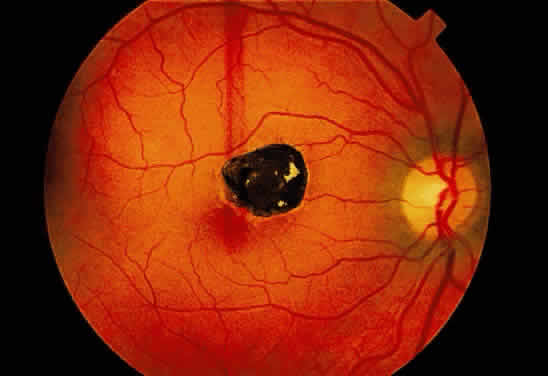 Fig. 18. Fundus photograph of pigmented scar secondary to retinal pigment epithelial
hyperplasia induced by injury. Fig. 18. Fundus photograph of pigmented scar secondary to retinal pigment epithelial
hyperplasia induced by injury.
|
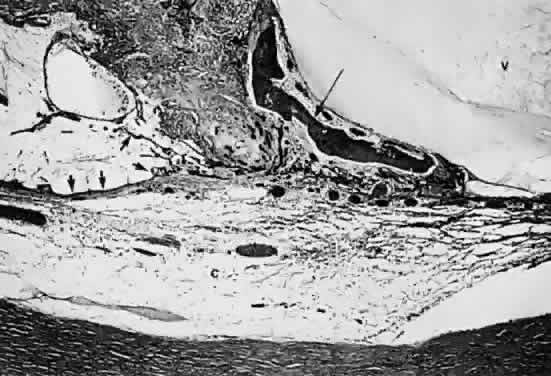 Fig. 19. Histologic section demonstrating response of the pigment epithelium to
injury. The sclera is at the bottom the micrograph; the choroid (c) contains scattered red blood cells and is edematous. The vitreous (v) also contains hemorrhage. Locate the pigment epithelium (short arrows) at the left third of the micrograph and notice the ribbons of pigment
epithelium that proliferate into the membrane, partially formed by fibrous
tissue (F). Bone (long arrow) also is present and probably was deposited by metaplastic pigment epithelium. Fig. 19. Histologic section demonstrating response of the pigment epithelium to
injury. The sclera is at the bottom the micrograph; the choroid (c) contains scattered red blood cells and is edematous. The vitreous (v) also contains hemorrhage. Locate the pigment epithelium (short arrows) at the left third of the micrograph and notice the ribbons of pigment
epithelium that proliferate into the membrane, partially formed by fibrous
tissue (F). Bone (long arrow) also is present and probably was deposited by metaplastic pigment epithelium.
|
Finally, fibrous tissue may enter the eye through a perforating wound. The
sclera does not heal itself by proliferating and bridging a dehiscence. Instead, healing
is achieved by tissue from the episclera and the
choroid. Occasionally, episcleral tissue enters the eye as a fibrous
ingrowth and may be seen even at the site of surgical scleral wounds, such
as sclerotomy portals for pars plana vitrectomy (Fig. 20).  Fig. 20. Histologic section of a vitrectomy wound in the pars plana region. The
wound is traced from the lower left of the illustration to the zone of
focal fibrous ingrowth (arrowhead). Suture material (s) is located on each side of the wound tract. Fig. 20. Histologic section of a vitrectomy wound in the pars plana region. The
wound is traced from the lower left of the illustration to the zone of
focal fibrous ingrowth (arrowhead). Suture material (s) is located on each side of the wound tract.
|
Gliosis is akin to fibrosis as a mechanism of repair. Repair in tissues
of the central nervous system is accomplished by gliosis. Since the neurosensory
retina originates embryologically from an outpouching of the
central nervous system, repair within the retina is accomplished by
gliosis. The Müller cell, thought to be responsible for this repair, also
may migrate through a break in the internal limiting membrane
and contribute to the formation of preretinal membranes. It also may
migrate into the retroretinal space to contribute to membranes in this
area. Glial membranes, if thin, may be transparent, but abundant glial
tissue may have a white color. In nonglaucomatous optic atrophy, such as the change that occurs after
an infarct of the optic nerve (i.e., arteritic or nonarteritic optic neuropathy), the loss of tissue substance
is accompanied by the proliferation of glial tissue. The combination
of gliosis and a decrease in the vascular supply to the nerve head
accounts for the whiteness of the disc (see Fig. 7). The pathologic correlate of the “waxy pallor” of the disc
in retinitis pigmentosa is similarly attributable to decreased vascular
supply and gliosis.5 Occasionally, gliosis and fibrosis occur together. Preretinal membranes
may be composed entirely of glial cells (Müller cell derivatives) or
may involve the proliferation of retinal pigment epithelial cells
and glial cells, especially in the presence of a retinal break, which
allows the pigment epithelial cells access to the preretinal space. Frequently, cells
with ultrastructural features of both fibroblasts and
smooth muscle cells, myofibroblasts, may be found in preretinal and subretinal
membranes.6 These cells are thought to originate from pigment epithelial cells. The
myofibroblasts are capable of secreting collagen as well as contracting, accounting
for the wrinkling of the internal limiting membrane, as
seen in “macular pucker” and proliferative vitreoretinopathy. Myofibroblasts
also have been implicated in the contraction of iris
neovascular membranes (ectropion uveae), cyclitic membranes, and the
organization of intraocular hemorrhage.7 The white “snow bank” seen on the inferior pars plana and retinal
periphery in chronic pars planitis represents organization of inflammatory
protein and cells with contributions from glial cells as well
as fibroblasts (presumably derived from nonpigmented ciliary epithelium).8 Organization of old vitreous hemorrhage also may appear white for similar
reasons. Myelin appears white (the difference between white and gray matter in the
central nervous system is myelin produced by oligodendroglia). Normally, myelinization
of the optic nerve axons proceeds centrifugally from
the nervous system and halts at the level of the lamina scleralis. Recall
that the axons of the optic nerve constitute the nerve fiber layer
of the retina and that the cell bodies of these axons reside in the
ganglion cell layer of the retina. Therefore, myelinated nerve fibers
in the retina have a superficial appearance ophthalmoscopically because
of their location in the nerve fiber layer. Their white appearance
results from myelin. They frequently have feathery edges because the
myelinization does not halt abruptly (Fig. 21). 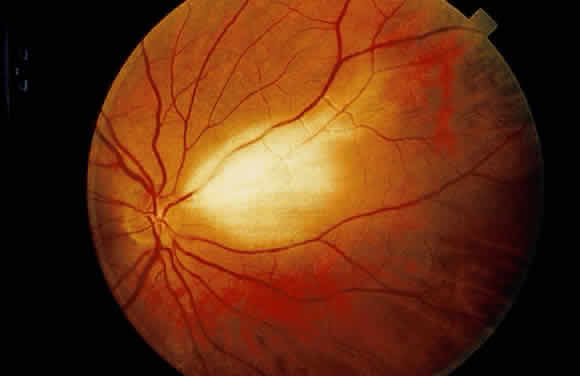 Fig. 21. Myelination of the nerve fibers extending beyond the disc. Notice the feathery
edges to the termination of the myelin. (Courtesy of William Tasman, MD, Philadelphia, PA) Fig. 21. Myelination of the nerve fibers extending beyond the disc. Notice the feathery
edges to the termination of the myelin. (Courtesy of William Tasman, MD, Philadelphia, PA)
|
Cotton-wool spots represent focal retinal infarcts at the level of the
nerve fiber layer. They result from ischemia-induced transection of the
axons of this layer. If axoplasmic transport continues to the point
of interruption, the material transported will accumulate at the zone
of transection in bulbous microscopic swellings, which, to early microscopists, resembled
cells and were called “cytoid [cell-like] bodies” (Fig. 22). 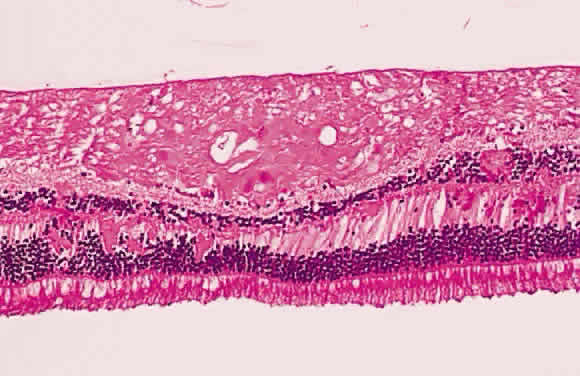 Fig. 22. Histologic section demonstrating retinal nerve fiber layer infarct. The
red dots within the infarct zone are bulbous swellings of nerve fiber
axons called “cytoid (cell-like) bodies.” The entire infarcted
zone is the histologic counterpart of the clinical cotton-wool spot. (Courtesy of Ralph C. Eagle Jr, MD, Philadelphia, PA) Fig. 22. Histologic section demonstrating retinal nerve fiber layer infarct. The
red dots within the infarct zone are bulbous swellings of nerve fiber
axons called “cytoid (cell-like) bodies.” The entire infarcted
zone is the histologic counterpart of the clinical cotton-wool spot. (Courtesy of Ralph C. Eagle Jr, MD, Philadelphia, PA)
|
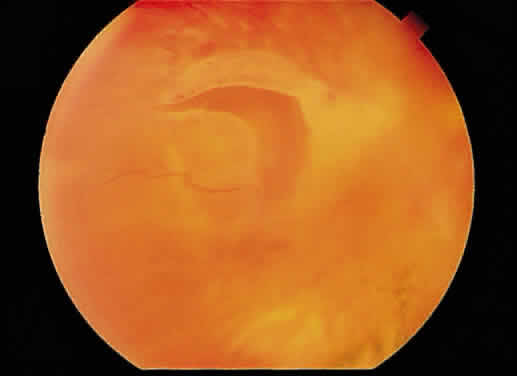
Cotton-wool spots are observed mainly in the posterior pole of the retina (Fig.23) The reason for this geographic restriction is not clear. Occlusion of
the most superficial radially oriented peripapillary capillaries (confined
in distribution to the posterior pole) has been implicated in the
pathogenesis of cotton-wool spots.9 It is also possible that nerve fiber infarcts in the periphery are not
visualized because there is insufficient inspissated axoplasmic material
in this location. 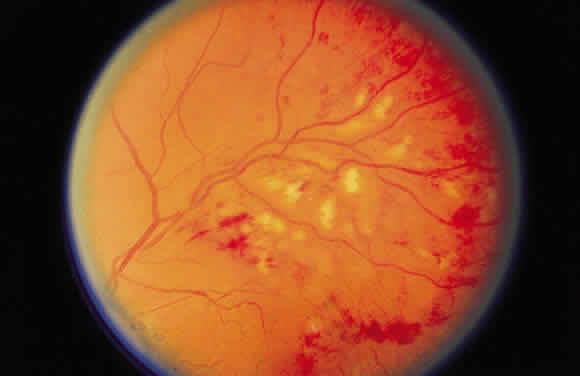 Fig. 23. Multiple cotton-wool spots along with retinal hemorrhages in a superior
temporal branch retinal vein occlusion. Fig. 23. Multiple cotton-wool spots along with retinal hemorrhages in a superior
temporal branch retinal vein occlusion.
|
Necrotic retina has a white appearance. Necrosis may result from a variety
of inflammatory conditions, including viral, fungal, and protozoal (Toxoplasma) retinitis. Each type of retinitis appears, in part, as a white retinal
lesion. Cytomegalovirus retinitis (Fig. 24) resembles a pizza pie with an admixture of white (retinal inflammation) and
red (hemorrhage) colors (Fig. 25). The retinal abscesses of fungal retinitis are white. Likewise, the satellite
lesion of retinochoroiditis caused by active Toxoplasma (the choroidal inflammation is merely in response to the primary retinal
infection) is white (Fig. 26). The white appearance of a lesion from inactive Toxoplasma results from the destruction of neurosensory retina, retinal pigment epithelium, and
choroid to permit a direct view of the sclera (Fig. 27). Necrosis in retinal-derived neoplasms is white; the appearance of regressed (necrotic) retinoblastoma often is described as “cottage
cheese.”10 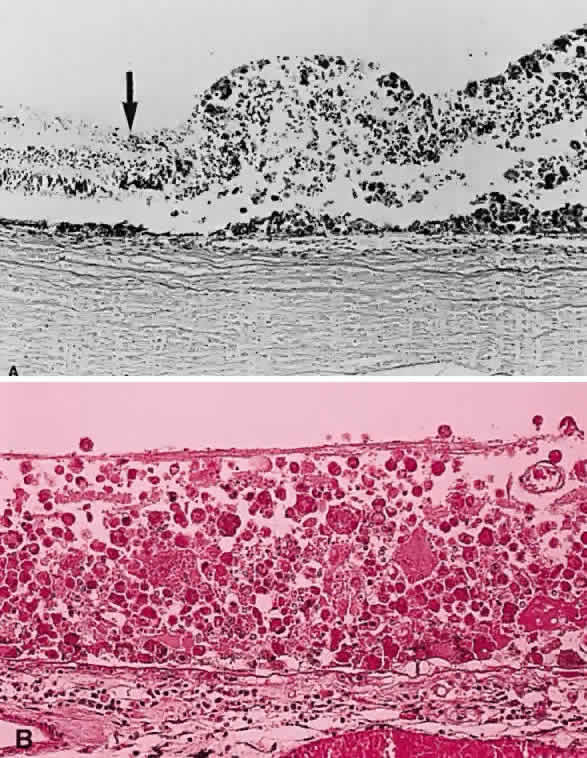 Fig. 24. A. Histologic section of cytomegalic inclusion retinitis. The characteristic
inclusions cannot be seen at this magnification. Compare the appearance
of viable healthy retina (left of arrow) with necrotic retina (right of arrow). The admixture of necrotic retina (clinically white) with hemorrhage (clinically
red) accounts for the ophthalmoscopic appearance of this entity. B. Cytomegalovirus retinitis. Histologic section of sensory retina demonstrating
massive necrosis involving all layers. (Courtesy of Ralph C. Eagle Jr, MD, Philadelphia, PA) Fig. 24. A. Histologic section of cytomegalic inclusion retinitis. The characteristic
inclusions cannot be seen at this magnification. Compare the appearance
of viable healthy retina (left of arrow) with necrotic retina (right of arrow). The admixture of necrotic retina (clinically white) with hemorrhage (clinically
red) accounts for the ophthalmoscopic appearance of this entity. B. Cytomegalovirus retinitis. Histologic section of sensory retina demonstrating
massive necrosis involving all layers. (Courtesy of Ralph C. Eagle Jr, MD, Philadelphia, PA)
|
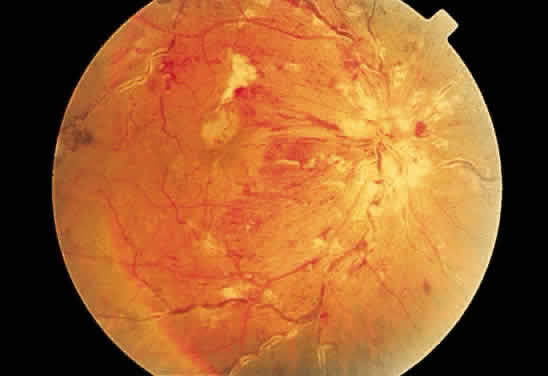 Fig. 25. Fundus photograph of cytomegalovirus retinitis with the classic admixture
of retinal infection (white) and hemorrhage, giving the so-called “pizza-pie” appearance. Fig. 25. Fundus photograph of cytomegalovirus retinitis with the classic admixture
of retinal infection (white) and hemorrhage, giving the so-called “pizza-pie” appearance.
|
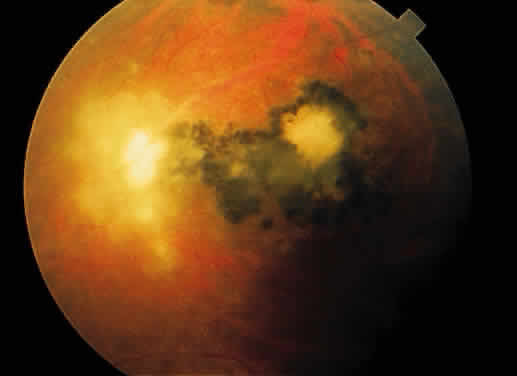 Fig. 26. Fundus photograph of toxoplasmosis chorioretinitis scar with white (active) satellite
lesion. Fig. 26. Fundus photograph of toxoplasmosis chorioretinitis scar with white (active) satellite
lesion.
|
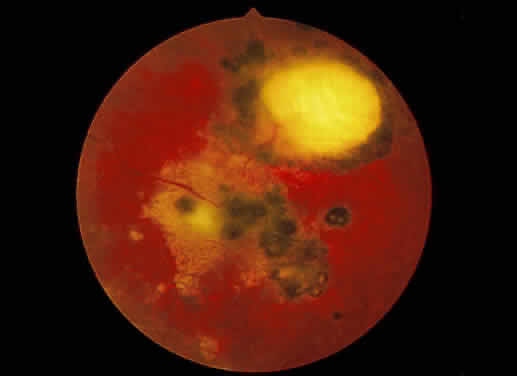 Fig. 27. Fundus photographs of quiescent toxoplasmosis chorioretinitis scar. The
white center is the result of destruction of the neurosensory retina, retinal
pigment epithelium, and choroid, leaving only the sclera in view. Fig. 27. Fundus photographs of quiescent toxoplasmosis chorioretinitis scar. The
white center is the result of destruction of the neurosensory retina, retinal
pigment epithelium, and choroid, leaving only the sclera in view.
|
Occasionally, accumulations of inflammatory cells may appear white even
in the absence of necrosis. Sheathing around retinal vessels (including
the “candle wax drippings” seen in retinal sarcoid) represents
a perivascular accumulation of inflammatory cells. Roth spots, which
are white-centered hemorrhages, as seen in patients with bacterial
endocarditis, are thought to represent the abscess within the hemorrhage. White-centered
hemorrhages may occur in other conditions, and
the explanation for the white center ranges from accumulations of leukemic
cells to cytoid bodies. Duane and associates found fibrin and platelet
aggregates in several patients with white-centered hemorrhages of
various etiologies.11 Granulomatous inflammatory deposits often appear yellow to yellow-white, the
clinical appearance of the Dalen-Fuchs nodule of sympathetic ophthalmia. Structures in the fundus may calcify, but the resultant white changes typical
of calcium may be difficult to discern ophthalmoscopically because
of the location of the calcium, viz., drusen of the optic nerve head. These calcific concretions are buried
within the substance of the optic nerve head, usually anterior to the
lamina scleralis (Fig. 28). They are covered by axonal and glial tissue together with the vascular
supply of the nerve head. They are recognizable because of distortions
in the shape of the disc, not the characteristic white color of the
calcified lesion (Fig. 29). Drusen of the optic nerve head must not be confused clinically with
papilledema (Figs. 30 and 31), with so-called “giant drusen,” which are glial hamartomata, or
with drusen of the pigment epithelium, which are deposits of basement
membrane material between the pigment epithelium and Bruch's
membrane. 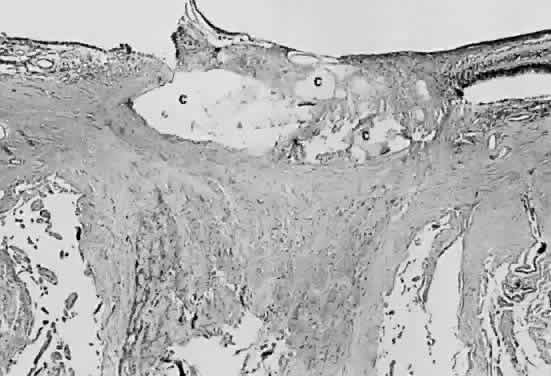 Fig. 28. Photomicrograph of drusen of the optic nerve head. Calcium (c) is deposited in the nerve anterior to the lamina scleralis. The optic
nerve in this case is markedly atrophic. The retina to the left of the
nerve is artifactually detached and missing from the plane of the section. Fig. 28. Photomicrograph of drusen of the optic nerve head. Calcium (c) is deposited in the nerve anterior to the lamina scleralis. The optic
nerve in this case is markedly atrophic. The retina to the left of the
nerve is artifactually detached and missing from the plane of the section.
|
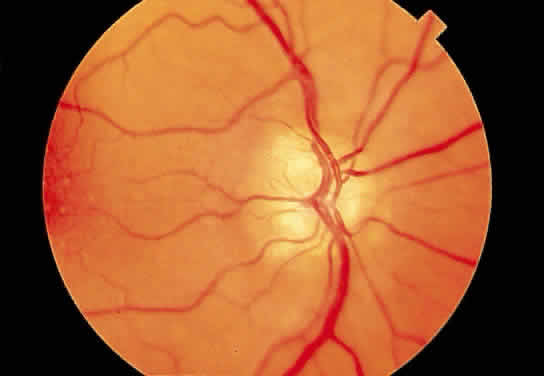 Fig. 29. Drusen of the optic disc. Fig. 29. Drusen of the optic disc.
|
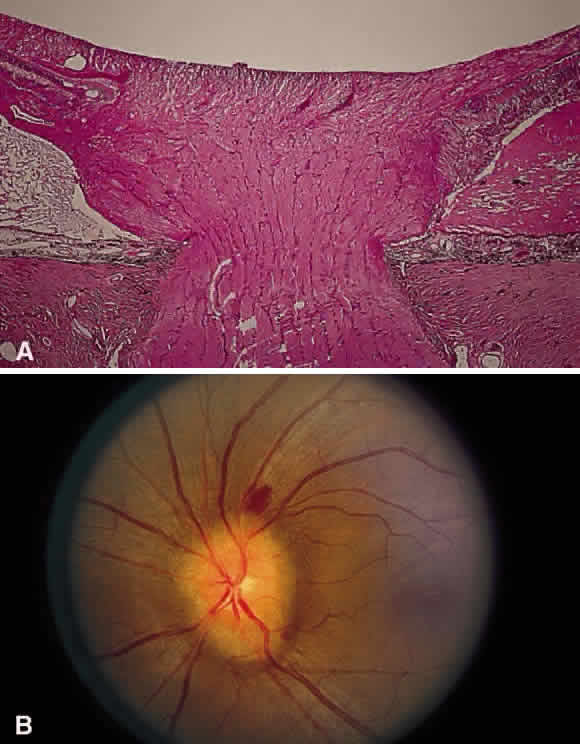 Fig. 30. A. Photomicrograph of an optic disc with papilledema. There is edema of the
disc surface, some engorgement of the vessels, and lateral displacement
of the photoreceptor elements, which results in enlargement of the
blind spot in papilledema. B. Early papilledema in a patient with pseudotumor cerebri. The disc margin
is blurred, the surface slightly elevated, and a small hemorrhage is
present superiorly. (A, courtesy of Ralph C. Eagle Jr, MD, Philadelphia, PA) Fig. 30. A. Photomicrograph of an optic disc with papilledema. There is edema of the
disc surface, some engorgement of the vessels, and lateral displacement
of the photoreceptor elements, which results in enlargement of the
blind spot in papilledema. B. Early papilledema in a patient with pseudotumor cerebri. The disc margin
is blurred, the surface slightly elevated, and a small hemorrhage is
present superiorly. (A, courtesy of Ralph C. Eagle Jr, MD, Philadelphia, PA)
|
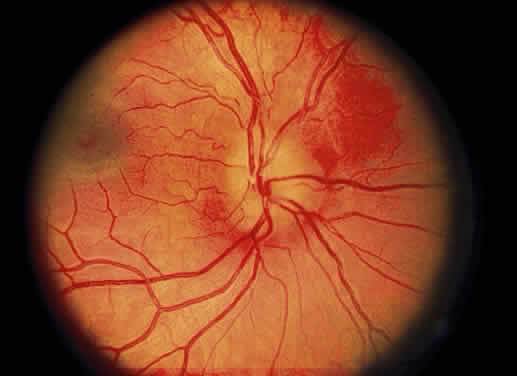 Fig. 31. Fundus photo of pseudopapilledema. The disc margin is blurred with slight
elevation of the disc surface in a 16-year-old hyperopic boy followed
for 10 years without change. Fig. 31. Fundus photo of pseudopapilledema. The disc margin is blurred with slight
elevation of the disc surface in a 16-year-old hyperopic boy followed
for 10 years without change.
|
It is difficult to visualize calcification at the level of Bruch's
membrane unless there is a focal disturbance in the distribution of melanin
in the overlying pigmented epithelium. For example, the calcification
of Bruch's membrane that occurs with age is not visible ophthalmoscopically
when the pigment epithelium is intact. Parenthetically, it
would be advantageous to detect this calcification because the
brittleness that it induces in Bruch's membrane often leads to breaks
in this barrier, creating an access path to the subretinal space
by choroidal neovascular membranes in the pathogenesis of exudative (wet) macular
degeneration.4 In the case of retinal pigment epithelial drusen, the presence of basement
membrane material (the substance of the drusen) atop Bruch's
membrane attenuates the overlying pigmented epithelium (Fig. 32), thinning out the normal distribution of melanin in these cells. Usually, these
basement membrane deposits appear yellow-white (Fig. 33), but when they calcify, they may appear ophthalmoscopically as white, elevated
dots. 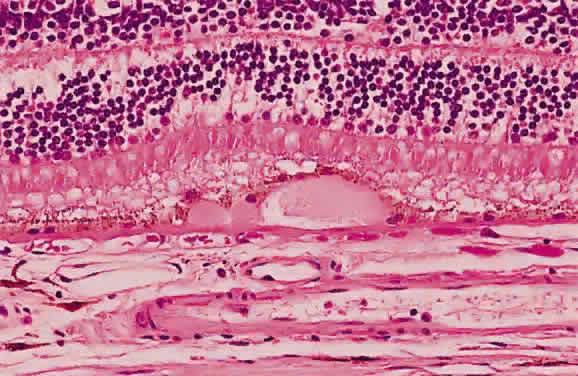 Fig. 32. Photomicrograph of a druse of the pigment epithelium. Notice the attenuation
of the overlying pigment epithelium. (Courtesy of Ralph C. Eagle Jr, MD, Philadelphia, PA) Fig. 32. Photomicrograph of a druse of the pigment epithelium. Notice the attenuation
of the overlying pigment epithelium. (Courtesy of Ralph C. Eagle Jr, MD, Philadelphia, PA)
|
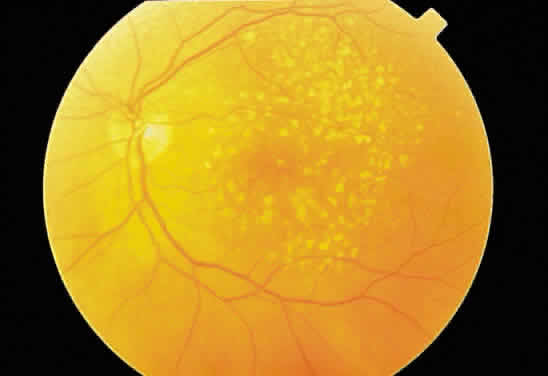 Fig. 33. Discrete and confluent drusen of the macula. Fig. 33. Discrete and confluent drusen of the macula.
|
GRAY-WHITE OPACIFICATION OF THE RETINA Transparent tissue, such as cornea or retina, when edematous, appears gray-white. A
typical example is the macular edema in diabetes, the leading
cause of visual impairment in diabetic patients (Fig. 34). Technically, the term edema refers to the extracellular accumulation of fluid. 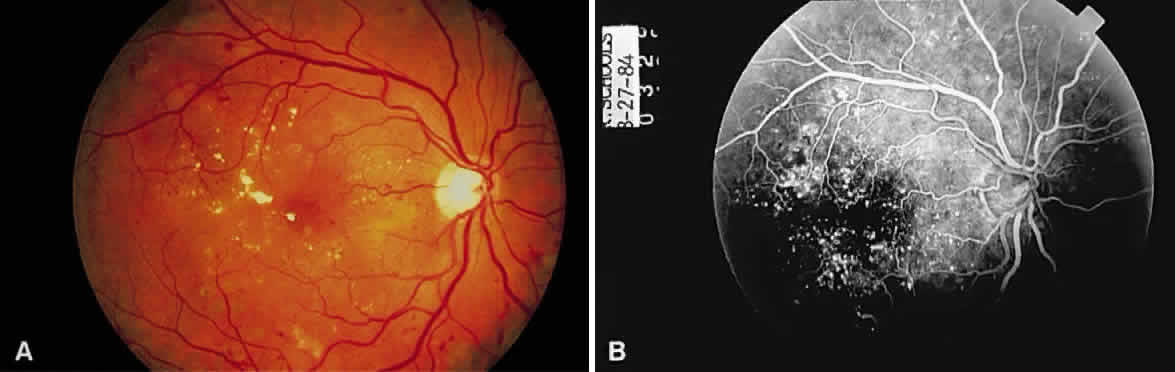 Fig. 34. A. Fundus photomicrograph of macular edema in a diabetic. Yellow-white edema
residues (hard exudates) and a few paramacular hemorrhages are present. B. Multiple microaneurysms are present on fluorescein angiography. Fig. 34. A. Fundus photomicrograph of macular edema in a diabetic. Yellow-white edema
residues (hard exudates) and a few paramacular hemorrhages are present. B. Multiple microaneurysms are present on fluorescein angiography.
|
Intracellular accumulation of fluid is designated by many as “hydropic
degeneration.” When a cell is about to die, there is a disturbance
at the level of the plasmalemma that renders the cell incapable
of separating the intracellular environment from the extracellular
environment. There is an influx of ions and fluid into the cell. Acute
ischemia of the retina, as reflected clinically by a central retinal
artery occlusion (Fig. 35), is seen ophthalmoscopically as a gray-white opacification of the retina
and likely reflects this metabolic disturbance. 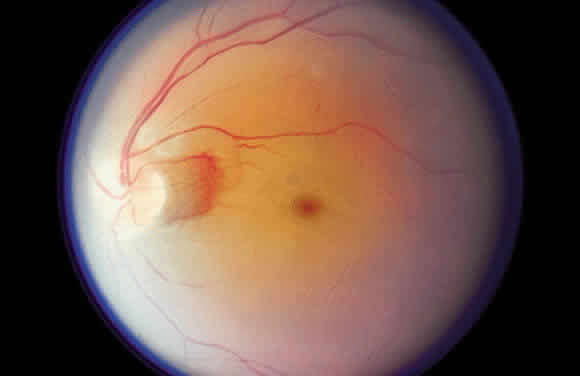 Fig. 35. Acute central retinal artery occlusion demonstrating generalized opacification
of the retina in the posterior pole except for the “cherry-red
spot” fovea and a small area of “normal” retina
adjacent to the disc supplied by a small cilioretinal artery. Fig. 35. Acute central retinal artery occlusion demonstrating generalized opacification
of the retina in the posterior pole except for the “cherry-red
spot” fovea and a small area of “normal” retina
adjacent to the disc supplied by a small cilioretinal artery.
|
Gray-white macular opacification may result from various pathogenetic mechanisms, to
which allusions have been made previously: (1) storage diseases, such
as Tay-Sachs disease (accumulation of storage material in
the ganglion cell layer); (2) accumulation of extracellular fluid (as
in diabetic retinopathy); and (3) early retinal necrosis (as in acute
central retinal artery occlusion). Notice that each of these entities
has a different pathologic basis yet their ophthalmoscopic presentation
is similar. Sometimes, even within a single clinicopathologic setting, a change within
the fundus may be explained by various mechanisms. For example, after
blunt trauma to the eye, the macula may undergo transient gray-white
opacification (Fig. 36) referred to as commotio retinae (also known as Berlin's edema). The
gray-white appearance of commotio retinae is explained by a disruption
at the cellular level of the photoreceptors.12 Presumably, as this disruption is reversed, the opacification resolves. This
explanation accounts for the clinical course of patients with commotio
retinae who show resolution of the fundus abnormality. Although
this theory has gained support, it does not explain completely the appearance
of a macular cyst or hole as possible sequelae of commotio retinae (Fig. 37). An older theory explained the gray-white appearance of commotio retinae
on the basis of transient vascular incompetence leading to the accumulation
of extracellular fluid in the macula (macular edema). According
to the “edema” theory, as the fluid was resorbed, the
gray-white opacification resolved. Although this edema theory has been
largely discredited,12 it offers an explanation for the development of a macular cyst or hole
after commotio retinae. Also, perhaps the same mechanism of Müller
cell damage that results in the lesion of cystoid macular edema1,2 may operate in some cases of blunt trauma to the eye and thus explain
the formation of a macular cyst or hole after commotio retinae. Finally, it
is possible that multiple pathogenetic mechanisms contribute to
the appearance of commotio retinae, depending on the violence of the blunt
injury to the eye (i.e., in milder blunt trauma, there is only photoreceptor damage and the gray-white
opacification resolves, whereas in more severe trauma, there is
true macular edema, Müller cell damage, or both, which may lead
to macular cyst or hole formation). 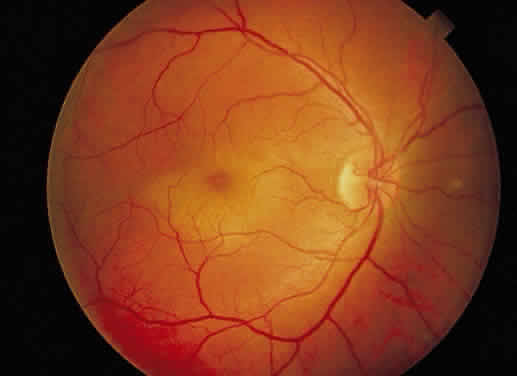 Fig. 36. Fundus photo of macular edema secondary to blunt trauma to the globe. This
is known as Berlin's edema or commotio retinae. (Courtesy of William Tasman, MD, Philadelphia, PA) Fig. 36. Fundus photo of macular edema secondary to blunt trauma to the globe. This
is known as Berlin's edema or commotio retinae. (Courtesy of William Tasman, MD, Philadelphia, PA)
|
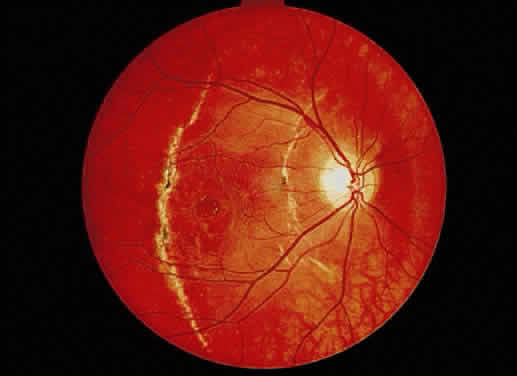 Fig. 37. Fundus photograph of traumatic macular hole. Notice the choroidal ruptures
and the pigment disturbance. The patient's VA has remained 20/40 for 11 years. Fig. 37. Fundus photograph of traumatic macular hole. Notice the choroidal ruptures
and the pigment disturbance. The patient's VA has remained 20/40 for 11 years.
|
YELLOW CHANGES IN THE FUNDUS The accumulation of lipid usually accounts for yellow deposits in the fundus. Lipid
may be derived from degenerated cells such as senescent erythrocytes
or from exudation. In the case of hemorrhage, the plasmalemma
of red blood cells frequently contributes to the formation of cholesterol
deposits, leading to the clinically observed condition of cholesterolosis
bulbi. The cholesterol crystals may be seen in the vitreous
by ophthalmoscopic examination or in the anterior chamber by slit-lamp
examination (Figs. 38 and 39). (Parenthetically, another byproduct of cell membrane degradation can
accumulate in the retinal pigment epithelium and appear orange. Lipofuscin
results from the accumulation of “residual bodies,” the
residua of phagolysosomes that have digested intracellular debris. This
orange color may be seen over the surface of choroidal nevi and
especially melanoma.13) 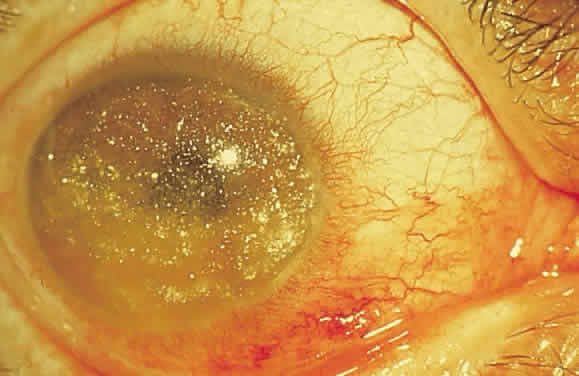 Fig. 38. Slit-lamp photograph of cholesterol crystals (cholesterolosis bulbi) in
anterior chamber. Similar crystals may be seen in the vitreous. Fig. 38. Slit-lamp photograph of cholesterol crystals (cholesterolosis bulbi) in
anterior chamber. Similar crystals may be seen in the vitreous.
|
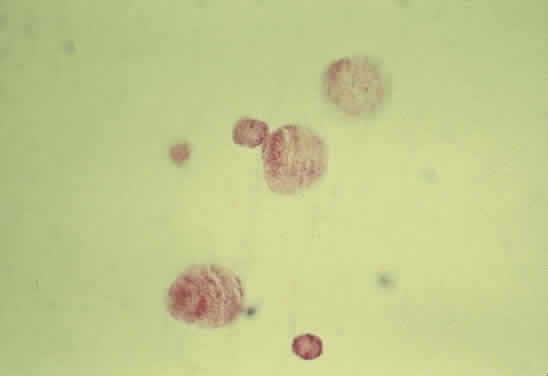 Fig. 39. Photomicrograph of cholesterol crystals filling the anterior chamber. The
crystals showed birefringence with polarized light and stained positive
with Schultz' method for cholesterol. Fig. 39. Photomicrograph of cholesterol crystals filling the anterior chamber. The
crystals showed birefringence with polarized light and stained positive
with Schultz' method for cholesterol.
|
Transudation and exudation imply vascular leakage. A transudate consists
of serum and has a low concentration of proteins and lipoproteins. Transudation
occurs when there is an imbalance in Starling's factors (the
intravascular pressure, the tissue turgor, and osmolarity of the
intravascular and tissue spaces). Exudation occurs when there is damage
to the vessel, allowing nonselective escape of plasma components
such as higher molecular weight proteins and lipoproteins. Therefore, whenever exudate is seen ophthalmoscopically, the examiner should be prepared
to attribute this finding to specific vascular abnormality. Exudates appear yellow or yellow-white, depending on the amount of lipid
present. When exudates are observed ophthalmoscopically over time, they
frequently appear to lose the yellow component as the content of lipid
in the deposit diminishes. Exudates are removed by vascular resorption
or by phagocytosis. Exudates tend to accumulate in the outer plexiform layer. Since this layer
is oriented perpendicularly to the internal limiting membrane, small
exudates assume a cylindrical shape in three dimensions. When small
exudates are viewed ophthalmoscopically, they appear round because the
cylinder is being viewed in a cross-section. Usually, these small exudates
can be distinguished from cotton-wool spots (which are not exudates
but are nerve fiber layer infarcts) by location, shape, and color; nerve
fiber layer infarcts occupy a more superficial location, tend
to have less distinct edges, and are more white than yellow. The amount and geographic location of the exudate may alter the appearance
of the lesion. Larger exudates appear more globoid (Fig. 40A) than the smaller exudates described earlier. Exudates that accumulate
in the macula still may occupy the outer plexiform layer, but because
this layer is oriented obliquely (see Fig. 40B), the examiner sees the cylindrical accumulations in profile rather than
in cross-section. These exudates resemble the spokes of a wheel radiating
from a central hub or the spokes of light radiating from a star (“macular
star”) (Fig. 41). 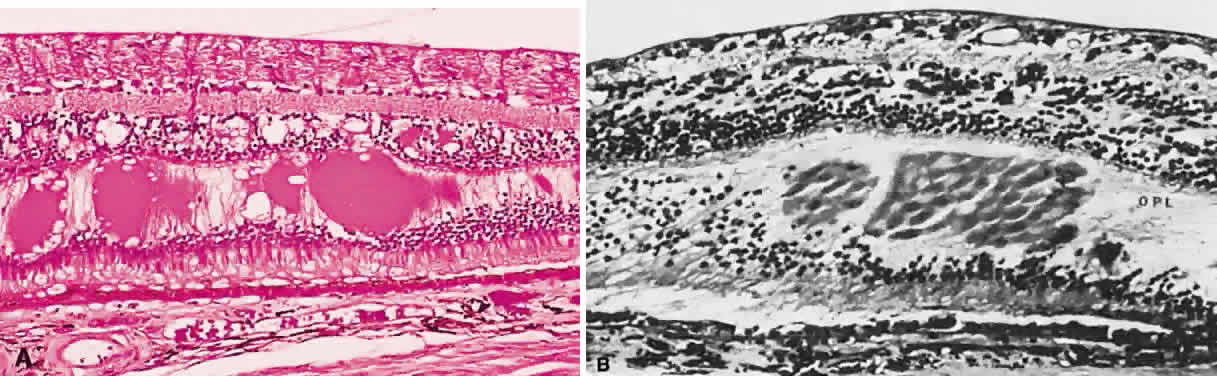 Fig. 40. A. Fundus photo demonstrating large globular exudates in the outer plexiform
layer. B. Photomicrograph of exudate in the macula. Notice the oblique orientation
of the fibers of the outer plexiform layer. (A, courtesy of Ralph C. Eagle Jr, MD, Philadelphia, PA) Fig. 40. A. Fundus photo demonstrating large globular exudates in the outer plexiform
layer. B. Photomicrograph of exudate in the macula. Notice the oblique orientation
of the fibers of the outer plexiform layer. (A, courtesy of Ralph C. Eagle Jr, MD, Philadelphia, PA)
|
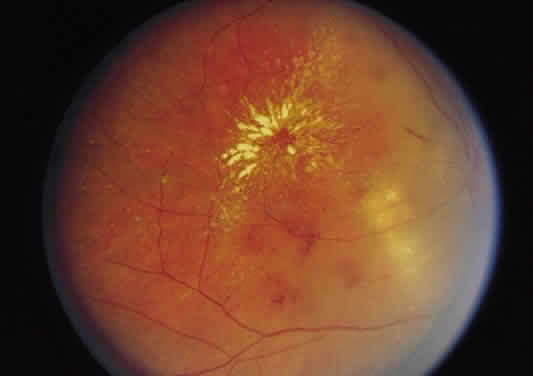 Fig. 41. Edema residues (hard exudates) in the form of a macular star in a patient
with severe hypertension secondary to renal disease. Fig. 41. Edema residues (hard exudates) in the form of a macular star in a patient
with severe hypertension secondary to renal disease.
|
Whereas the presence of an exudate demands a search for vascular pathologic
changes, the shape of the exudate may help to localize the vascular
abnormality. A star-shaped macular exudate may result from local vascular
disturbances such as hypertensive retinopathy. If the lipid accumulation
in the macula is abundant, the exudates may appear globular
and may be arranged in a circinate or garland-like pattern. Macular exudates
may accumulate not only because of local vascular abnormality, but
also as a result of a vascular lesion in the retinal periphery, such
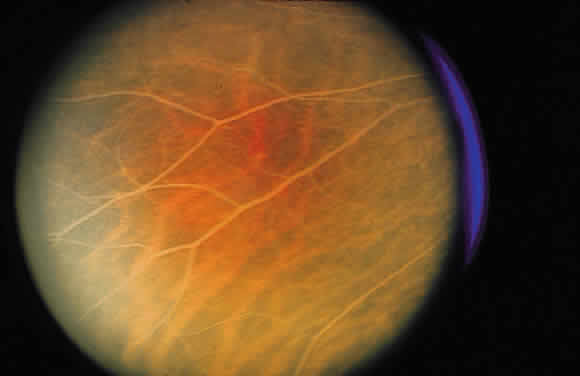
as the vascular retinal lesions of Von Hippel (Fig. 42) or Coats (Fig. 43), inflammation in the optic nerve head, or hypertensive retinopathy (Fig. 44). 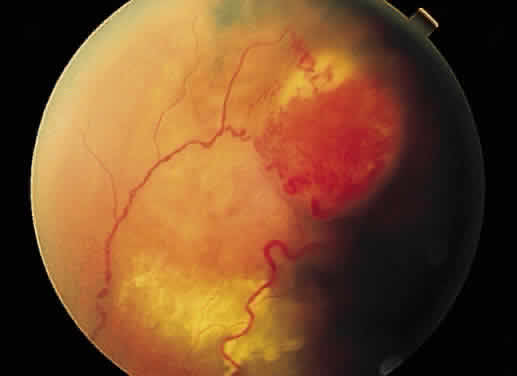 Fig. 42. Fundus photograph of Von Hippel lesion. Notice the dilated, tortuous vessels
leading to and from the tumor and the exudate just visible in the
macular region at the bottom of the photo. Fig. 42. Fundus photograph of Von Hippel lesion. Notice the dilated, tortuous vessels
leading to and from the tumor and the exudate just visible in the
macular region at the bottom of the photo.
|
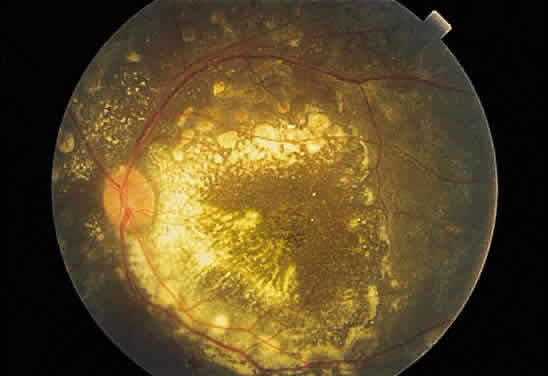 Fig. 43. Fundus photograph of Coats' disease. Dense exudate is present in the posterior
pole. The peripheral lesions, the source of the exudates, are
not visible in the photograph. Fig. 43. Fundus photograph of Coats' disease. Dense exudate is present in the posterior
pole. The peripheral lesions, the source of the exudates, are
not visible in the photograph.
|
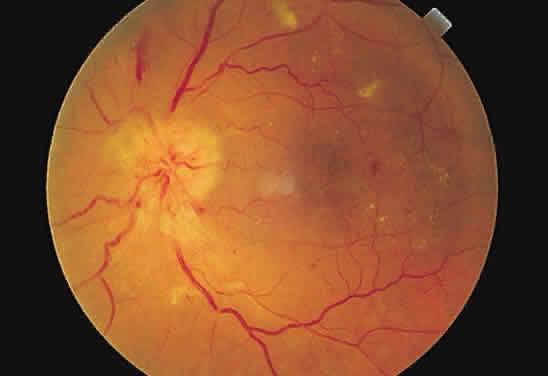 Fig. 44. Fundus photograph of hypertensive retinopathy, grade IV. The disc is swollen
with surrounding “splinter” hemorrhages. The arterioles
are narrowed throughout, the circumpapillary retina is edematous, and
macular exudates are present. Fig. 44. Fundus photograph of hypertensive retinopathy, grade IV. The disc is swollen
with surrounding “splinter” hemorrhages. The arterioles
are narrowed throughout, the circumpapillary retina is edematous, and
macular exudates are present.
|
When an exudate is present in a ring configuration outside of the macula, the
vascular disease is most commonly found within the ring. Retinal
macroaneurysm is an example (Fig. 45) of a focal retinal vascular lesion surrounded frequently by exudate. 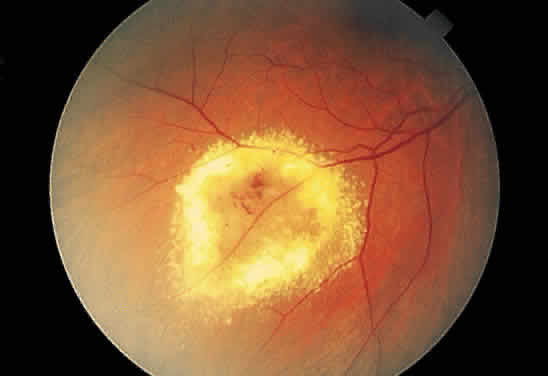 Fig. 45. Fundus photograph of circinate exudate. Notice the macroaneurysm at the
center of the exudate ring. Fig. 45. Fundus photograph of circinate exudate. Notice the macroaneurysm at the
center of the exudate ring.
|
BLACK CHANGES IN THE FUNDUS Black changes in the fundus usually are the result of changes occurring
in the retinal pigment epithelium, which is capable of a variety of responses
to injury. It may lose pigment, enlarge and gain pigment (hypertrophy), or replicate (hyperplasia). It also may undergo change to another type of adult tissue such as fibrous
tissue or bone (metaplasia) (see Fig. 19). The attenuation of the pigment epithelium overlying drusen is an example
of relative loss of pigment by the cell, better revealing the appearance
of the basement membrane accumulation that represents the lesion. Another
example of pigment loss is seen in the “salt and pepper” fundus
of congenital rubella, in which focal patches of retinal
pigment epithelial hypopigmentation (salt) alternate with hyperpigmentation (pepper)14 (Fig. 46). 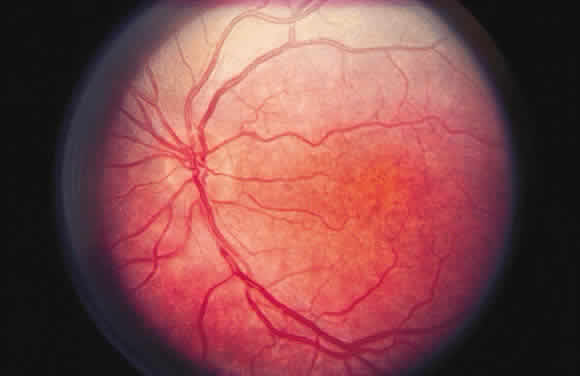 Fig. 46. Fundus photograph of rubella retinopathy. Notice the fine “salt and
pepper” pattern resulting from pigment loss and microclumping. Despite
extensive pigmentary changes, the visual acuity and electroretinogram
may be within normal limits. Fig. 46. Fundus photograph of rubella retinopathy. Notice the fine “salt and
pepper” pattern resulting from pigment loss and microclumping. Despite
extensive pigmentary changes, the visual acuity and electroretinogram
may be within normal limits.
|
The accumulation of additional pigmentation by the pigment epithelium frequently
is accompanied by enlargement of the cell. This alteration may
be seen as a normal anatomical variant in the macula, in which the
pigment epithelium is taller and more pigmented than in other retinal
zones. The physiologic hypertrophy of the macula accounts for some darkening
of the fundus color in this area. A similar response is seen in congenital hypertrophy of the pigment epithelium15 (Fig. 47). The hypertrophy may be so prominent as to impart a jet black appearance
to the fundus focally. Congenital hypertrophy of the pigment epithelium
may be mistaken clinically for choroidal nevi and melanomas, but
the two groups of lesions are distinct ophthalmoscopically. Congenital
hypertrophy appears jet black, whereas choroidal melanocytic lesions
appear slate gray to brown-black (Fig. 48). In addition, the pigmentation within congenital hypertrophy of the pigment
epithelium may not be uniform, accounting for focal “lacunae” of “depigmentation.” Also, the borders of the congenital
pigment epithelial hypertrophy frequently are distinct and scalloped. Grouped
pigmentation of the fundus (“bear tracks”) is
another manifestation of hypertrophy of the pigment epithelium.16 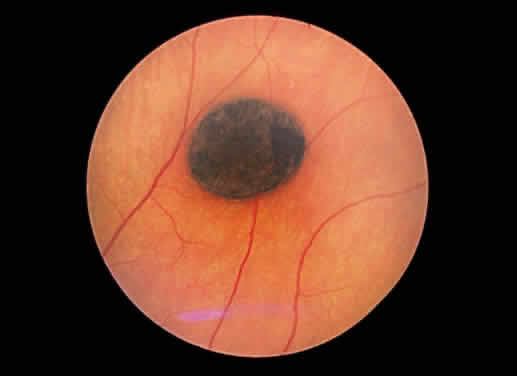 Fig. 47. Fundus photograph of congenital hypertrophy of the pigment epithelium. Notice
the sharp delineation of the lesion and its jet black color. Fig. 47. Fundus photograph of congenital hypertrophy of the pigment epithelium. Notice
the sharp delineation of the lesion and its jet black color.
|
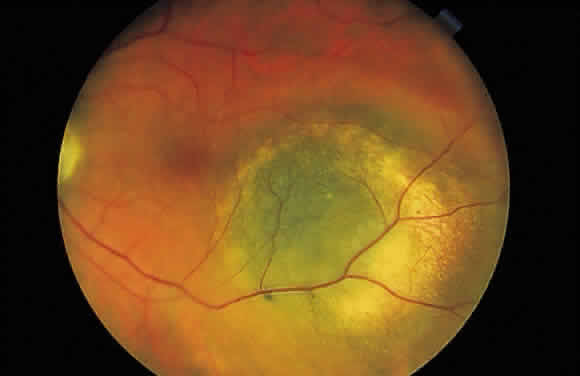 Fig. 48. Fundus photograph of choroidal melanocytic lesion. Notice its slate gray
color, which differentiates it from the jet black congenital hypertrophy
of the pigment epithelium. Fig. 48. Fundus photograph of choroidal melanocytic lesion. Notice its slate gray
color, which differentiates it from the jet black congenital hypertrophy
of the pigment epithelium.
|
Notice that all of the examples of hypertrophy of the retinal pigment epithelium
cited earlier refer to conditions present at birth. Since the
retinal pigment epithelial cells are dopa-negative postnatally, it is
doubtful that they synthesize additional pigment in adulthood.17 When injury induces pigment epithelial hyperplasia, tight adhesions may
be formed between this layer and the neurosensory retina. The black demarcation
lines seen in detachments of the neurosensory retina is an
example of this type of pigment epithelial response (Fig. 49). The seal thus created tends not be as tight as one created therapeutically
when performing retinopexy, and the detachment is more likely to
advance beyond the demarcation line. 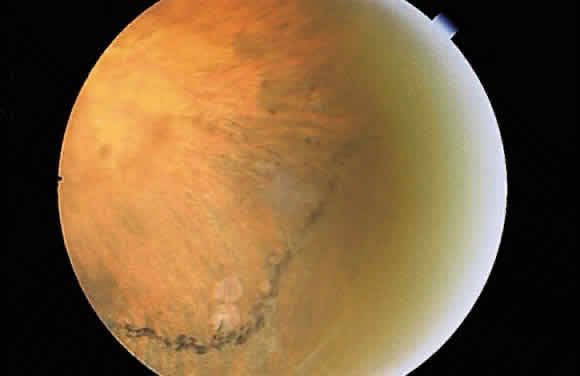 Fig. 49. Fundus photograph of a demarcation line, the result of pigment epithelial
hyperplasia “walling off” a neurosensory detachment. Fig. 49. Fundus photograph of a demarcation line, the result of pigment epithelial
hyperplasia “walling off” a neurosensory detachment.
|
Clumps of black pigmentation may also be seen in chorioretinal scars. The
lesion of quiescent toxoplasmosis may appear white centrally (bare
sclera) but may be rimmed by clumps of black pigmentation, reflecting
the response of the pigment epithelium to prior inflammation or trauma (see Figs. 19 and 37). Another example of pigment epithelial response to injury is seen in
the ophthalmoscopic appearance of some photocoagulation spots. Whereas
the heat generated by the laser may destroy the cells focally, pigment
epithelium adjacent to the treatment zone frequently responds by hyperplasia. Destruction
of the pigment epithelium may result in dispersion
of melanin, which is taken up by other pigment epithelial cells or
macrophages, thus imparting a black color to the edge of the injured zone. When the pigment epithelium responds by hyperplasia, it deposits basement
membrane and frequently deposits collagen (metaplasia). Thus, pigment
epithelial hyperplasia, often accompanied by metaplasia, may result
in a lesion that is mildly elevated. The bone that appears in phthisical
globes is presumed to be derived from metaplastic pigmented epithelium. Pigment epithelial melanin may accumulate within the retina, especially
around retinal vessels (Fig. 50). Because the small retinal vessels branch frequently, the pigment appears
to have the “bone-corpuscular” appearance typically seen
in retinitis pigmentosa (Fig. 51) but also after blunt ocular trauma and intraocular inflammation. 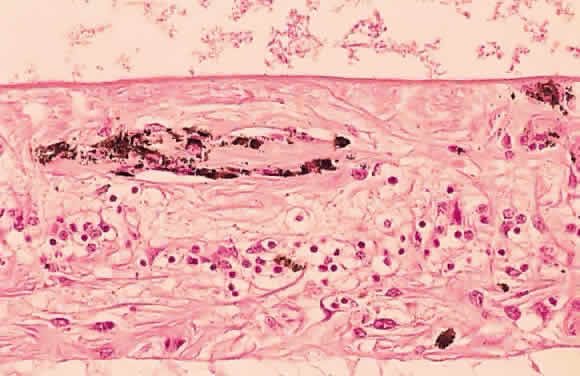 Fig. 50. Photomicrograph of primary retinitis pigmentosa demonstrating pigment accumulation
surrounding a superficial blood vessel and absence of the
rods and cones in the posterior retinal layers. (Courtesy of Ralph C. Eagle Jr, MD, Philadelphia, PA) Fig. 50. Photomicrograph of primary retinitis pigmentosa demonstrating pigment accumulation
surrounding a superficial blood vessel and absence of the
rods and cones in the posterior retinal layers. (Courtesy of Ralph C. Eagle Jr, MD, Philadelphia, PA)
|
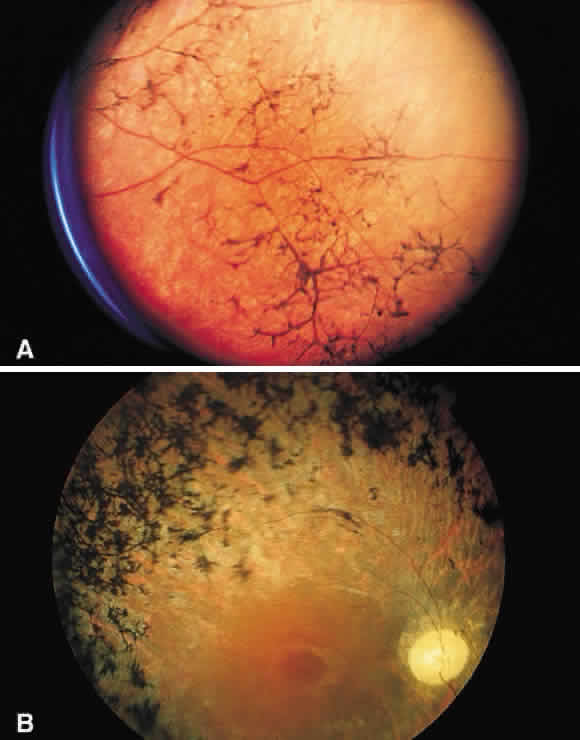 Fig. 51. A. Fundus photograph of retinitis pigmentosa with bone spicule pigmentation. The “bone-corpuscular” appearance is the result of pigment
epithelial melanin accumulation around the small retinal vessel branches. B. Advanced retinitis pigmentosa. There is marked pigment clumping along
with waxy pallor of the disc and attenuated arterioles. Fig. 51. A. Fundus photograph of retinitis pigmentosa with bone spicule pigmentation. The “bone-corpuscular” appearance is the result of pigment
epithelial melanin accumulation around the small retinal vessel branches. B. Advanced retinitis pigmentosa. There is marked pigment clumping along
with waxy pallor of the disc and attenuated arterioles.
|
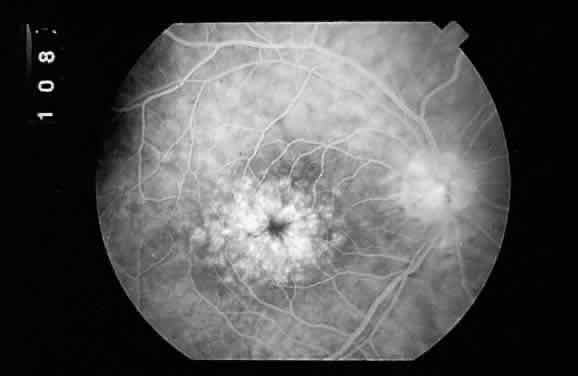
RED CHANGES IN THE FUNDUS Red lesions in the fundus indicate the presence of blood or abnormal blood
vessels in an abnormal location. The blood may be intravascular, implying
a vascular pathologic process, or extravascular, as in hemorrhage. Microaneurysms, such as those seen in diabetic retinopathy (Fig. 52), are reflective of vascular abnormality, which has been demonstrated
histologically using trypsin-digestion techniques (Fig. 53). Notice that many microaneurysms measure less than 60 μm in diameter. Since
the maximum resolving power of the direct ophthalmoscope is 60 μm, many
of the small red dots observed in the fundus are not microaneurysms
but microhemorrhages. In fact, alterations in vascular permeability
are seen early in background diabetic retinopathy, and the
microaneurysms frequently leak fluorescein during angiographic studies. Therefore, fluorescein
angiography may be a more sensitive technique
for the detection of microaneurysms than is direct fundus observation.18 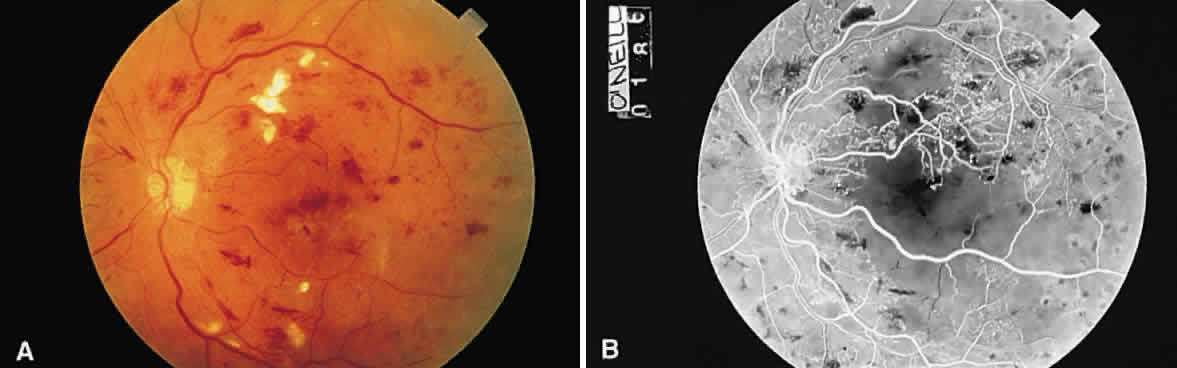 Fig. 52. A. Fundus photograph of nonproliferative diabetic retinopathy. Notice the
dilated veins, the small exudate clusters, the ischemic infarct (cotton-wool
spot), and the numerous microaneurysms and small hemorrhages. B. Mid-venous phase fluorescein angiogram of the same eye. Notice the focal
area of nonperfusion in the superior temporal arcade (ischemic areas), the
fluorescing microaneurysms, and the hemorrhages, which appear
dark in contrast to the background fluorescence. (Courtesy of William Tasman, MD, Philadelphia, PA) Fig. 52. A. Fundus photograph of nonproliferative diabetic retinopathy. Notice the
dilated veins, the small exudate clusters, the ischemic infarct (cotton-wool
spot), and the numerous microaneurysms and small hemorrhages. B. Mid-venous phase fluorescein angiogram of the same eye. Notice the focal
area of nonperfusion in the superior temporal arcade (ischemic areas), the
fluorescing microaneurysms, and the hemorrhages, which appear
dark in contrast to the background fluorescence. (Courtesy of William Tasman, MD, Philadelphia, PA)
|
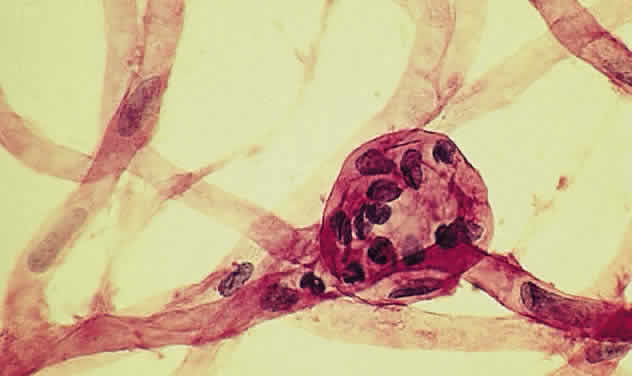 Fig. 53. Trypsin digest of the retinal capillary circulation demonstrating microaneurysm
formation. (Courtesy of Ralph C. Eagle Jr, MD, Philadelphia, PA) Fig. 53. Trypsin digest of the retinal capillary circulation demonstrating microaneurysm
formation. (Courtesy of Ralph C. Eagle Jr, MD, Philadelphia, PA)
|
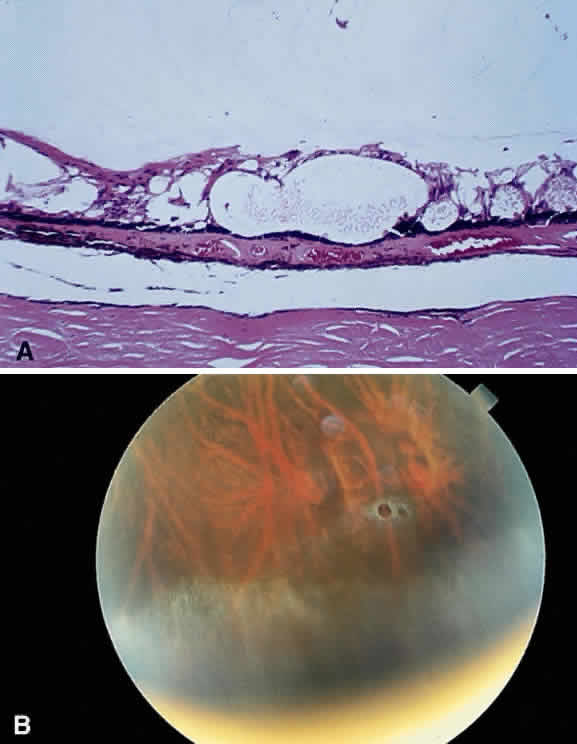
The term retinal neovascularization refers to new vessels originating within the retina that have broken through
the internal limiting membrane. In early retinal neovascularization, the
vessels do not invade the vitreous but lie between the internal
limiting membrane and the hyaloid face (Fig. 54). These new vessels are fragile and prone to rupture and bleed. The blood
may remain localized between the retina and the hyaloid (preretinal
or subhyaloid hemorrhage), but with time, many of these hemorrhages
break through into the vitreous itself (Fig. 55). 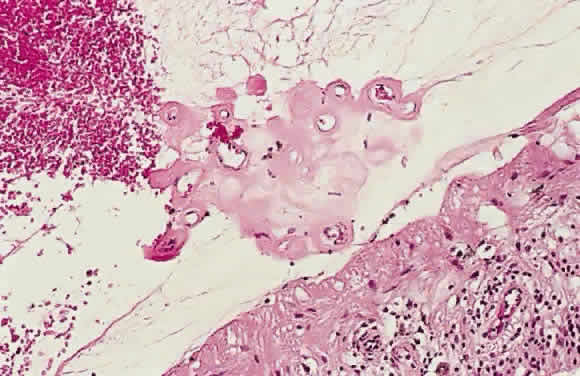 Fig. 54. Photomicrograph of retinal neovascularization. A tuft of new vessels is
seen anterior to the plane of the internal limiting membrane. The detached
vitreous contains old hemorrhage. (Courtesy of Ralph C. Eagle Jr, MD, Philadelphia, PA) Fig. 54. Photomicrograph of retinal neovascularization. A tuft of new vessels is
seen anterior to the plane of the internal limiting membrane. The detached
vitreous contains old hemorrhage. (Courtesy of Ralph C. Eagle Jr, MD, Philadelphia, PA)
|
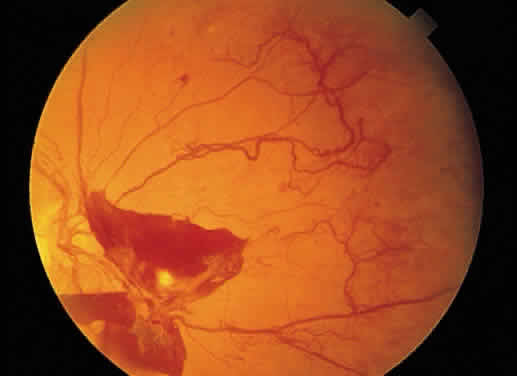 Fig. 55. Fundus photograph of early proliferative diabetic retinopathy. The new
vessels have not invaded the vitreous, and the hemorrhage has remained
between the retina and the hyaloid (preretinal/subhyaloid). Notice the
vascular loops and vascular duplication. Fig. 55. Fundus photograph of early proliferative diabetic retinopathy. The new
vessels have not invaded the vitreous, and the hemorrhage has remained
between the retina and the hyaloid (preretinal/subhyaloid). Notice the
vascular loops and vascular duplication.
|
When new vascular tissue invades the vitreous, the resulting hemorrhage, secondary
to vitreous traction on the fronds, tends to be more extensive. The
ophthalmoscopic appearance of vitreous hemorrhage depends on
the amount of blood present and its duration. If a small amount of blood
is present, the view of the fundus may be only slightly clouded and
red tinged. In massive acute hemorrhage, the view of the fundus is obscured
by bright red blood. With time, the blood may settle to the inferior
portion of the eye. If there is no additional hemorrhage, sequential
color changes may be observed in the vitreous. Since the red blood
cell has a finite life span, the cell eventually loses its metabolic
apparatus and its shape as a biconcave disc. With the plasmalemma no
longer intact, hemoglobin may seep out of the cells. The erythrocytes
no longer appear red but rather khaki or tan. These are ghost erythrocytes or ghost cells (Fig. 56). They begin to appear within 2 weeks after vitreous hemorrhage and may
be mistaken for inflammatory cells by vitreous biomicroscopic examination. Ghost
cells remain confined to the vitreous unless the anterior
hyaloid has been disrupted, in which case they invade the anterior chamber
and clog the trabecular meshwork. The result is a secondary open-angle
glaucoma (“ghost cell glaucoma”).19 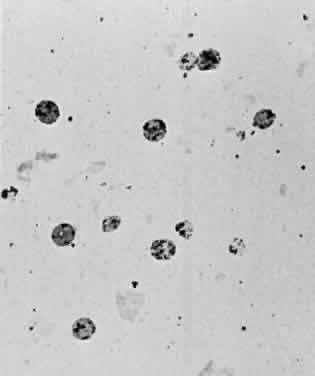 Fig. 56. Photomicrograph of a Millipore filter preparation of a vitrectomy specimen
taken from a patient with a vitreous hemorrhage of greater than 2 weeks' duration. Numerous
ghost erythrocytes are present. The dark dots
just inside the red cell membrane represent Heinz bodies. Fig. 56. Photomicrograph of a Millipore filter preparation of a vitrectomy specimen
taken from a patient with a vitreous hemorrhage of greater than 2 weeks' duration. Numerous
ghost erythrocytes are present. The dark dots
just inside the red cell membrane represent Heinz bodies.
|
The example of vitreous hemorrhage illustrates the color changes that may
occur with time in any intraocular hemorrhage. To further emphasize
this point, the blood settled inferiorly may become organized and eventually
appear white. Additionally, with repeated vitreous hemorrhages, enough
iron may be deposited within the eye to result in ocular hemosiderosis, similar
to a retained iron intraocular foreign body. The iron
may be deposited in the retina, imparting a brown tint to this otherwise
transparent and colorless tissue. Likewise, an old intraretinal
hemorrhage may appear brown because of the presence of hemosiderin within
it. Occasionally, intraretinal hemosiderin appears granular. These
deposits are thought to account for the refractile bodies seen in the
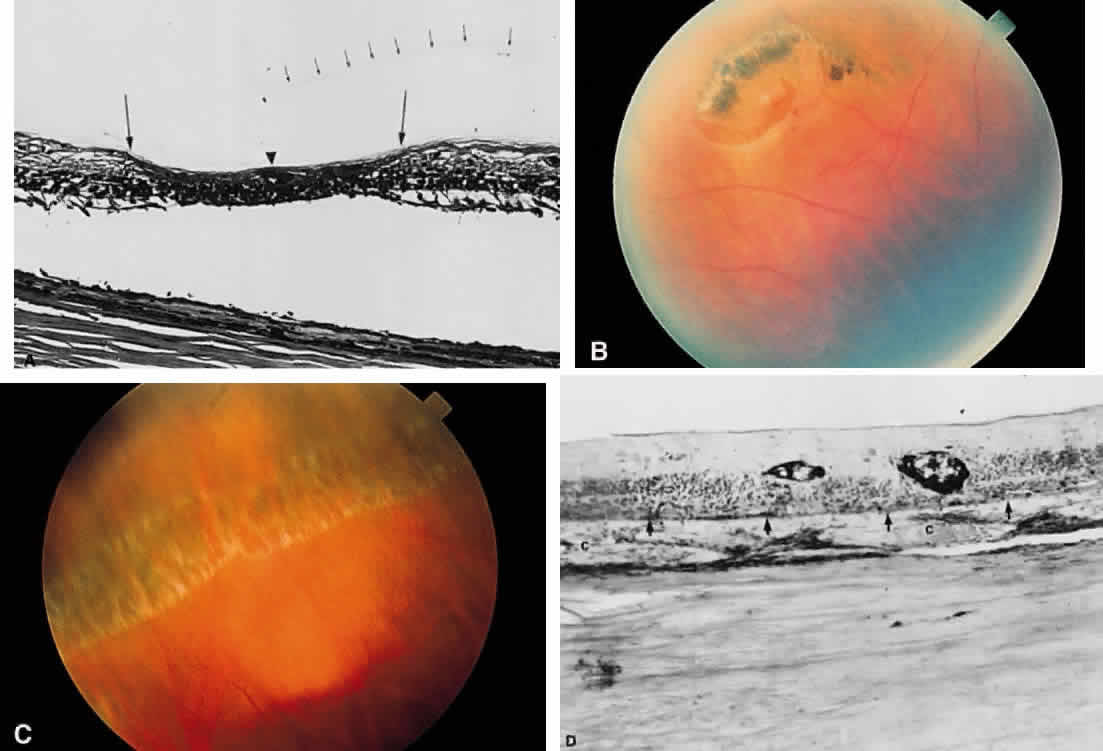
sunburst lesion of sickle cell retinopathy.20 The location of the hemorrhage also may affect is ophthalmoscopic appearance. Fresh hemorrhage between the retinal pigment epithelium and Bruch's membrane
may appear brown or red-black, in contrast with the bright red color
seen in fresh hemorrhages anterior to the pigment epithelium (see Fig. 13A). The location of hemorrhages within the retina also accounts for the shape of the lesion. Blood that accumulates between the nerve fiber layer and
the internal limiting membrane (subinternal limiting membrane hemorrhage) (Fig. 57) assumes a shape defined by gravity, meniscus or boat-shaped, when the
patient is upright (Fig. 58). The name subinternal limiting membrane hemorrhage is technically ambiguous, since
a hemorrhage in the outer plexiform layer also is below (sub) the
internal limiting membrane. 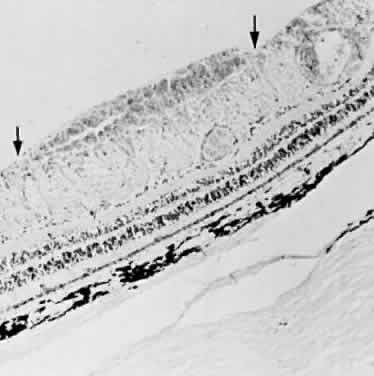 Fig. 57. Photomicrograph of a hemorrhage present just beneath the internal limiting
membrane. The hemorrhage is delimited on the photomicrograph by arrows. Fig. 57. Photomicrograph of a hemorrhage present just beneath the internal limiting
membrane. The hemorrhage is delimited on the photomicrograph by arrows.
|
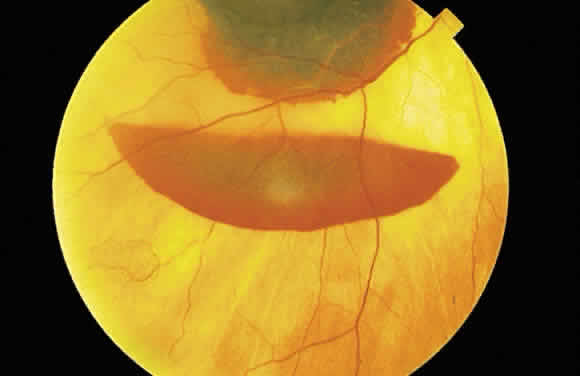 Fig. 58. Hemorrhage located just beneath the internal limiting membrane with the
patient in the upright position. The retinal vessels are visible on the
surface of the layered hemorrhage. Fig. 58. Hemorrhage located just beneath the internal limiting membrane with the
patient in the upright position. The retinal vessels are visible on the
surface of the layered hemorrhage.
|
The shape of other retinal hemorrhages depends on the layer of the retina
affected. The hemorrhage spreads along the plane defined by the orientation
of retinal structures. Thus, a hemorrhage within the nerve fiber
layer is oriented in the direction of the nerve fibers, parallel to
the internal limiting membrane (seeFig. 11A) and is seen in the posterior pole as a flame-shaped hemorrhage (see Fig. 11B). In the retinal periphery, however, nerve fiber layer hemorrhages are
not flame shaped but appear ophthalmoscopically as dots or blots. The
explanation of this phenomenon also is related to the orientation of
the nerve fiber layer. In the retinal periphery, the nerve fibers become
separated and are oriented so as to form a network of round or polygonal
spaces between the fibers, whereas in the posterior pole, the tight
fascicular orientation of the fibers forms potential trough-like planes. The orientation of the processes of retinal cells deep to the nerve fiber
layer is perpendicular to the plane of the internal limiting membrane. Therefore, hemorrhages
in the deeper (or outermost) retinal layer
are oriented in a cylindrical column. The ophthalmoscopist views a cross-section
of this column (the cylinder end-on) and sees a dot or blot
hemorrhage (see Fig. 23). |