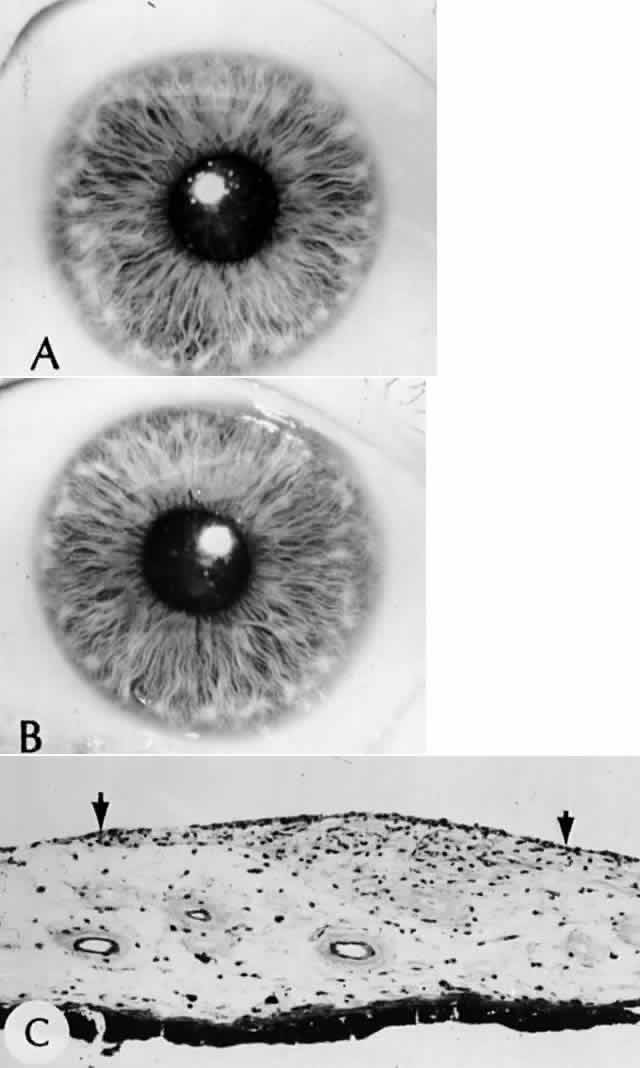
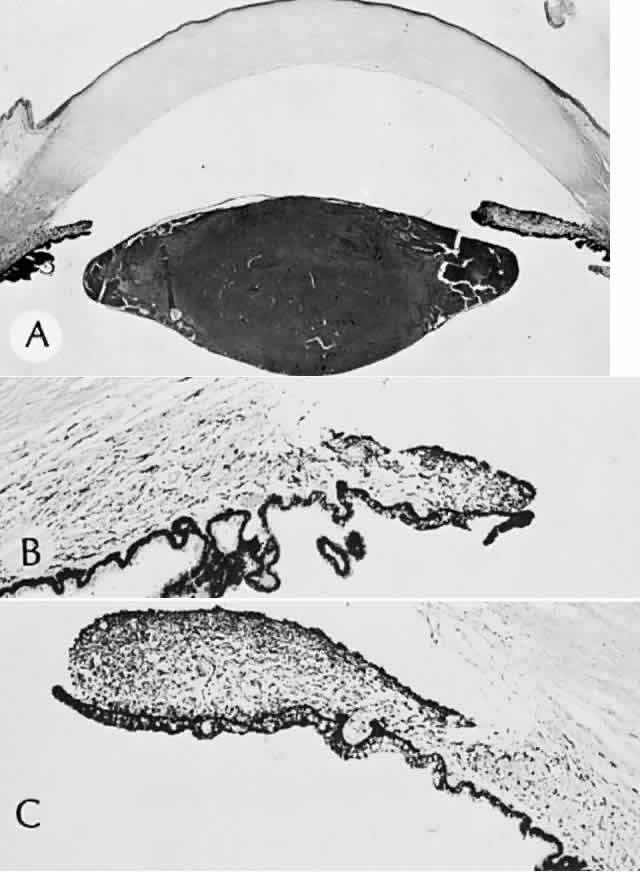
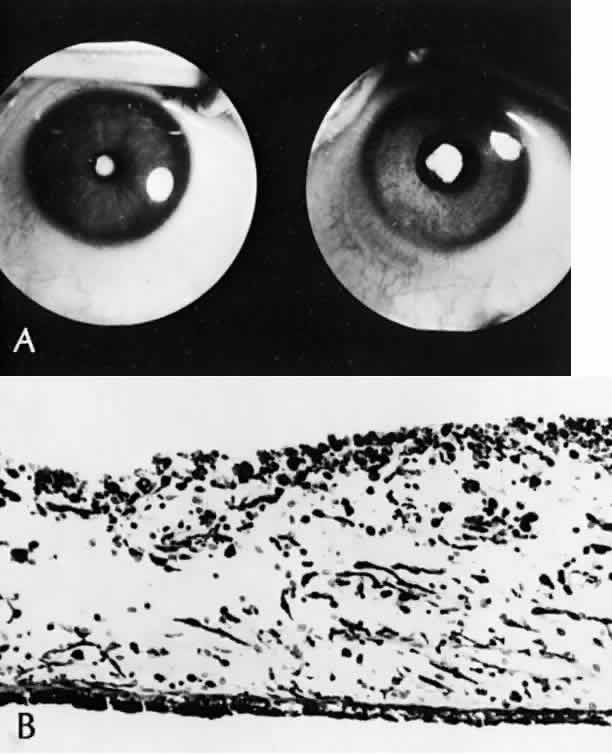
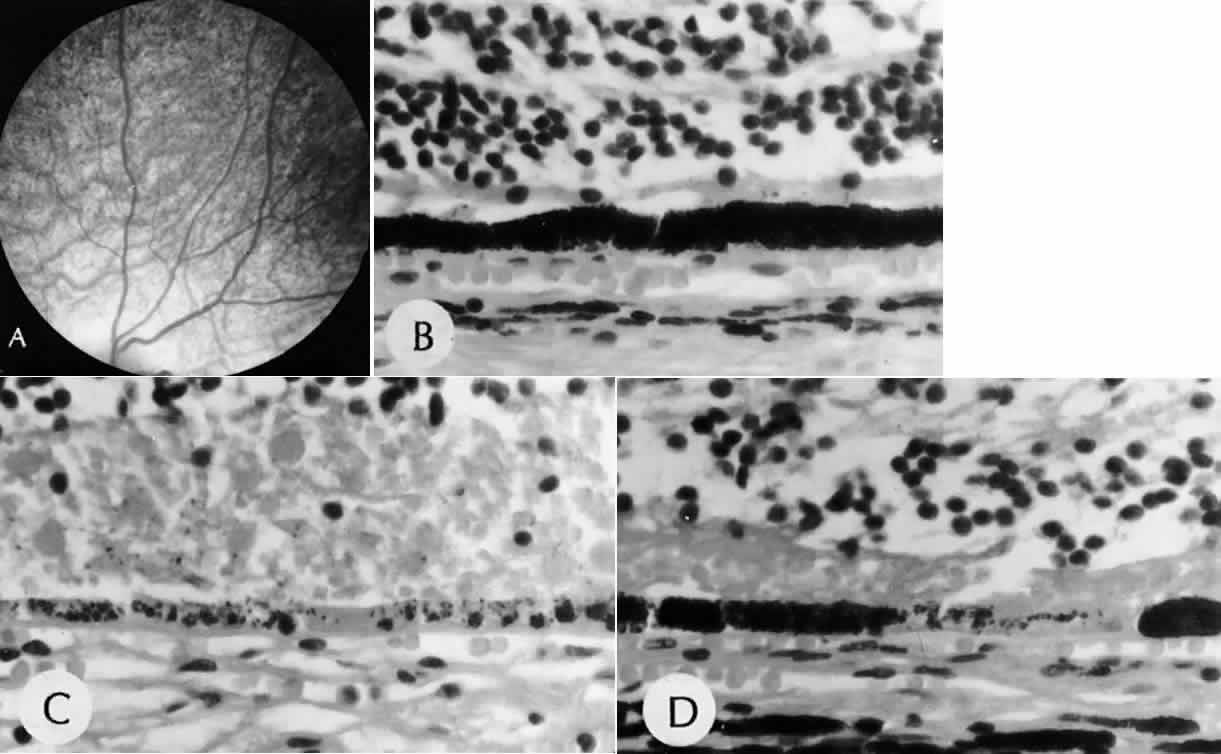
1. Mann I: The Development of the Human Eye. New York, Grune & Stratton, 1964 2. Zimmerman LE, Font RL: Congenital malformations of the eye. JAMA 196:684, 1966 3. Graham CA, Redmond RM, Nevin NC: X-linked clinical anophthalmos: Localization of the gene to Xq27-Xq28. Ophthal Paed Genet 12:43, 1991 4. Marcus DM, Shore JW, Albert DM: Anophthalmia in the focal dermal hypoplasia syndrome. Arch Ophthalmol 108:96, 1990 5. Sassani JW, Yanoff M: Anophthalmos in an infant with multiple congenital anomalies. Am J Ophthalmol 83:43, 1977 6. Haberland C, Perou M: Primary bilateral anophthalmia. J Neuropathol Exp Neurol 28:337, 1969 7. Brunquell PJ, Papale JH, Horton JC et al: Sex-linked hereditary bilateral anophthalmos. Arch Ophthalmol 102:108, 1984 8. Guyer DR, Green WR: Bilateral extreme microphthalmos. Ophthal Paed Genet 4:81, 1984 9. Calhoun FP Jr: The management of glaucoma in nanophthalmos. Trans Am Ophthalmol Soc 73:97, 1975 10. Brockhurst RJ: Nanophthalmos with uveal effusion: A new clinical entity. Trans Am Ophthalmol Soc 72:371, 1974 11. Weiss AH, Kousseff BG, Ross EA et al: Simple microphthalmos. Arch Ophthalmol 107:1625, 1989 12. Waring GO III, Roth AM: Clinicopathologic correlation of microphthalmos with cyst. Am J Ophthalmol 82:714, 1976 13. François J, Pallota R, Gallenza PE: Microphthalmos and malformative syndromes. Ophthal Paed Genet 2:201, 1983 14. Pasquale LR, Romayananda N, Kubacki J et al: Congenital cystic eye with multiple ocular and intracranial anomalies. Arch Ophthalmol 109:985, 1991 15. Torczynski E, Jacobiec FA, Johnston MC et al: Synophthalmia and cyclopia: A histopathologic, radiographic and organogenetic
analysis. Doc Ophthalmol 44:311, 1977 16. Vare AM: Cyclopia. Am J Ophthalmol 75:880, 1973 17. Yanko L, Zaifrani S: Synophthalmos in a full-term newborn child: An anatomic and pathologic
study. J Pediatr Ophthalmol 10:65, 1973 18. Howard RO: Chromosomal abnormalities associated with cyclopia and synophthalmia. Trans Am Ophthalmol Soc 75:505, 1977 19. Stefani FH, Hausmann N, Lund O-E: Unilateral diplophthalmos. Am J Ophthalmol 112:581, 1991 20. Andersen SR, Bro-Rasmussen F, Tygstrup I: Anencephaly related to ocular development and malformation. Am J Ophthalmol 64:559, 1967 21. Boniuk V, Ho PK: Ocular findings in anencephaly. Am J Ophthalmol 88:613, 1979 22. Rootman J, Carvounis EP: Vasculature of the optic nerve in anencephaly. Br J Ophthalmol 63:188, 1979 23. Addison DJ, Font RL, Manschot WA: Proliferative retinopathy in anencephalic babies. Am J Ophthalmol 74:967, 1972 24. Font RL, Ferry AP: The phakomatoses. Int Ophthalmol Clin 12(1):1, 1972 25. Beck RO, Hanno R: The phakomatoses. Int Ophthalmol Clin 25(1):97, 1985 26. Jakobiec FA, Font RL, Johnson FB: Angiomatosis retinae: An ultrastructural study and lipid analysis. Cancer 38:2042, 1976 27. Jurco S, III, Nadji M, Harvey DG et al: Hemangioblastomas: Histogenesis of the stromal cell studied by immunocytochemistry. Hum Pathol 13:13, 1982 28. Grossniklaus HE, Thomas JW, Vigneswaran N et al: Retinal hemangioblastoma: A histologic, immunohistochemical, and ultrastructural
evaluation. Ophthalmology 99:140, 1992 29. Chan C-C, Vortmeyer AO, Chew EY et al: VHL gene deletion and enhanced VEGF gene expression detected in the stromal
cells of retinal angioma. Arch Ophthalmol 117:625, 1999 30. Mottow-Lippa L, Tso MOM, Peyman GA et al: von Hippel angiomatosis: A light, electron microscopic, and immunoperoxidase
characterization. Ophthalmology 90:848, 1983 31. Nicholson DH, Green WR, Kenyon KR: Light and electron microscopic study of early lesions in angiomatosis retinae. Am J Ophthalmol 82:193, 1976 32. Jesberg DO, Spencer WH, Hoyt WF: Incipient lesions of von Hippel-Lindau disease. Arch Ophthalmol 80:632, 1968 33. Whitson JT, Welch RB, Green WR: Von Hippel-Lindau disease: Case report of a patient with spontaneous regression
of a retinal angioma. Retina 6:253, 1986 34. Wing GL, Weiter JJ, Kelly PJ et al: von Hippel-Lindau disease: Angiomatosis of the retina and central nervous
system. Ophthalmology 88:1311, 1981 35. Witschel H, Font RL: Hemangioma of the choroid. A clinicopathologic study of 71 cases and a
review of the literature. Surv Ophthalmol 20:415, 1976 36. Phelps CD: The pathogenesis of glaucoma in Sturge-Weber syndrome. Ophthalmology 85:276, 1978 37. Cibis GW, Tripathi RC, Tripathi BJ: Glaucoma in Sturge-Weber syndrome. Ophthalmology 91:1061, 1984 38. Destro M, D'Amico DJ, Gragoudas ES et al: Retinal manifestations of neurofibromatosis: Diagnosis and management. Arch Ophthalmol 109:662, 1991 39. Gutmann DH, Aylsworth A, Carey JC et al: The diagnostic evaluation and multidisciplinary management of neurofibromatosis 1 and
neurofibromatosis 2. JAMA 278:51, 1997 40. Lewis RA, Riccardi VM: von Recklinghausen neurofibromatosis: Incidence of iris hamartomata. Ophthalmology 88:348, 1981 41. Perry HD, Font RL: Iris nodules in von Recklinghausen's neurofibromatosis: Electron microscopic
confirmation of their melanocytic origin. Arch Ophthalmol 100:1635, 1982 42. Weleber RG, Zonana J: Iris hamartomas (Lisch nodules) in a case of segmental neurofibromatosis. Am J Ophthalmol 96:740, 1983 43. Savino PJ, Glaser JS, Luxenberg MN: Pulsating enophthalmos and choroidal hamartomas: Two rare stigmata of neurofibromatosis. Br J Ophthalmol 61:483, 1977 44. Riccardi VM: Van Recklinghausen neurofibromatosis. N Engl Med J 305:1617, 1981 45. La Piana FG: Sectoral retinal pigmentation in neurofibromatosis. Ann Ophthalmol 9:413, 1977 46. Spector B, Klintworth GK, Wells SA Jr: Histologic study of the ocular lesions in multiple endocrine neoplasia
syndrome type IIb. Am J Ophthalmol 91:204, 1981 47. Grant WM, Walton DS: Distinctive gonioscopic findings in glaucoma due to neurofibromatosis. Arch Ophthalmol 79:127, 1968 48. Rettele GA, Brodsky MC, Merin LM et al: Blindness, deafness, quadriparesis, and a retinal malformation: The ravages
of neurofibromatosis 2. Surv Ophthalmol 41:135, 1996 49. Ragge NK, Baser ME, Klein J et al: Ocular abnormalities in neurofibromatosis 2. Am J Ophthalmol 120:634, 1995 50. Williams R, Taylor D: Tuberous sclerosis. Surv Ophthalmol 30:143, 1985 51. Hered RW: Tuberous sclerosis. Arch Ophthalmol 110:410, 1992 52. Lucchese NJ, Goldberg MF: Iris and fundus pigmentary changes in tuberous sclerosis. J Pediatr Ophthalmol Strabismus 18:45, 1981 53. Bansley D, Wolter JR: The retinal lesion in tuberous sclerosis. J Pediatr Ophthalmol 8:261, 1971 54. Nyboer JH, Robertson DM, Gomez MR: Retinal lesions in tuberous sclerosis. Arch Ophthalmol 94:1277, 1976 55. de Juan E, Green WR, Gupta PK et al: Vitreous seeding by retinal astrocytic hamartoma in a patient with tuberous
sclerosis. Retina 4:100, 1984 56. Kinder RSL: The ocular pathology of tuberous sclerosis. J Pediatr Ophthalmol 9:106, 1972 57. Shields JA, Shields CL, Ehya H et al: Atypical retinal astrocytic hamartoma diagnosed by fine-needle biopsy. Ophthalmology 103:949, 1996 58. Boder E, Sedgwick RP: Ataxia-telangiectasia: A familial syndrome of progressive cerebellar ataxia, oculo-cutaneous
telangiectasia and frequent pulmonary infections. Pediatrics 21:526, 1970 59. Harley RD, Baird HW, Craven EM: Ataxia-telangiectasia: Report of seven cases. Arch Ophthalmol 77:582, 1967 60. Savitsky K, Bar-Shira A, Gilad S et al: A single ataxia-telangiectasia gene with a product similar to PI-3 kinase. Science 268:1749, 1995 61. Wyburn-Mason R: Arteriovenous aneurysm of mid-brain and retina, facial naevi, and mental
changes. Brain 66:163, 1943 62. Brown DG, Hilal SH, Tenner HS: Wyburn-Mason syndrome. Arch Neurol 28:67, 1973 63. Bernth-Petersen P: Racemose haemangioma of the retina: Report of three cases with long-term
follow-up. Acta Ophthalmol 57:669, 1979 64. Mansour AM, Wells CG, Jampol LM et al: Ocular complications of arteriovenous communications of the retina. Arch Ophthalmol 107:232, 1989 65. Archer DB, Deutman A, Ernest JT et al: Arteriovenous communications of the retina. Am J Ophthalmol 75:224, 1973 66. Bellhorn RW, Friedman AH, Henkind P: Racemose (cirsoid) hemangioma in Rhesus monkey retina. Am J Ophthalmol 74:517, 1972 67. Cameron ME, Greer CH: Congenital arteriovenous aneurysm of the retina. A postmortem report.Br J Ophthalmol 52:768, 1968 68. Horiuchi T, Gass JDM, David NJ: Arteriovenous malformation of the retina of a monkey. Am J Ophthalmol 82:896, 1976 69. Mets MB, Maumenee IH: The eye and the chromosome. Surv Ophthalmol 28:20, 1983 70. Howard RO, Boué J, Deluchat C et al: The eyes of embryos with chromosomal abnormalities. Am J Ophthalmol 78:167, 1974 71. Taylor AI: Autosomal trisomy syndromes: A detailed study of 27 cases of Edwards' syndrome
and 27 cases of Patau's syndrome. J Med Genet 5:227, 1968 72. Yanoff M, Frayer WC, Scheie HG: Ocular findings in a patient with 13–15 trisomy. Arch Ophthalmol 70:372, 1963 73. Karseras AG, Laurence KM: Eyes in arhinencephalic syndromes. Br J Ophthalmol 59:462, 1975 74. Ginsberg J, Bove M: Ocular pathology of trisomy 13. Ann Ophthalmol 6:113, 1974 75. Hoepner J, Yanoff M: Ocular anomalies in trisomy 13–15: An analysis of 13 eyes with two
new findings. Am J Ophthalmol 74:729, 1972 76. Rodrigues MM, Valdes-Dapena M, Kistenmacher M: Ocular pathology in a case of 13 trisomy. J Pediatr Ophthalmol 10:54, 1973 77. Cogan DG, Kuwabara T: Ocular pathology of the 13–15 trisomy syndrome. Arch Ophthalmol 72:246, 1964 78. Yanoff M, Font RL, Zimmerman LE: Intraocular cartilage in a microphthalmic eye of an otherwise healthy girl. Arch Ophthalmol 81:238, 1969 79. Hinzpeter EN, Naumann G, Steidinger J: Buphthalmus bei Trisomie-13 Syndrome. Ophthalmologica 170:381, 1975 80. Lichter PR, Scmickel RD: Posterior vortex vein and congenital glaucoma in a patient with trisomy-13 syndrome. Am J Ophthalmol 80:939, 1975 81. Calderone JP, Chess J, Borodic G et al: Intraocular pathology of trisomy 18 (Edwards's syndrome): Report of
a case and review of the literature. Br J Ophthalmol 67:162, 1983 82. Pe'er J, Braun JT: Ocular pathology in trisomy 18 (Edwards' syndrome). Ophthalmologica 192:176, 1986 83. Holmes JM, Coates CM: Assessment of visual acuity in children with trisomy 18. Ophthalmic Genetics 15:115, 1994 84. Ginsberg J, Bove K, Nelson R et al: Ocular pathology of trisomy 18. Ann Ophthalmol 3:273, 1971 85. Rodrigues MM, Punnett HH, Valdas-Dapena M et al: Retinal pigment epithelium in a case of trisomy 18. Am J Ophthalmol 76:265, 1973 86. Mullaney J: Ocular pathology in trisomy 18 (Edwards' syndrome). Am J Ophthalmol 76:246, 1973 87. Garcia-Castro JM, Reyes de Torres LC: Nictitating membrane in trisomy 18 syndrome. Am J Ophthalmol 80:550, 1975 88. Christianson RE: Down's syndrome and maternal age. Lancet 2:1198, 1976 89. Gaynon MW, Schimek RA: Down's syndrome: A ten-year group study. Ann Ophthalmol 9:1493, 1977 90. Jaeger EA: Ocular findings in Down's syndrome. Trans Am Ophthalmol
Soc 78:1980 91. Williams EJ, McCormick AQ, Tischler B: Retinal vessels in Down's syndrome. Arch Ophthalmol 89:269, 1973 92. Ahmad A, Pruett RC: The fundus in mongolism. Arch Ophthalmol 94:772, 1976 93. Orellana J, Palumbo J, Ritch R: Mesenchymal dysgenesis in a patient with Down's syndrome. J Pediatr Ophthalmol Strabismus 19:144, 1982 94. Ginsberg J, Bofinger MK, Roush JR: Pathologic features of the eye in Down's syndrome with relationship
to other chromosomal anomalies. Am J Ophthalmol 83:874, 1977 95. Robb RM, Marchevsky A: Pathology of the lens in Down's syndrome. Arch Ophthalmol 96:1039, 1978 96. Schwinger E, Wiebusch D: Coloboma of iris and choroid in XYY syndrome. Klinische Monatsblaetter fuer Augenheilkunde 156:873, 1970 97. Cameron JD, Yanoff M, Rayer WC: Turner's syndrome and Coats's disease. Am J Ophthalmol 78:852, 1974 98. Ginsberg J, Ballard ET, Soukup S: Pathologic features of the eye in triploidy. J Pediatr Ophthalmol Strabismus 18:48, 1981 99. Fulton AB, Howard RO, Albert DM et al: Ocular findings in triploidy. Am J Ophthalmol 84:859, 1977 100. Peterson RA: Schmid-Fraccaro syndrome (“cat's eye” syndrome): Partial
trisomy of G chromosome. Arch Ophthalmol 90:287, 1973 101. Weleber RG, Walknowska J, Peakman D: Cytogenetic investigations of “cat eye” syndrome. Am J Ophthalmol 84:477, 1977 102. Ginsberg J, Soukup S, Ballard ET: Pathologic features of the eye in trisomy 9. J Pediatr Ophthalmol Strabismus 19:37, 1982 103. Kohn G, Mayall BH, Miller ME et al: Tetraploid-diploid mosaicism in a surviving infant. Pediatr Res 1:461, 1967 104. Yanoff M, Rorke LB: Ocular and central nervous system findings in tetraploid-diploid mosaicism. Am J Ophthalmol 75:1036, 1973 105. Howard RO: Ocular abnormalities in the cri du chat syndrome. Am J Ophthalmol 73:949, 1972 106. Grotsky H, Hsu LYP, Hirschhorn K: A case of cri-du-chat associated with cataracts and transmitted from a
mother with a 4/5 translocation. J Med Genet 8:369, 1971 107. Schechter RJ: Ocular findings in a newborn with cri du chat syndrome. Ann Ophthalmol 10:339, 1978 108. Wilcox LM, Bercovitch L, Howard RO: Ophthalmic features of chromosome deletion 4p- (Wolf-Hirschhorn syndrome). Am J Ophthalmol 86:834, 1978 109. Mayer UM, Bialasiewicz AA: Ocular findings in a 4p- deletion syndrome (Wolf-Hirschhorn). Ophthal Paed Genet 10:69, 1989 110. Lurie IW, Samochvalov VA: Trisomy 4p- and ocular defects. Br J Ophthalmol 78:415, 1994 111. Andersen SR, Geetinger P, Larsen HW et al: Aniridia, cataract and gonadoblastoma in a mentally retarded girl with
deletion of chromosome 11. Ophthalmologica 176:171, 1978 112. Margo CE: Congenital aniridia: A histopathologic study of the anterior segment in
children. J Pediatr Ophthalmol Strabismus 20:192, 1983 113. Nelson LB, Spaeth GL, Nowinski TS et al: Aniridia: A review. Surv Ophthalmol 28:621, 1984 114. Yanoff M, Rorke LB, Niederer BS: Ocular and cerebral abnormalities in chromosome 18 deletion defect. Am J Ophthalmol 70:391, 1970 115. Boniuk M: Rubella and other intraocular viral diseases in infancy. Int Ophthalmol Clin 12(2):3, 1972 116. Givens KT, Lee DA, Jones T et al: Congenital rubella syndrome: Ophthalmic manifestations and associated systemic
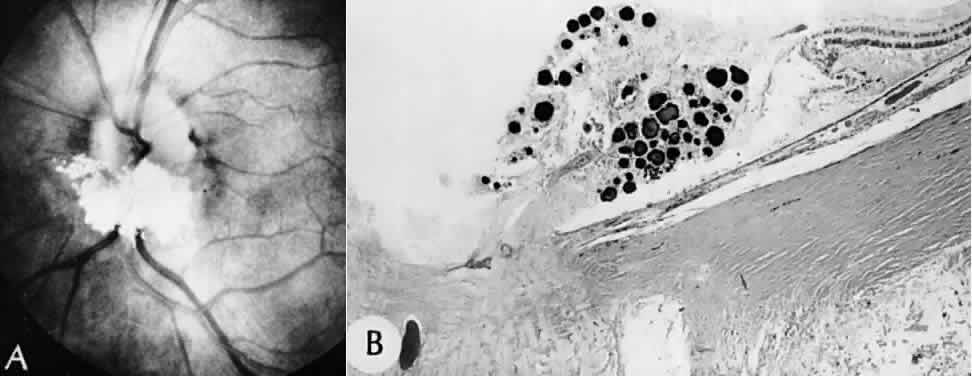
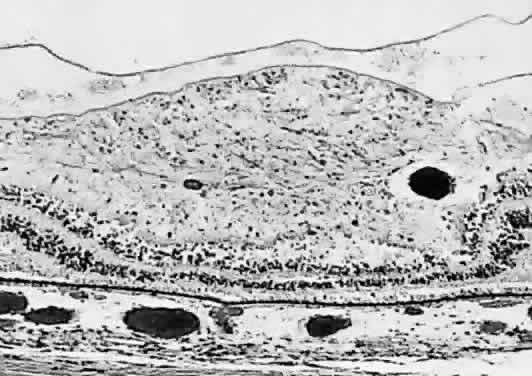
disorders. Br J Ophthalmol 77:358, 1993 117. Forrest JM, Menser MA, Burgess JA: High frequency of diabetes mellitus in young adults with congenital rubella. Lancet 2:332, 1971 118. Boniuk M, Zimmerman LE: Ocular pathology in the rubella syndrome. Arch Ophthalmol 77:455, 1967 119. Yanoff M, Schaffer DB, Scheie HG: Rubella ocular syndrome: Clinical significance of viral and pathologic
studies. Trans Am Acad Ophthalmol Otolaryngol 72:896, 1968 120. Yanoff M: The retina in rubella. In Tasman W (ed): Retinal Diseases in
Children. New York, Harper & Row, 1971 121. Frank KE, Purnell EW: Subretinal neovascularization following rubella retinopathy. Am J Ophthalmol 86:462, 1978 122. Slusher MM, Tyler ME: Rubella retinopathy and subretinal neovascularization. Ann Ophthalmol 14:292, 1982 123. Boger WP III: Late ocular complications in congenital rubella syndrome. Ophthalmology 87:1244, 1980 | 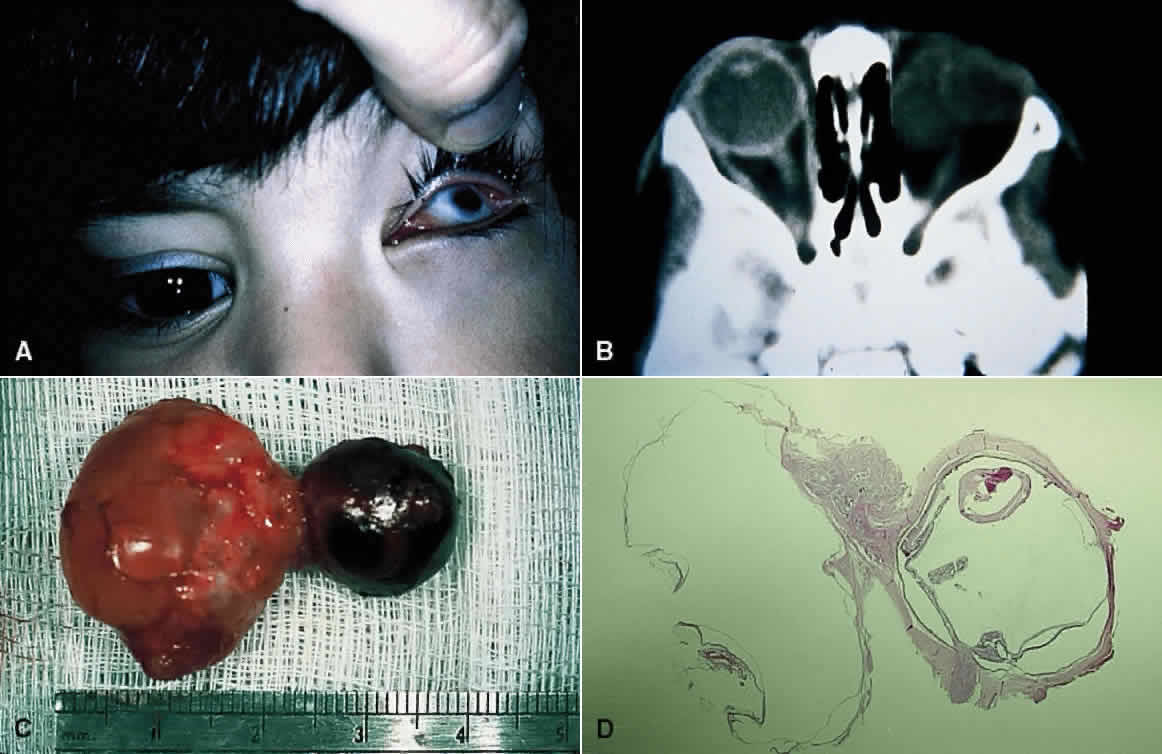 Fig. 1. Microphthalmia with cyst. A. The microphthalmic left eye in a 2-year-old girl. B. Computed tomographic scan showing the cyst nasal to the microphthalmic
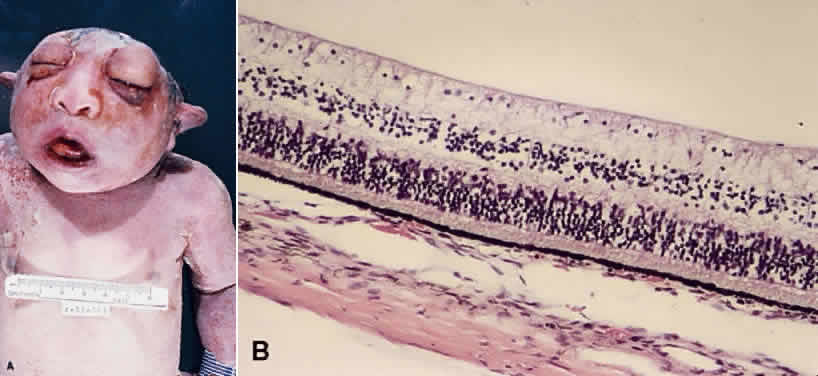
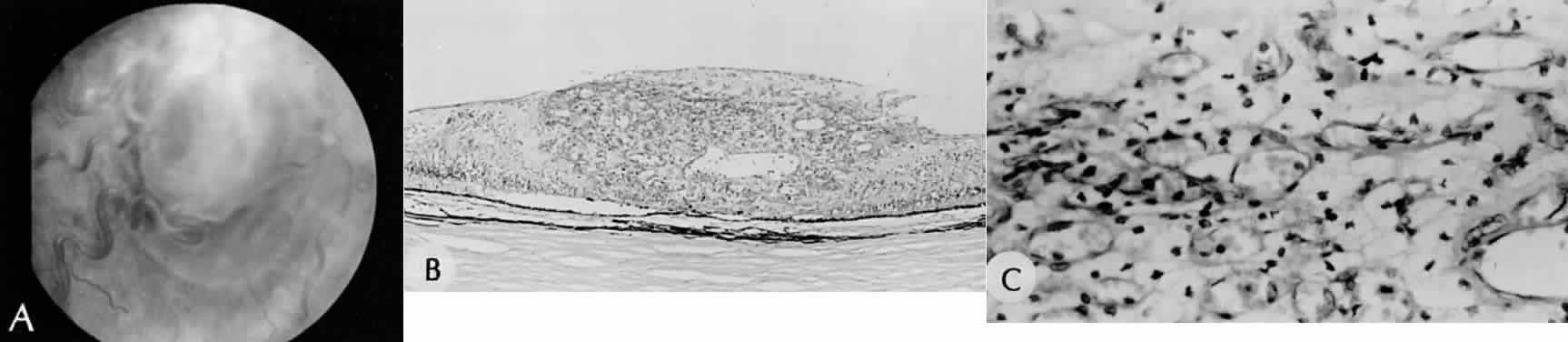
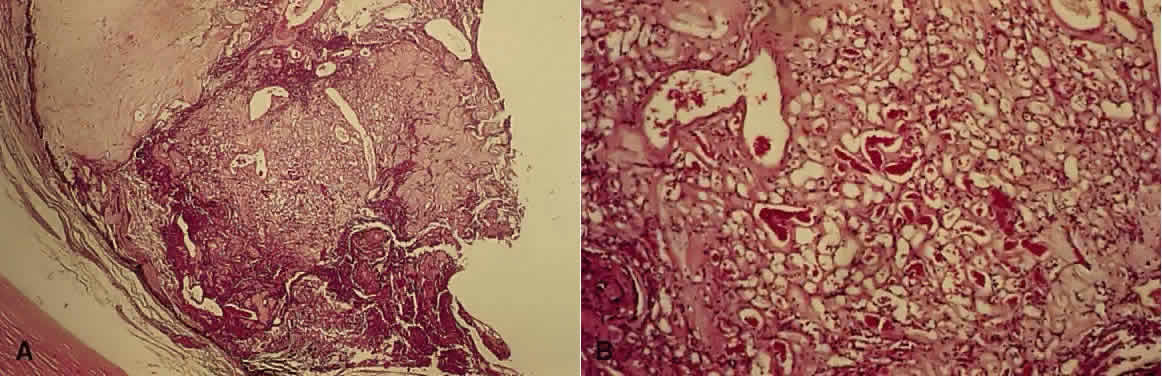
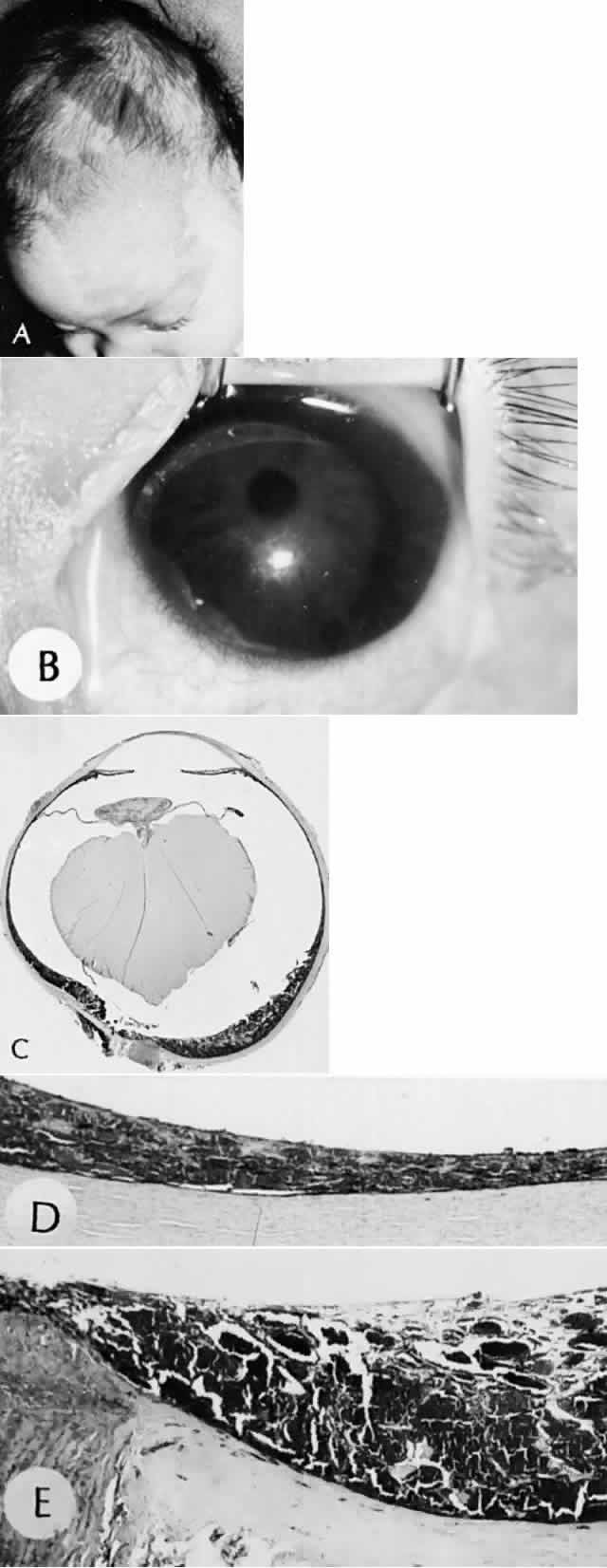
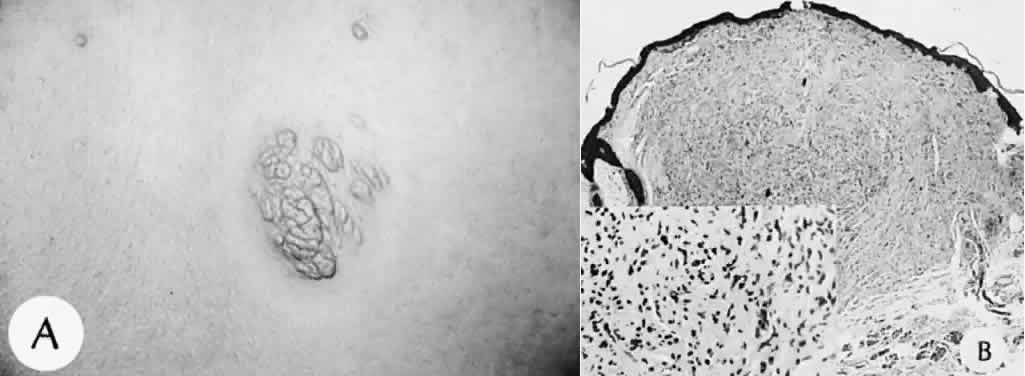
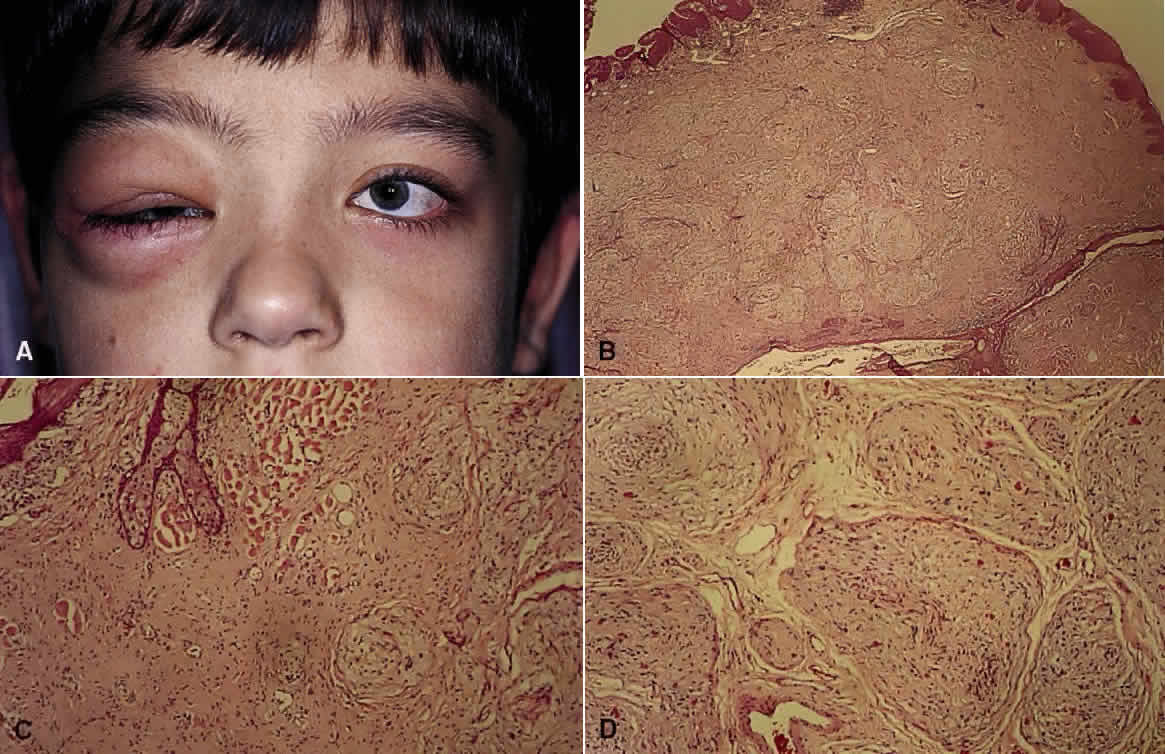
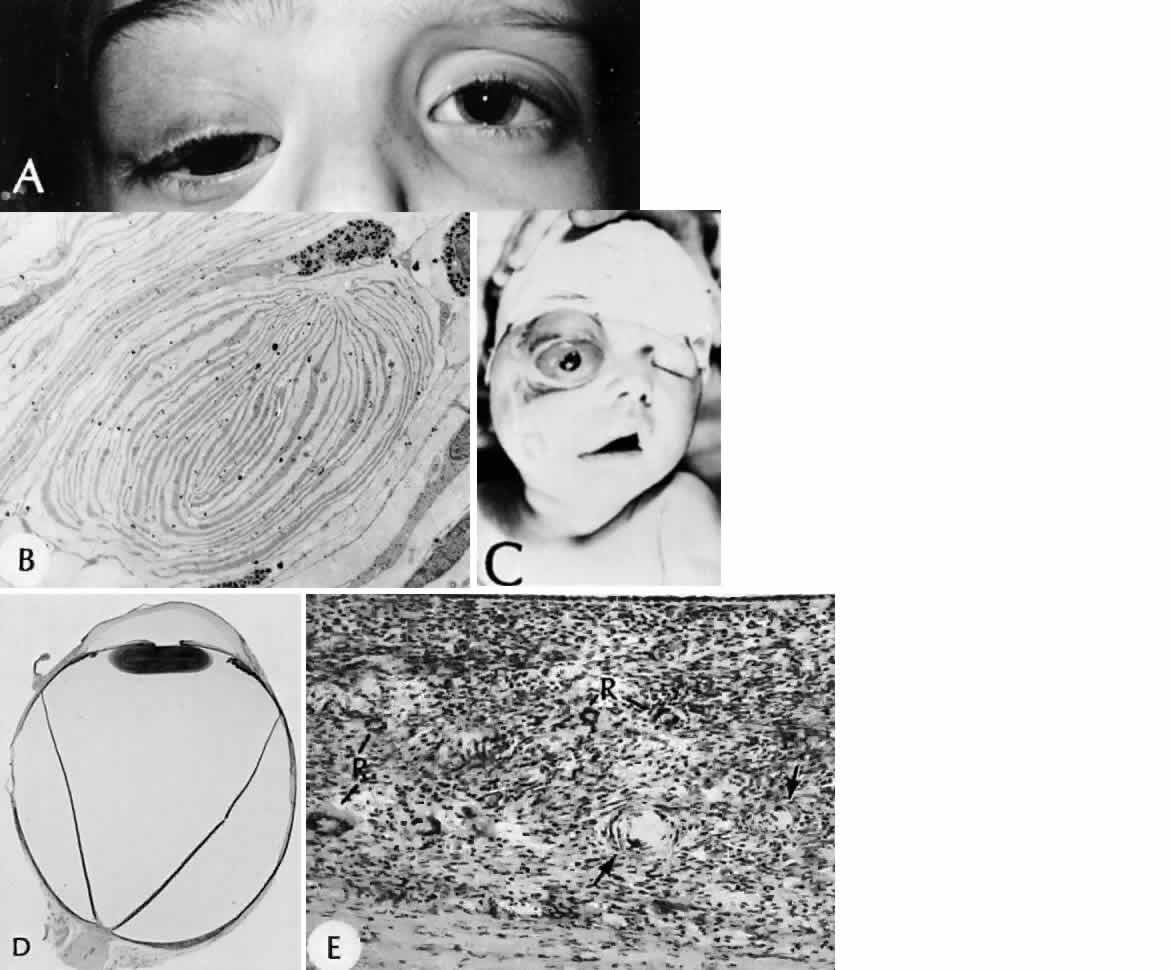
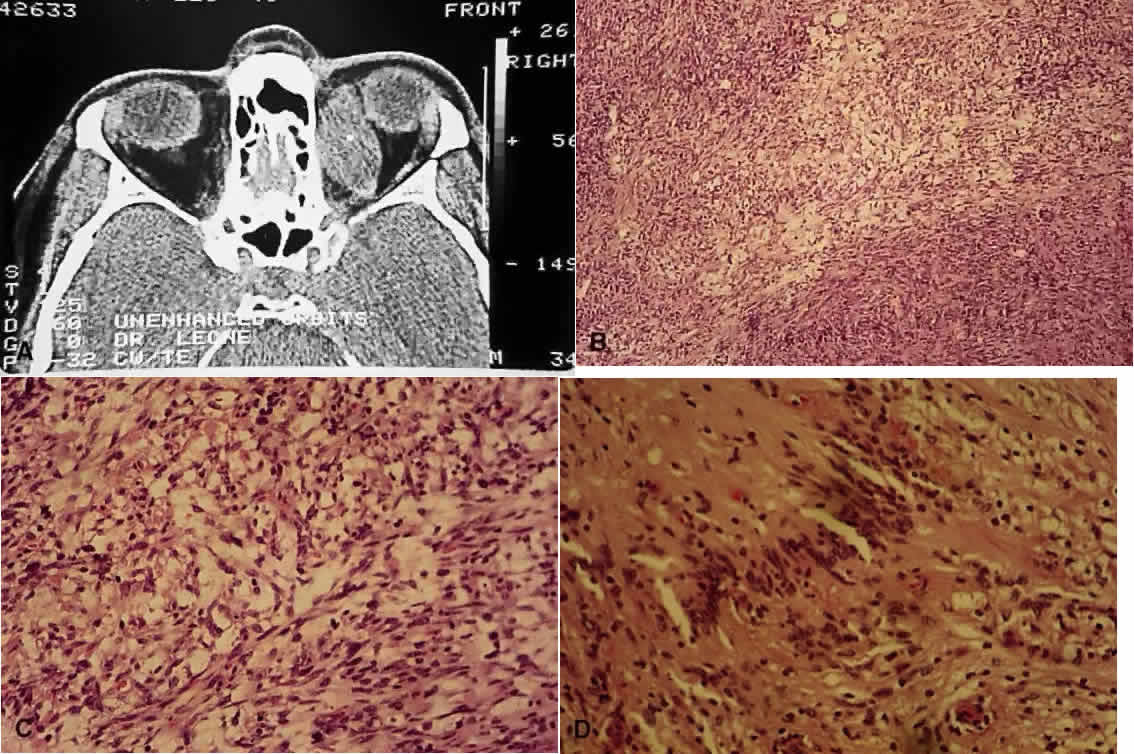
eye. C. Gross specimen, showing that the cyst is larger than the eye. D. Low-power photomicrograph showing the relatively well-formed eye and large
cyst (H&E, × 1). There is focal retinal dysplasia. (UTHSC-SA EP 169. Courtesy of Charles R. Leone, MD)
Fig. 1. Microphthalmia with cyst. A. The microphthalmic left eye in a 2-year-old girl. B. Computed tomographic scan showing the cyst nasal to the microphthalmic
eye. C. Gross specimen, showing that the cyst is larger than the eye. D. Low-power photomicrograph showing the relatively well-formed eye and large
cyst (H&E, × 1). There is focal retinal dysplasia. (UTHSC-SA EP 169. Courtesy of Charles R. Leone, MD)