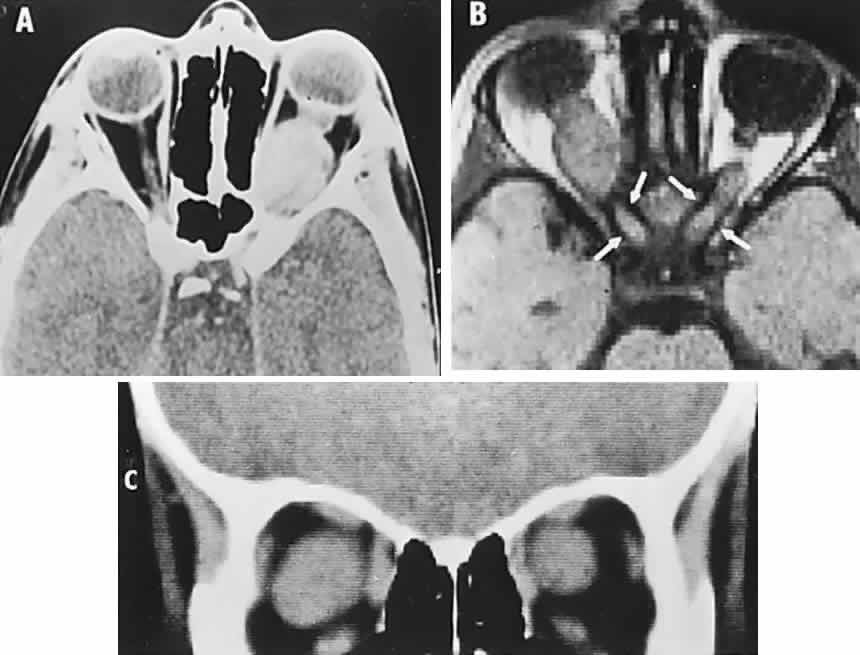
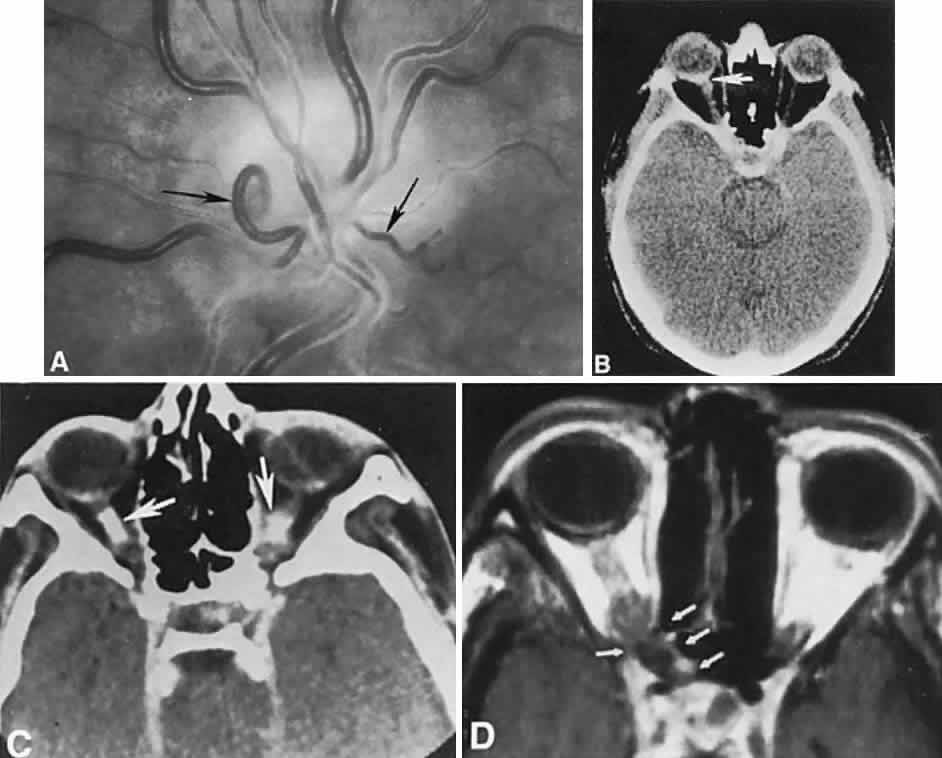
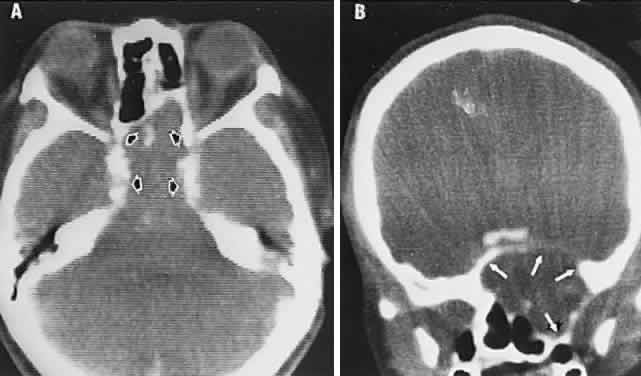
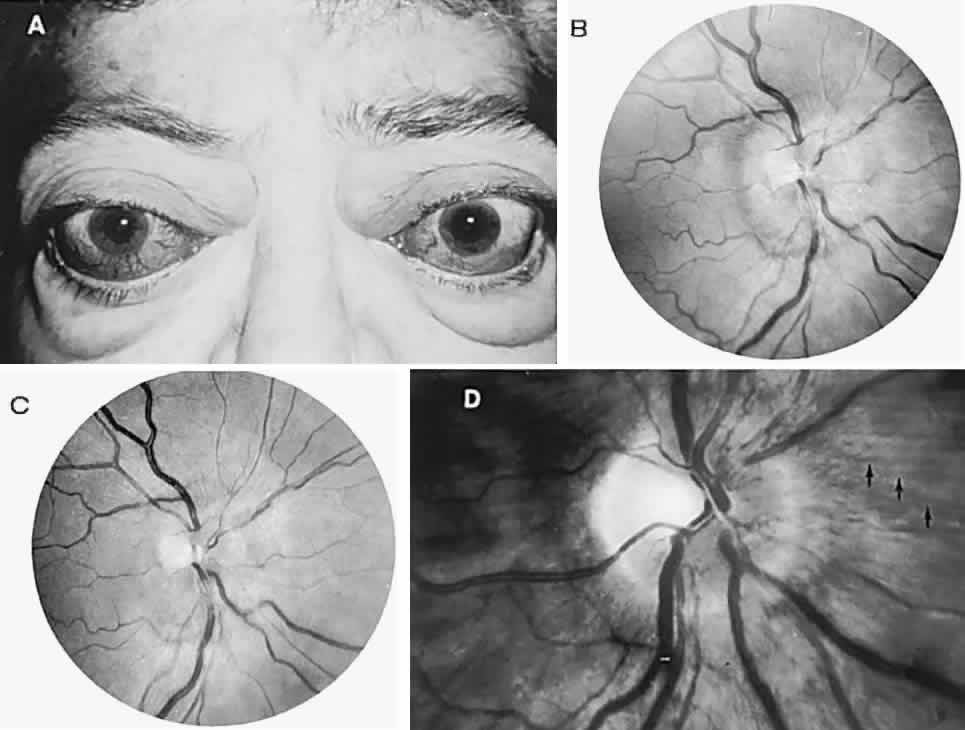
1. Lee MS, Gonzalez C: Unilateral peripapillary myelinated retinal nerve fibers associated with
strabismus, amblyopia and myopia. Am J Ophthalmol 125:554, 1998 2. Lambert SR, Hoyt CS, Narahara MH: Optic nerve hypoplasia. Surv Ophthalmol 32:1, 1987 3. Brodsky MC: Congenital optic disk anomalies. Surv Ophthalmol 39:89, 1994 4. Skarf B, Hoyt CS: Optic nerve hypoplasia in children: association with anomalies of the endocrine
and CNS. Arch Ophthalmol 102:62, 1984 5. Alvarez E, Wakakura M, Khan Z et al: J Pediatr Ophthalmol Strab 25:151, 1988 6. Jonas JB, Papastathopoulos K: Ophthalmoscopic measurement of the optic disc. Ophthalmology 102:1102, 1995 7. Zeki SM, Dudgeon J, Dutton GN: Reappraisal of the ratio of disc to macula/disc diameter in optic nerve
hypoplasia. Br J Ophthalmol 75:538, 1991 8. Peterson RA, Walton DS: Optic nerve hypoplasia with good visual acuity and visual field defect: a
study of children of diabetic mothers. Arch Ophthalmol 95:254, 1977 9. Buchanan TA, Hoyt WF: Temporal visual field defects associated with nasal hypoplasia of the optic
disc. Br J Ophthalmol 65:636, 1981 10. Brodsky MC, Schroeder GT, Ford R: Superior segmental optic hypoplasia in identical twins. J Clin Neuroophthalmol 13:152, 1993 11. Good WV, Ferriero DM, Golabi M et al: Abnormalities of the visual system in infants exposed to cocaine. Ophthalmology 99:341, 1992 12. Novakovic P, Taylor DSI, Hoyt WF: Localising patterns of optic nerve hypoplasiaretina to occipital lobe. Br J Ophthalmol 72:176, 1988 13. Hoyt WF, Rios-Montenegro EN, Behrens MM et al: Homonymous hemioptic hypoplasia: funduscopic features in standard and red-free
illumination in three patients with congenital hemiplegia. Br J Ophthalmol 56:537, 1972 14. Brodsky MC, Conte FA, Taylor D et al: Sudden death in septo-optic dysplasia: report of of 5 cases. Arch Ophthalmol 115:66, 1997 15. Margalith D, Tze WJ, Jan JE: Congenital optic nerve hypoplasia with hypothalamic-pituitary dysplasia. Am J Dis Child 139:361, 1985 16. Brodsky MC, Glasier CM: Optic nerve hypoplasia: clinical significance of associated central nervous
system abnormalities on magnetic resonance imaging. Arch Ophthalmol 111:66, 1993 17. Taylor D: Congenital tumours of the anterior visual system with dysplasia of the
optic disc. Br J Ophthalmol 66: 455, 1982 18. Grimson BS, Perry DD: Enlargement of the optic disc in childhood optic nerve tumors. Am J Ophthalmol 97:627, 1984 19. Howard MA, Thompson JT, Howard RO: Aplasia of the optic nerve. Trans Am Ophthalmol Soc 91:267, 1993 20. Scott IU, Warman R, Nolan A: Bilateral aplasia of the optic nerves, chiasm, and tracts in an otherwise
healthy infant. Am J Ophthalmol 124:409, 1997 21. Apple DJ, Rabb MF, Walsh PM: Congenital anomalies of the optic disc. Surv Ophthalmol 27:3, 1982 22. Pollock S: The morning glory disc anomaly: contractile movement, classification, and
embryogenesis. Doc Ophthalmol 65:439, 1987 23. Vuori M-L: Morning glory disc anomaly with pulsating peripapillary staphyloma: a case
history. Acta Ophthalmol 65:602, 1987 24. Savell J, Cook JR: Optic nerve colobomas of autosomal dominant heredity. Arch Ophthalmol 94:395, 1976 25. Gopal L, Badrinath SS, Kumar KS et al: Optic disc in fundus coloboma. Ophthalmology 103:2120, 1996 26. Slamovits TL, Kimball GP, Friberg TI et al: Bilateral optic disc colobomas with orbital cysts and hypoplastic optic
nerves and chiasm. J Clin Neuroophthalmol 9:172, 1989 27. Orcutt JC, Bunt AH: Anomalous optic discs in a patient with Dandy-Walker cyst. J Clin Neuroophthalmol 2:43, 1982 28. Hanson MR, Price RL, Rothner AD et al: Developmental anomalies of the optic disc and carotid circulation. J Clin Neuroophthalmol 5:3, 1985 29. Eustis HS, Sanders MR, Zimmerman T: Morning glory syndrome in children. Arch Ophthalmol 112:204, 1994 30. Theodossiadis GP, Kollia AK, Theodossiadis PG: Cilioretinal arteries in conjunction with a pit of the optic disc. Ophthalologica 204:115, 1992 31. Ragge NK, Ravine D, Wilkie AOM: Dominant inheritance of optic pits. Am J Ophthalmol 125:124, 1998 32. Brown GC: Congenital pits of the optic nerve head. II. Clinical studies in humans. Ophthalmology 87:51, 1980 33. Adelung K, Aulhorn E, Thiel H-J: Funktionsstorungen bei Grubenpapille. Klin Monatsbl Augenheilk 191:1, 1987 34. Cashwell LF, Ford JG: Central visual field changes associated with acquired pits of the optic
nerve. Ophthalmology 102:1270, 1995 35. Friberg TR, McLellan TG: Vitreous pulsations, relative hypotony, and retrobulbar cyst associated
with a congenital optic pit. Am J Ophthalmol 114:767, 1992 36. Giuffre G: The spectrum of the visual field defects in the tilted disc syndrome: clinical
study and review. Neuroophthalmology 6:239, 1986 37. Riise D: The nasal fundus ectasia. Acta Ophthalmol Suppl 126, 1975 38. Margolis S, Siegel IM: The tilted disc syndrome in craniofacial diseases. In
Smith JL (ed), Neuro-ophthalmology Focus 1980, p 97. Masson, 1980 39. Spencer WH: Drusen of the optic disc and aberrant axoplasmic transport. Am J Ophthalmol 85:1, 1978 40. Tso MOM: Pathology and pathogenesis of drusen of the optic nerve head. Ophthalmology 88:1066, 1981 41. Hoover DL, Robb RM, Peterson RA: Optic disc drusen in children. J Pediatr Ophthalmol Strab 25:191, 1988 42. Mansour AM, Hamed LM: Racial variation of optic nerve diseases. Neuroophthalmology 11:319, 1991 43. Lorentzen SE: Drusen of the optic disc: a clinical and genetic study. Acta Ophthalmol 90:1, 1966 44. Rosenberg MA, Savino PJ, Glaser JS: A clinical analysis of pseudopapilledema. I. Population, laterality, acuity, refractive
error, ophthalmoscopic characteristics, and coincident
disease. Arch Ophthalmol 97:65, 1979 45. Mustonen E: Pseudopapilledema with and without verified optic disc drusen: a clinical
analysis I. Acta Ophthalmol 61:1037, 1983 46. Novack RL, Foos RY: Drusen of the optic disc in retinitis pigmentosa. Am J Ophthalmol 103:44, 1987 47. Pierro L, Brancato R, Minicucci M et al: Echographic diagnosis of drusen of the optic nerve head in patients with
angioid streaks. Ophthalmologica 208:239, 1994 48. Brown SM, Del Monte MA: Choroidal neovascular membrane associated with optic nerve head drusen
in a child. Am J Ophthalmol 121:215, 1996 49. Savino PJ, Glaser JS, Rosenberg MA: A clinical analysis of pseudopapilledema. II. Visual field defects. Arch Ophthalmol 97:71, 1979 50. Mustonen E: Pseudopapilledema with and without verified optic disc drusen: a clinical
analysis II. Visual fields. Acta Ophthalmol 61:1057, 1983 51. Gittinger JW, Lessell S, Bondar RL: Ischemic optic neuropathy associated with disc drusen. J Clin Neuroophthalmol 4:79, 1984 52. Beck RW, Corbett JJ, Thompson HS et al: Decreased visual acuity from optic disc drusen. Arch Ophthalmol 103:1155, 1985 53. Moody TA, Irvine AR, Cahn PH et al: Sudden visual field constriction associated with optic disc drusen. J Clin Neuroophthalmol 13:8, 1993 54. Sadun A, Currie JN, Lessell S: Transient visual obscurations with elevated optic discs. Ann Neurol 16:489, 1984 55. Sarkies NJC, Sanders MD: Optic disc drusen and episodic visual loss. Br J Ophthalmol 71:537, 1987 56. Friedman AH, Beckerman B, Gold DH et al: Drusen of the optic disc. Surv Ophthalmol 21:375, 1977 57. Mullie MA, Sanders MD: Computed tomographic diagnosis of buried drusen of the optic nerve head. Can J Ophthalmol 20:114, 1985 58. Moller HU: Recessively inherited, simple optic atrophy: does it exist? Ophthalmic Paediatr Genet 13:31, 1992 59. Francois J: Heredity in Ophthalmology. St. Louis, CV Mosby, 1961 60. Horoupian DS, Zucker DK, Moshe S et al: Behr syndrome: a clinicopathologic report. Neurology 29:323, 1979 61. Costeff H, Elpeleg O, Apter N et al: 3-Methylglutaconic aciduria in “optic atrophy plus.”Ann Neurol 33:103, 1993 62. Chalmers RM, Riorden-Eva P, Wood NW: Autosomal recessive inheritance of hereditary motor and sensory neuropathy
with optic atrophy. J Neurol Neurosurg Psychiatry 62:385, 1997 63. Shevell MI, Colangelo P, Treacy E et al: Progressive encephalopathy with edema, hypsarryhthmia, and optic atrophy (PEHO
syndrome). Pediatr Neurol 15:337, 1996 64. Rizzo JF, Lessell S, Liebman SD: Optic atrophy in familial dysautonomia. Am J Ophthalmol 102:463, 1986 65. Cremers CWRJ, Wijdeveld PGAB, Pinkers AJLG: Juvenile diabetes, optic atrophy, hearing
loss, diabetes insipidus, atonia of the urinary tract and
bladder, and other abnormalities (Wolfram syndrome): a review of 88 cases
from the literature with personal observations on 3 new patients. Acta
Paediatr Scand Suppl 264, 1977 66. Lessell S, Rosman NP: Juvenile diabetes mellitus and optic atrophy. Arch Neurol 34:759, 1977 67. Barrett TG, Bundey SE, Macleod AF: Neurodegeneration and diabetes: UK nationwide study of Wolfram (DIDMOAD) syndrome. Lancet 346:1458, 1995 68. Genis D, Davalos A, Molins A, Ferrer I: Wolfram syndrome: a neuropathologic study. Acta Neuropathol (Berl) 93:426, 1997 69. Scolding NJ, Kellar-Wood HF, Shaw C et al: Wolfram syndrome: hereditary diabetes mellitus with brainstem and optic
atrophy. Ann Neurol 39:352, 1996 70. Pilz D, Quarell OW, Jones EW: Mitochondrial mutation commonly associated with Leber's hereditary
optic neuropathy observed in a patient with Wolfram syndrome (DIDMOAD). J Med Genet 31:328, 1994 71. Barrientos A, Casademont J, Saiz A et al: Autosomal recessive Wolfram syndrome associated with an 8.5-kb mtDNA single
deletion. Am J Genet 58:963, 1996 72. Hofmann S, Bezold R, Jaksch M et al: Wolfram (DIDMOAD) syndrome and Leber hereditary optic neuropathy (LHON) are
associated with distinct mitochondrial haplotypes. Genomics 39:8, 1997 73. Kjer P: Infantile optic atrophy with dominant mode of inheritance: a clinical
and genetic study of 19 Danish families. Acta Ophthalmol Suppl 54, 1959 74. Smith DP: Diagnostic criteria in dominantly inherited juvenile optic atrophy: a report
of 3 new families. Am J Optom 49:183, 1972 75. Votruba M, Fitzke FW, Holder GE et al: Clinical features in affected individuals from 21 pedigrees with dominant
optic atrophy. Arch Ophthalmol 116:351, 1998 76. Brown J, Fingert JH, Taylor CM et al: Clinical and genetic analysis of a family affected with dominant optic
atrophy (OPA1). Arch Ophthalmol 115:95, 1997 77. Johnston RL, Seller MJ, Behman JT et al: Dominant optic atrophy. Refining the clinical diagnostic criteria in light
of genetic linkage studies. Ophthalmol 106:123, 1999 78. Kjer B, Eiberg H, Kjer P, Rosenberg T: Dominant optic atrophy mapped to chromosome 3q region. II. Clinical and
epidemiologic aspects. Acta Ophthalmol Scand 74:3, 1996 79. Hoyt CS: Autosomal dominant optic atrophy: a spectrum of disability. Ophthalmology 87:245, 1980 80. Grehn F, Kommerell G, Ropers H-H et al: Dominant optic atrophy with sensorineural hearing loss. Ophthalmic Paediatr Genet 1:77, 1982 81. Treft RL, Sanborn GE, Carey J et al: Dominant optic atrophy, deafness, ptosis, ophthalmoplegia, dystaxia, and
myopathy: a new syndrome. Ophthalmology 91:908, 1984 82. Chalmers RM, Bird AC, Harding AE: Autosomal dominant optic atrophy with asymptomatic peripheral neuropathy. J Neurol Neurosurg Psychiatry 60:195, 1996 83. Johnston PB, Gaster RN, Smith VC et al: A clinicopathologic study of autosomal dominant optic atrophy. Am J Ophthalmol 88:868, 1979 84. Kjer P, Jensen OA, Klinken L: Histopathology of eye, optic nerve and brain in a case of dominant optic
atrophy. Acta Ophthalmol 61:300, 1983 85. Kellar-Wood H, Robertson N, Govan GG et al: Leber's hereditary optic neuropathy mitochondrial DNA mutations in
multiple sclerosis. Ann Neurol 36:109, 1994 86. Newman NJ: Optic neuropathy. Neurology 46:315, 1996 87. Mashima Y, Oshitari K, Imamura Y, Momoshima S et al: Orbital high resolution magnetic resonance imaging with fast spin echo
in the acute stage of Leber's hereditary optic neuropathy. J Neurol Neurosurg Psychiat 64:124, 1998 88. Nikoskelainen EK, Huoponen K, Juvonen V et al: Ophthalmologic findings in Leber hereditary optic neuropathy, with special
reference to mtDNA mutations. Arch Ophathalmol 103:504, 1996 89. Nikoskelainen E, Hoyt WF, Nummelin K et al: Fundus findings in Leber's hereditary optic neuroretinopathy. III. Fluorescein
angiographic studies. Arch Ophthalmol 102:981, 1984 90. Newman NJ: Leber's hereditery optic neuropathy: new genetic considerations. Arch Neurol 50:540, 1993 91. Newman NJ, Lott MT, Wallace DC: The clinical characteristics of pedigrees of Lebers' hereditary optic neuropathy
with the 11778 mutation. Am J Ophthalmol 111:750, 1991 92. Riordan-Eva P, Sanders MD, Govan GG et al: The clinical features of Leber's hereditary optic neuropathy defined
by the presence of a pathogenic mitochondrial DNA mutation. Brain 118:319, 1995 93. Johns DR, Heher KL, Miller NR et al: Leber's hereditary optic neuropathy: clinical manifestations of the 14484 mutation. Arch Ophthalmol 111:495, 1993 94. Mashima Y, Hiida Y, Saga M et al: Risk of false-positive molecular genetic diagnosis of Leber's hereditary
optic neuropathy. Am J Ophthalmol 119:245, 1995 95. Johns DR, Smith KH, Miller NR et al: Identical twins who are discordant for Leber's hereditary optic neuropathy. Arch Ophthalmol 111:1491, 1993 96. Hoyt WF: Charcot-Marie-Tooth disease with primary optic atrophy: report of a case. Arch Ophthalmol 64:925, 1960 97. Bird TD, Griep E: Pattern reversal visual evoked potentials: studies in Charcot-Marie-Tooth
hereditary neuropathy. Arch Neurol 38:739, 1981 98. Paradiso G, Micheli F, Taratuto AL et al: Familial bulbospinal neuronopathy with optic atrophy: a distinct entity. J Neurol Neurosurg Psychiatry 61:196, 1996 99. Nicolaides P, Appleton RE, Fryer A: Cerebellar ataxia, areflexia, pes cavus, optic atrophy, and sensorineural
hearing loss (CAPOS): a new syndrome. J Med Genet 33:419, 1996 100. Groom M, Kay MD, Corrent GF: Optic neuropathy in familial dysautonomia. J Neuroophthalmol 17:101, 1977 101. Livingston IR, Mastaglia FL, Edis R et al: Visual involvement in Friedreich's ataxia and hereditary spastic ataxia: a
clinical and visual evoked response study. Arch Neurol 38:75, 1981 102. Frisen L, Claesson M: Narrowing of the retinal arterioles in descending optic atrophy: a quantitative
clinical study. Ophthalmology 94:1020, 1984 103. Papastathopoulos K, Jonas JB: Focal narrowing of retinal arterioles in optic nerve atrophy. Ophthalmology 102: 1706, 1995 104. Ettl A, Kramer J, Daxer A et al: High resolution magnetic resonance imaging of neurovascular orbital anatomy. Ophthalmology 104:869, 1997 105. Mukhenji SK, Tart RP, Fitzsimmons J et al: Fat-suppressed MR of the orbit and cavernous sinus: comparison of fast
spin-echo and conventional spin-echo. AJNR Am J Neuroradiol 15:1707, 1994 106. Davis PC, Newman NJ: Advances in neuro-imaging of the visual pathways. Am J Ophthalmol 121:690, 1996 107. Byrne SF, Green RL: Ultrasound of the Eye and Orbit. St. Louis, CV Mosby, 1992 108. Katz B, Hoyt WF: Intrapapillary and peripapillary hemorrhage in young patients with incomplete
posterior vitreous detachment. Ophthalmology 102:349, 1995 109. Kokame GT: Intrapapillary, papillary, and vitreous hemorrhage: letters, and reply. Ophthalmology 102:1003, 1995 110. Sher NA, Wirtschafter J, Shapiro SK et al: Unilateral papilledema in “benign” intracranial hypertension (pseudotumor
cerebri). JAMA 250:2346, 1983 111. Liu D, Michon J: Measurement of the subarachnoid pressure of the optic nerve in human subjects. Am J Ophthalmol 119:81, 1994 112. Hayreh SS: Pathogenesis of optic disc oedema. In Kennard C, Rose FC (eds): Physiological
Aspects of Clinical Neuro-Ophthalmology, p 431. Chicago, Year
Book, 1988 113. Liu D, Kahn M: Measurement and relationship of subarachnoid pressure of the optic nerve
to intracranial pressure in fresh cadavers. Am J Ophthalmol 116:548, 1993 114. Peterson RA, Rosenthal A: Retinopathy and papilledema in cyanotic congenital heart disease. Pediatrics 49:243, 1972 115. Bucci FA, Krohel GB: Optic nerve swelling secondary to the obstructive sleep apnea syndrome. Am J Ophthalmol 105:428, 1988 116. Hardten DR, Wen DY, Wirtschafter JD et al: Papilledema and intraspinal lumbar paraganglioma. J Clin Neuroophthalmol 12:158, 1992 117. Michowiz SD, Rappaport HZ, Shaked I et al: Thoracic disc herniation associated with papilledema. J Neurosurg 61:1132, 1984 118. Marks SJ, Schick A, Charney JZ et al: The association of papilledema with syringomyelia: case report. Mt Sinai J Med 55:333, 1988 119. Ropper AH, Marmarou A: Mechanism of pseudotumor in Guillain-Barré syndrome. Arch Neurol 41:259, 1984 120. Stern BJ, Gruen R, Koeppel J et al: Recurrent thyrotoxicosis and papilledema in a patient with communicating
hydrocephalus. Arch Neurol 41:65, 1984 121. Scharf D: Neurocysticercosis: two hundred thirty-eight cases from a California hospital. Arch Neurol 45:777, 1988 122. Chimowitz MI, Little JR, Awad IA et al: Intracranial hypertension associated with unruptured cerebral arteriovenous
malformations. Ann Neurol 27:474, 1990 123. Verm A, Lee AG: Bilateral optic disk with macular exudates as the manifesting sign of a
cerebral arteriovenous malformation. Am J Ophthalmol 123:422, 1997 124. Karahalios DG, Rekate HL, Khayata MH et al: Elevated intracranial venous pressure as a universal mechanism in pseudotumor
cerebri of varying etiologies. Neurology 46:198, 1996 125. Marr WG, Chambers RG: Pseudotumor cerebri syndrome following unilateral neck dissection. Am J Ophthalmol 51:605, 1961 126. Graus F, Slatkin NE: Papilledema in the metastatic jugular foramen syndrome. Arch Neurol 40:816, 1983 127. Strominger MB, Weiss GB, Mehler MF: Asymptomatic unilateral papilledema in pseudotumor cerebri. J Clin Neuroophthalmol 12:238, 1992 128. Muci-Mendoza R, Arruga J, Hoyt WF: Distensión bilateral del espacio subaracnoideo perioptico en el
pseudotumor cerebral con papiledema unilateral: su demonstración
a traves de la tomografia computarizada de la orbita. Rev Neurol 39:11, 1981 129. Pagani LF: The rapid appearence of papilledema. J Neurosurg 30:247, 1969 130. Steffen H, Eifert B, Aschoff A et al: The diagnostic value of optic disc elevation in acute elevated intracranial
pressure. Ophthalmology 103:1229, 1996 131. Hilton-Jones D, Ponsford JR, Graham N: Transient visual obscurations, without papilledema. J Neurol Neurosurg Psychiatry 45:832, 1982 132. Hedges TR, Baron EM, Hedges TR et al: The retinal venous pulse: its relation to optic disc characteristics and
choroidal pulse. Ophthalmology 101:542, 1994 133. Carter SR, Seifrf SR: Macular changes in pseudotumor cerebri before and after optic nerve sheath
fenestration. Ophthalmology 102:937, 1995 134. Jacobson DM: Intracranial hypertension and syndrome of acquired hyperopia and choroidal
folds. J Neuroophthalmol 15:178, 1995 135. Corbett JJ, Savino PJ, Thompson HS et al: Visual loss in pseudotumor cerebri: follow-up of 57 patients from five
to 41 years and a profile of 14 patients with permanent severe visual
loss. Arch Neurol 39:461, 1982 136. Baker RS, Buncic JR: Sudden visual loss in pseudotumor cerebri due to central retinal artery
occlusion. Arch Neurol 41:1274, 1984 137. Beck RW, Greenberg HS: Post-decompression optic neuropathy. J Neurosurg 63:196, 1985 138. Mashima Y, Oshitari K, Imamura Y et al: High-rsolution magnetic resonance imaging of the intraorbital optic nerve
and sub-arachnoid space in patients with papilledema and optic atrophy. Arch Ophthalmol 114:1197, 1996 139. Lessell S: Pediatric pseudotumor cerebri (idiopathic intracranial hypertension). Surv Ophthalmol 37:155, 1992 140. Cinciripini GS, Donahue S, Borchert MS: Idiopathic intracranial hypertension in prepubertal pediatric patients: characteristics, treatment, and outcome. Am J Ophthalmol 127:178, 1999 141. Sorensen PS, Gjerris F, Svenstrup B: Endocrine studies in patients with pseudotumor cerebri: estrogen levels
in blood and cerebrospinal fluid. Arch Neurol 43:902, 1986 142. Radhakrishnan K, Ahlskog JE, Garrity JA et al: Idiopathic intracranial hypertension. Mayo Clin Proc 69:169, 1994 143. Digre KB, Corbett JJ: Pseudotumor cerebri in men. Arch Neurol 45:866, 1988 144. Borruat FX, Regli F: Pseudotumor cerebri as a complication of amiodarone therapy. Am J Ophthalmol 116:776, 1993 145. Hamed LM, Glaser JS, Schatz NJ et al: Danazol-induced pseudotumor cerebri. Arch Ophthalmol 100:1000, 1989 146. Sanborn GE, Selhorst JB, Calabrese VP et al: Pseudotumor cerebri and insecticide intoxication. Neurology 29: 1222, 1979 147. Saul RF, Hamburger HA, Selhorst JB: Pseudotumor cerebri secondary to lithium carbonate. JAMA 253: 2869, 1985 148. Green JP, Newman NJ, Stowe ZN et al: “Normal pressure” pseudotumor cerebri. J Neuroophthalmol 16:241, 1996 149. Friedman DI, Forman S, Levi L, Lavin PJ et al: Unusual ocular motility disturbances with increased intracranial pressure. Neurol 50:1893, 1998 150. Moser FG, Hilal SK, Abrams G et al: MR imaging of pseudotumor cerebri. AJNR Am J Neuroradiol 9:39, 1988 151. Wang SJ, Silberstein SD, Patterson S, Young WB: Idiopathic intracranial hypertension without papilledema. A case-control
study in a headache center. Neurol 51:245, 1998 151a. Corbett JJ: Problems in the diagnosis and treatment of pseudotumor cerebri. Can J Neurol Sci 10:221, 1983 152. Wall M, George D: Visual loss in pseudotumor cerebri: incidence and defects related to visual
field strategy. Arch Neurol 44:170, 1987 153. Hedges TR, Legge RH, Peli E et al: Retinal nerve fiber layer loss in idiopathic intracranial hypertension. Ophthalmology 102:1242, 1995 154. Gu XZ, Tsai JC, Wurdeman A et al: Pattern of axonal loss in longstanding papilledema due to idiopathic intracranial
hypertension. Curr Eye Res 14:173, 1995 155. Fan JT, Johnson DH, Burk RR: Transient myopia, angle closure and choroidal detachments after oral acetazolamide. Am J Ophthalmol 115:813, 1993 156. Liu GT, Glaser JS, Schatz NJ: High-dose methylprednisolone and acetazolamide for visual loss in pseudotumor
cerebri. Am J Ophthalmol 118:88, 1994 157. Johnson LN, Krohel GB, Madsen RW et al: The role of weight loss and acetazolamide in the treatment of idiopathic
intracranial hypertension (pseudotumor cerebri). Ophthalmology 105:2313, 1998 158. Burgett RA, Purvin VA, Kawasaki A: Lumboperitoneal shunting for pseudotumor cerebri. Neurology 49:734, 1997 159. Liu GT, Volpe NJ, Schatz NJ et al: Severe sudden visual loss caused by pseudotumor cerebri and lumboperitoneal
shunt failure. Am J Ophthalmol 122:129, 1996 160. Lee AG: Visual loss as the manifesting symptom of ventriculoperitoneal shunt malfunction. Am J Ophthalmol 122:127, 1996 161. Corbett JJ, Nerad JA, Tse DT et al: Results of optic nerve sheath fenestration for pseudotumor cerebri: the
lateral orbitotomy approach. Arch Ophthalmol 106:1391, 1988 162. Spoor TC, McHenry JG: Long-term effectiveness of optic nerve sheath decompression for pseudotumor
cerebri. Arch Ophthalmol 111:632, 1993 163. Rizzo JF, Lessell S: Choroidal infarction after optic nerve sheath fenestration. Ophthalmology 101:1622, 1994 164. Mauriello JA, Shaderowsky P, Gizzi M et al: Management of visual loss after optic nerve sheath decompression in patients
with pseudotumor cerebri. Ophthalmology 102:441, 1995 165. Mathew NT, Ravishankar K, Sanin LC: Coexistence of migraine and idiopathic intracranial hypertension without
papilledema. Neurology 46:1226, 1996 166. Magnante DO, Bullock JD: Familial pseudotumor cerebri: occurrence in a
mother while pregnant with a subsequently affected daughter. (personal
communication) 167. Lam BL, Schatz NJ, Glaser JS et al: Pseudotumor cerebri from cranial venous obstruction. Ophthalmology 99:706, 1992 168. Purvin VA, Trobe JD, Kosmorsky G: Neuro-ophthalmic features of cerebral venous obstruction. Arch Neurol 52:880, 1995 169. Hauser D, Barzilai N, Zalish M et al: Bilateral papilledema with retinal hemorrhages in association with cerebral
venous sinus thrombosis and paroxysmal nocturnal hemoglobinuria. Am J Opthalmol 122:592, 1996 170. Ireland B, Corbett JJ, Wallace R: The search for causes of idiopathic intracranial hypertension. Arch Neurol 47:315, 1990 171. FDA Medwatch and Spontaneous Reporting System. Norplant 1991-1993. Obstet
Gynecol 85:538, 1995 172. Malozowski S, Tanner LA, Wysowski DK et al: Benign intracranial hypertension in children with growth hormone deficiency
treated with growth hormone. J Pediatr 126: 996, 1995 173. Alexandrakis G, Filatov, Walsh T: Pseudotumor cerebri in a 12-year-old boy with Addison's disease. Am J Ophthalmol 116:650, 1993 174. Liu GT, Kay MD, Bienfang DC et al: Pseudotumor cerebri associated with corticosteroid withdrawal in inflammatory
bowel disease. Am J Ophthalmol 117:352, 1994 175. Sirdofsky M, Kattah J, Macedo P: Intracranial hypertension in a dieting patient. J Neuro-ophthalmol 14:9, 1994 175a. Chiu AM, Chuenkongkaew WL, Cornblath W, et al: Minocycline treatment and pseudotumor cerebri syndrome. Am J Ophthalmol 126:116, 1998 176. Jennum P, Borgesen SE: Intracranial pressure and obstructive sleep apnea. Chest 95:279, 1989 177. Landau K, Gloor BP: Therapy resistant papilledema in achondroplasia. J Neuroophthalmol 14:24, 1994 178. Stavrou P, Sgouros S, Willshaw HE et al: Visual failure caused by raised intracranial pressure in craniosynostosis. Childs Nerv Syst 13:64, 1997 179. Johnston I, Hawke S, Halmagyi M et al: The pseudotumor syndrome: disorders of cerebrospinal fluid circulation
causing intracranial hypertension without ventriculomegaly. Arch Neurol 48:740, 1991 180. Katz B: The dyschromatopsia of optic neuritis. Trans Am Ophthalmol Soc 93:685, 1995 181. Mojon DS, Rösler KM, Oetliker H: A bedside test to determine motion stereopsis using the Pulfrich phenomenon. Ophthalmol 105:1337, 1998 182. Selhorst JB, Saul RF: Uhthoff and his symptom. J Neuroophthalmol 15:63, 1995 183. Keltner JL, Johnson CA, Spurr JO et al: Visual field profile of optic neuritis: one-year follow-up in the optic
neuritis treatment trial. Arch Ophthalmol 112:946, 1994 184. Kennedy C, Caroll FD: Optic neuritis in children. Trans Am Acad Ophthalmol Otolaryngol 64:700, 1960 185. Riikonen R: The role of infection and vacination in the genesis of optic neuritis and
multiple sclerosis in children. Acta Neurol Scand 80:425, 1989 186. Lucchinetti CF, Kiers L, O'Duffy A et al: Risk factors for developing multiple sclerosis after childhood optic neuritis. Neurology 49:1413, 1997 187. Hamed LM, Silbiger J, Guy J et al: Parainfectious optic neuritis and encephalomyelitis: a report of two cases
with thalamic involvement. J Clin Neuroophthalmol 13:18, 1993 188. Purvin VA, Chioran G: Recurrent neuroretinitis. Arch Ophthalmol 112:365, 1994 189. Beck RW, Cleary PA, Trobe JD et al: The effect of corticosteroids for acute optic neuritis on the subsequent
development of multiple sclerosis. N Engl J Med 329:1764, 1993 190. Warner JEA, Lessell S, Rizzo JF et al: Does optic disc appearance distinguish ischemic optic neuropathy from optic
neuritis. Arch Ophthalmol 115:1408, 1997 191. Guy J, Mao J, Bidgood WD et al: Enhancement and demyelination of the intraorbital optic nerve: fat suppression
magnetic resonance imagining. Ophthalmology 99:713, 1992 192. Dunker S, Wiegand W: Prognostic value of magnetic resonance imaging in monosymptomatic optic
neuritis. Ophthalmology 103:1768, 1996 193. Beck RW, Kupersmith MJ, Cleary PA et al: Fellow eye abnormalities in acute unilateral optic neuritis. Ophthalmology 100:691, 1993 194. Cleary PA, Beck RW, Bourque LB et al: Visual symptoms after optic neuritis: results from the Optic Neuritis Treatment
Trial. J Neuroophthalmol 17:18, 1997 195. Jacobs L, Karpik A, Bozian D et al: Auditory-visual synesthesia. Arch Neurol 38:211, 1981 196. Safran AB, Kine LB, Glaser JS: Positive visual phenomena in optic nerve
and chiasm disease: photopsias and photophobia. In Glaser JS (ed): Neuro-ophthalmology, Vol
X, p 225. St. Louis, CV Mosby, 1980 197. Kaufman DI, Beck R, ONTT Study Group: The 5-year risk of MS after optic
neuritis: experience of the Optic Neuritis Treatment Trial. Neurology 49:1404, 1997 198. Jacobs LD, Kaba SE, Miller CM et al: Correlation of clinical: magnetic resonance imaging, and cerebrospinal
fluid findings in optic neuritis. Ann Neurol 41:392, 1997 199. Francis DA, Compston DA, Batchelor JR et al: A reassessment of the risk of multiple sclerosis developing in patients
with optic neuritis after extended follow-up. J Neurol Neurosurg Psychiatry 50:6, 1987 200. Soderstom M, Ya-Ping J, Hillert J et al: Optic neuritis. Prognosis for multiple sclerosis from MRI, CSF, and HLA
findings. Neurology 50:708, 1998 201. Lightman S, McDonald WI, Bird AC et al: Retinal venous sheathing in optic neuritis: its significance for pathogenesis
of multiple sclerosis. Brain 100:405, 1987 202. Engell T: Neurological disease activity in multiple sclerosis patients with periphlebitis
retinae. Acta Neurol Scand 73:168, 1986 203. Malinowski SM, Pulido JS, Folk JC: Long-term visual outcome and complications associated with pars planitis. Ophthalmology 100:818, 1993 204. Gass A, Graham E, Moseley IF et al: Cranial MRI in idiopathic retinal vasculitis. J Neurol 242:174, 1995 205. Cole SR, Beck RW, Moke PS et al: The predictive value of CSF oligoclonal banding for MS 5 years after optic
neuritis. Optic Neuritis Study Group. Neurol 51:885, 1998 206. Mandler RN, Davis LE, Jeffrey DR et al: Devic's neuromyelitis optica: a clinocopathologic study of 8 patients. Ann Neurol 34:162, 1993 207. O'Riordan JI, Gallagher HL, Thompson AJ et al: Clinical, CSF and MRI findings in Devic's neuromyelitis optica. J Neurol Neurosurg Psychiatry 60:382, 1996 208. Brazis PW, Lee AG: Optic disk edema with a macular star. Mayo Clin Proc 71:1162, 1996 209. Ellis BD, Kosmorsky GS, Cohen BH. Medical and surgical management of acute
disseminated encephalomyelitis. J Neuroophthalmol 14:210, 1994 210. Hamed LM, Silbiger J, Guy J et al: Parainfectious optic neuritis and encephalomyelitis. J Neuroophthalmol 13:18, 1993 211. Kawasaki A, Purvin VA, Tang R: Bilateral anterior ischemic optic neuropathy following influenza vaccination. J Neuroophthalmol 18:56, 1998 212. Hull TP, Bates JH: Optic neuritis after influenza vaccination. Am J Ophthalmol 124:703, 1997 213. Pall HS, Williams AC: Subacute polyradiculopathy with optic and auditory nerve involvement. Arch Neurol 44:885, 1987 214. Borruat FX, Schatz NJ, Glaser JS, Forteza A: Central nervous system involvement in Guillain-Barré-like syndrome: clinical
and magnetic resonance imaging evidence. Eur Neurol 38:129, 1997 215. Miller DH, Kay R, Schon F et al: Optic neuritis following chickenpox in adults. J Neurol 233:182, 1986 216. Selbst RG, Selhorst JB, Harbison JW et al: Parainfectious optic neuritis. Arch Neurol 40:347, 1983 217. Berrios RR, Serrano LA: Bilateral optic neuritis after a bee sting. Am J Ophthalmol 117:677, 1994 218. Keane J. Neuro-ophthalmologic signs of AIDS: 50 patients. Neurology 41:841, 1991 219. Nichols JW, Goodwin JA: Neuro-ophthalmologic complications of AIDS. Semin Ophthalmol 7:24, 1992 220. Tenhula WN, Xu SZ, Madigan MC et al: Morphometric comparisons of optic nerve axon loss in acquired immunodeficiency
syndrome. Am J Ophthalmol 113:14, 1992 221. Patel SS, Rutzen AR, Marx JL et al: Cytomegalovirus papillitis in patients with acquired immune deficiency
syndrome. Ophthalmology 103:1476, 1996 222. Cohen DB, Glasgow BJ: Bilateral optic nerve cryptococcosis in sudden blindness in patients with
acquired immune deficiency syndrome. Ophthalmology 100:1689, 1993 223. Garrity JA, Herman DC, Imes R et al: Optic nerve sheath decompression for visual loss in patients with acquired
immunodeficiency syndrome and cryptococcal meningitis with papilledema. Am J Ophthalmol 116:472, 1993 224. Grossniklaus HE, Specht CS, Allaire G et al: Toxoplasma gondii retinochoroiditis and optic neuritis in acquired immune deficiency syndrome: report
of a case. Ophthalmology 97:1342, 1990 225. Shayegani A, Odel JG, Kazim M et al: Varicella-zoster virus optic neuritis in a patient with human immunodeficiency
virus. Am J Ophthalmol 122:586, 1996 226. Lee MS, Cooney EL, Stoessel KM et al: Varicella zoster virus retrobulbar optic neuritis in patients with acquired
immunodeficiency syndrome. Ophthalmology 105:467, 1998 227. Yau TH, Rivera-Velazquez, Mark AS et al: Unilateral optic neuritis in a patient with the acquired immunodeficiency
syndrome. Am J Ophthalmol 121:324, 1996 228. Gabuzda DH, Hirsch MS: Neurologic manifestations of infection with human immunodeficiency virus. Ann Intern Med 107:383, 1987 229. Burton BJL, Leff AP: Plant steroid-responsive HIV optic neuropathy. J Neuroophthalmol 18:25, 1998 230. Margo CE, Hamed LM: Ocular syphilis. Surv Ophthalmol 37:203, 1992 231. Zambrano W, Perez GM, Smith JL: Acute syphilitic blindness in AIDS. J Clin Neuroophthalmol 7:1, 1987 232. Toshniwal P: Optic perineuritis with secondary syphilis. J Clin Neuroophthalmol 7:6, 1987 233. Arruga J, Valentines J, Mauri F et al: Neuroretinitis in acquired syphilis. Ophthalmology 92:262, 1985 234. Balcer LJ, Winterkorn JMS, Galetta SL: Neuro-ophthalmic manifestations of Lyme disease. J Neuroophthalmol 17:108, 1997 235. Schmutzhard E, Pohl P, Stanek G: Involvement of Borrelia burgdorferi in cranial nerve affection. Zentrabl Bakt Hyg A 263:328, 1986 236. Pachner AR, Steere AC: The triad of neurologic manifestations of Lyme disease: meningitis, cranial
neuritis, and radiculoneuritis. Neurology 35:47, 1985 237. Wu G, Lincoff H, Ellsworth RM et al: Optic disc edema and Lyme disease. Ann Ophthalmol 18:252, 1986 238. Strominger MB, Slamovits TL, Herskovitz S et al: Transient worsening of optic neuropathy as a sequela of the Jarisch-Herxheimer
reaction in the treatment of Lyme disease. J Neuroophthalmol 14:77, 1994 239. Lyme disease: Connecticut. JAMA 259:1147, 1988 240. Wong MT, Dolan MJ, Lattuada CP et al: Neuroretinitis, aseptic meningitis, and lymphadenitis associated with Bartonella henselae infection in immunocompetent patients and patients infected with human
immunodeficiency virus type 1. Clin Infect Dis 21:352, 1995 241. Golnik KC, Marotto ME, Fanous MM et al: Ophthalmic manifestations of Rochalimaea species. Am J Ophthalmol 118:145, 1994 242. Reed JB, Scales DK, Wong MT et al: Bartonella henselae neuro-retinitis in cat scratch disease: diagnosis, management, and sequelae. Ophthalmology 105:459, 1998 243. Cox TA, Haskins GE, Gangitano JL et al: Bilateral Toxacara optic neuropathy. J Clin Neuroophthalmol 3:267, 1983 244. Fish RH, Hoskins JC, Kline LB: Toxoplasmosis neuroretinitis. Ophthalmology 100:1177, 1993 245. Lossos A, Eliashiv S, Ben-Chetrit E: Optic neuritis associated with familial Mediterranean fever. J Clin Neuroophthalmol 13:141, 1993 246. Ormerod IEC, McDonald WI: Multiple sclerosis presenting with progressive visual failure. J Neurol Neurosurg Psychiatry 47:943, 1984 247. Eidelberg D, Newton MR, Johnson G et al: Chronic unilateral optic neuropathy: a magnetic resonance study. Ann Neurol 24:3, 1988 248. Fay AM, Kane SA, Kazim M et al: Magnetic resonance imaging of optic perineuritis. J Neuroophthalmol 17: 247, 1997 249. Dutton JJ, Anderson RL: Idiopathic inflammatory perioptic neuritis simulating optic nerve sheath
meningioma. Am J Ophthalmol 100:424, 1985 250. Hykin PG, Spalton DJ: Bilateral perineuritis of the optic nerves. J Neurol Neurosurg Psychiatry 54:375, 1991 251. Winterkorn JM, Odel JG, Behrens MM et al: J Neuroophthalmol 14:157, 1994 252. Frohman L, Wolansky L: Magnetic resonance imaginging of syphilitic optic perineuritis. J Neuroophthalmol 17:57, 1997 253. Beardsley TL, Brown SVL, Sydnor CF et al: Eleven cases of sarcoidosis of the optic nerve. Am J Ophthalmol 97:62, 1984 254. Vrabec TR, Augsburger JJ, Fisccher DH et al: Taches de bougie. Ophthalmology 102:1712, 1995 255. Akova YA, Kansu T, Duman S: Pseudotumor cerebri secondary to dural sinus thrombosis in neurosarcoidosis. J Clin Neuroophthalmol 13:188, 1993 256. Beck AD, Newman NJ, Grossniklaus HE et al: Optic nerve enlargement and chronic visual loss. Surv Ophthalmol 38:555, 1994 257. Newman LS, Rose CS, Maier LA: Sarcoidosis. N Engl J Med 336:1224, 1997 258. Belden CJ, Hamed L, Mancuso AA: Bilateral isolated retrobulbar optic neuropathy in limited Wegener's
granulomatosis. J Clin Neuroophthalmol 13:119, 1993 259. Slavin M, Glaser JS: Acute severe irreversible visual loss with sphenoethmoiditis: “posterior” orbital
cellulitis. Arch Ophthalmol 105:345, 1987 260. Kodsi SR, Younge BR, Leavitt JA et al: Intracranial plasma cell granuloma presenting as an optic neuropathy. Surv Ophthalmol 38:70, 1993 261. Ofner S, Baker RS: Visual loss in cryptococcal meningitis. J Clin Neuropophthalmol 7:45, 1987 262. Lee MS, Cooney EL, Stoessel KM et al: Varicella zoster virus retrobulbar optic neuritis preceding retinitis in
patients with acquired immunodeficiency syndrome. Ophthalmology 105:467, 1998 263. Borruat FX, Herbort CP: Herpes zoster ophthalmicus: anterior ischemic optic neuropathy and acyclovir. J Clin Neuroophthalmol 12:37, 1992 264. Lam BL, Barrett DA, Glaser JS et al: Visual loss from idiopathic intracranial pachymeningitis. Neurology 44: 694, 1994 265. Hayreh SS: The optic nerve head circulation in health and disease. Exp Eye Res 61:259, 1995 266. Onda E, Ciofi GA, Bacon DR, van Buskirk EM: Microvasculature of the human optic nerve. Am J Ophthalmol 120:92, 1995 267. Hattenhauer MG, Leavitt JA, Hodge DO et al: Incidence of nonarteritic anterior ischemic optic neuropathy. Am J Ophthalmol 123:103, 1997 268. IONDT Study Group: Characteristics of patients with nonarteritic anterior
ischemic neuropathy eligible for the ischemic optic neuropathy decompression
trial. Arch Ophthalmol 114:1366, 1996 269. Hayreh SS, Joos KM, Podhajsky PA et al: Systemic diseases associated with nonarteritic anterior ischemic optic
neuropathy. Am J Ophthalmol 118:766, 1994 270. Kline LB: Progression of visual field defects in ischemic optic neuropathy. Am J Ophthalmol 106:199, 1988 271. Movsas T, Kelman SE, Elman MJ et al: The natural course of non-arteritic ischemic optic neuropathy. Invest Ophthalmol Vis Sci 42:951, 1991 272. Borchert M, Lessell S: Progressive and recurrent nonarteritic anterior ischemic optic neuropathy. Am J Ophthalmol 106:443, 1988 273. Arnold AC, Hepler RS: Natural history of nonarteritic anterior ischemic optic neuropathy. J Neuroophthalmol 14:66, 1994 274. Gordon RN, Burde RM, Slamovits T: Asymptomatic optic disc edema. J Neuroophthalmol 17:29, 1997 275. Hayreh SS: Acute ischemic disorders of the optic nerve: pathogenesis, clinical manifestations, and
management. Ophthalmol Clin North Am 9:407, 1996 276. Olver JM, Spalton DJ, McCartney ACE: Microvascular study of the retrolaminar optic nerve in man: the possible
significance in anterior ischemic optic neuropathy. Eye 4:7, 1990 277. Arnold AC, Hepler RS: Fluorescein angiography in acute nonar-teritic anterior ischemic optic
neuropathy. Am J Ophthalmol 117:222, 1994 278. Kalenak JW, Kosmorsky GS, Rockwood EJ: Nonarteritic anterior ischemic optic neuropathy and intraocular pressure. Arch Ophthalmol 109:660, 1991 279. Hayreh SS, Podhajsky PA, Zimmerman B: Nonarteritic anterior ischemic optic neuropathy: time of onset of visual
loss. Am J Ophthalmol 124:641, 1997 280. Rizzo JF, Lessell S: Optic neuritis and ischemic optic neuropathy: overlapping clinical profiles. Arch Ophthalmol 109:1668, 1991 281. Trobe JD, Glaser JS, Cassady JC et al: Nonglaucomatous excavation of the optic disc. Arch Ophthalmol 98:1046, 1980 282. Trobe JD, Glaser JS, Cassady JC: Optic atrophy: differential diagnosis by fundus observation alone. Arch Ophthalmol 98:1040, 1980 283. Frisen L, Claesson M: Narrowing of the retinal arterioles in descending optic atrophy: a quantitative
clinical study. Ophthalmology 91:1342, 1984 284. Beri M, Klugman MR, Kohler JA et al: Anterior ischemic optic neuropathy. VII. Incidence of bilaterality and
various influencing factors. Ophthalmology 94:1020, 1987 285. Boone MI, Massry GG, Frankel RA et al: Visual outcome in bilateral nonarteritic anterior ischemic optic neuropathy. Ophthalmology 103:1223, 1996 286. WuDunn D, Zimmerman K, Sadun AA, Feldon SE: Comparison of visual function in fellow eyes after bilateral nonarteritic
anterior ischemic optic neutropathy. Ophthalmology 104:104, 1997 287. Lepore FE, Yarian DL: A mimic of the “exact diagnostic sign” of Foster Kennedy. Ann Ophthalmol 17:411, 1985 288. Fry CL, Carter JE, Kanter MC et al: Anterior ischemic optic neuropathy is not associated with carotid artery
atherosclerosis. Stroke 24:539, 1993 289. Arnold AC, Hepler RS, Hamilton DR, Lufkin RB: Magnetic resonance imaging of the brain in nonarteritic ischemic optic
neuropathy. J Neuroophthalmol 15:158, 1995 290. Giuffre G: Hematologic risk factors for anterior ischemic optic neuropathy. Neuroophthalmology 10:197, 1990 291. Watts MT, Greaves M, Rennie IG et al: Antiphospholipid antibodies in the aetiology of of ischaemic optic neuropathy. Eye 5:75, 1991 292. Worrall BB, Moazami G, Odel JG et al: Anterior ischemic optic neuropathy and activated protein C resistance: a
case report and review of the literature. J Neuroophthalmol 17:162, 1997 293. Talks SJ, Chong NH, Gibson JM, Dodson PM: Fibrinogen, cholesterol and smoking as risk factors for non-arteritic anterior
ischemic optic neuropathy. Eye 9:85, 1995 294. Beck RW, Servais GE, Hayreh SS: Anterior ischemic optic neuropathy. IX. Cup-to-disc ratio and its role
in pathogenesis. Ophthalmology 94:1503, 1987 295. Mansour AM, Shoch D, Logani S: Optic disk size in ischemic optic neuropathy. Am J Ophthalmol 106:587, 1988 296. Katz B, Spencer WH: Hyperopia as a risk factor for nonarteritic anterior ischemic optic neuropathy. Am J Ophthalmol 116:754, 1993 297. Arnold AC, Hepler RS, Lieber M, Alexander JM: Hyperbaric oxygen therapy for nonarteritic anterior ischemic optic neuropathy. Am J Ophthalmol 122:535, 1996 298. Johnson LN, Gould TJ, Krohel GB: Effect of levodopa and carbidopa on recovery of visual function in patients
with nonar-teritic anterior ischemic optic neuropathy of longer than
six months' duration. Am J Ophthalmol 121:77, 1996 299. Beck RW, Hayreh SS, Podhajsky PA et al: Aspirin in nonarteritic anterior ischemic optic neuropathy. Am J Ophthalmol 123:212, 1997 300. Kupersmith MJ, Frohman L, Sanderson M et al: Aspirin reduces the incidence of second eye NAION: a retrospective study. J Neuroophthalmol 17:250, 1997 301. Ischemic Optic Neuropathy Decompression Trial Research Group: Optic nerve
decompression surgery for nonarteritic anterior ischemic optic neuropathy (NAION) is
not effective and may be harmful. JAMA 273:625, 1995 302. Glaser JS, Teimory M, Schatz NJ: Optic nerve sheath fenestration for progressive ischemic optic neuropathy. Arch Ophthalmol 112:1047, 1994 303. Yee RD, Selky AK, Purvin VA: Outcomes of surgical and nonsurgical management of nonarteritic ischemic
optic neuropathy. Trans Am Ophthmalol Soc 91:227, 1993 304. Johnson LN, Arnold AC: Incidence of nonarteritic and arteritic anterior ischemic optic neuropathy. J Neuroophthalmol 14:38, 1994 305. Chung SM, Gay CA, McCrary JA: Nonarteritic ischemic optic neuropathy: the impact of tobacco use. Ophthalmology 101:779, 1994 306. Hayreh SS, Podhajsky PA, Zimmerman B: Ocular manifestations of cranial arteritis. Am J Ophthalmol 125:509, 1998 307. Tomlinson FH, Lie JT, Nienhuis BJ et al: Juvenile temporal arteritis revisited. Mayo Clin Proc 69:445, 1994 308. Weyand CM, Bartley GB: Giant cell arteritis: new concepts in pathogenesis and implications for
management. Am J Ophthalmol 123:392, 1997 309. Wagner AD, Bjornsson J, Bartley GB et al: Interferon gamma-producing T cells in giant cell vasculitis represent a
minority of tissue-infiltrating cells and located distant from the site
of pathology. Am J Pathol 148:1925, 1996 310. Ghanci FD, Dutton GN: Current concepts in giant cell (temporal) arteritis. Surv Ophthalmol 42:99, 1997 311. Hauser WA, Ferguson RH, Holley KE et al: Temporal arteritis in Rochester, Minnesota, 1951 to 1967. Mayo Clin Proc 46:597, 1971 312. Turnbull J: Temporal arteritis and polymyalgia rheumatica: nosographic and nosologic
considerations. Neurology 46:901, 1996 313. Hayreh SS, Podhajsky P: Visual field defects in anterior ischemic optic neuropathy. Doc Ophthalmol Proc Ser 19:53, 1979 314. Beri M, Klugman MR, Kohler JA et al: Anterior ischemic optic neuropathy. VII. Incidence of bilaterality and
various influencing factors. Ophthalmology 94:1020, 1987 315. Sebag J, Thomas JV, Epstein EL et al: Optic disc cupping in arteritic anterior ischemic optic neuropathy resembles
glaucomatous cupping. Ophthalmology 93:357, 1986 316. Melberg NS, Grand MG, Diekert JP et al: Cotton-wool spots and early diagnosis of giant cell arteritis. Ophthalmology 102:1611, 1995 317. Barricks ME, Traviesa DB, Glaser JS, Levy IS: Ophthalmoplegia in cranial arteritis. Brain 100:209, 1977 318. Bienfang DC: Loss of the ocular pulse in the acute phase of temporal arteritis. Acta Ophthalmol Suppl 191:35, 1989 319. Bosley TM, Savino PJ, Sergott RC et al: Ocular pneumoplethysmography can help in the diagnosis of giant-cell arteritis. Arch Ophthalmol 107:379, 1989 320. Siatkowski RM, Gass JDM, Glaser JS et al: Fluorescein angiography in the diagnosis of giant cell arteritis. Am J Ophthalmol 115:57, 1993 321. Slavin ML, Barondes MJ: Visual loss caused by choroidal ischemia preceding anterior ischemic optic
neuropathy in giant cell arteritis. Am J Ophthalmol 117:81, 1994 322. Ho AC, Sergott RC, Regillo CD et al: Color Doppler hemodynamics of giant cell arteritis. Arch Ophthalmol 112:938, 1994 323. Schmidt WA, Kraft HE, Vorpahl K et al: Color duplex ultrasonography in the diagnosis of temporal arteritis. N Engl J Med 337:1336, 1997 324. Andersson R, Malvall BE, Bengtsson BA: Long-term survival in giant cell arteritis including remporal arteritis
and polymyalgia rheumatica. Acta Med Scand 220:361, 1986 325. Aiello PD, Trautman JC, McPhee TJ et al: Visual prognosis in giant cell arteritis. Ophthalmology 100:550, 1993 326. Liu GT, Glaser JS, Schatz NJ, Smith JL: Visual morbidity in giant cell arteritis: clinical characteristics and
prognosis for vision. Ophthalmology 101:1779, 1994 327. Hayreh SS, Podhajsky PA, Raman R, Zimmerman B: Giant cell arteritis: validity and reliability of various diagnostic criteria. Am J Ophthalmol 123:285, 1997 328. Cullen JF: Ischemic optic neuropathy. Trans Ophthalmol Soc UK 87:759, 1967 329. Miller A, Green M: Simple rule for calculating normal erythrocyte sedimentation rate. BMJ 286:266, 1983 330. Gruener G, Merchut MP: Renal causes of elevated sedimentation rate in suspected temporal arteritis. J Clin Neuroophthalmol 12:272, 1992 331. Klein RG, Campbell RJ, Hunder GG, Carney JA: Skip lesions in temporal arteritis. Mayo Clin Proc 51:504, 1976 332. Nishino H, DeRemee RA, Rubino FA et al: Wegener's granulomatosis associated with vasculitis of the temporal
artery: report of five cases. Mayo Clin Proc 68:115, 1993 333. Levy MH, Margo CE: Temporal artery biopsy and sarcoidosis. Am J Ophthalmol 117:409, 1994 334. Goodman BW: Temporal arteritis. Am J Med 67:839, 1979 335. Wilkinson IMS, Russell RWR: Arteries of the head and neck in giant cell arteritis: a pathological study
to show the pattern of arterial involvement. Arch Neurol 27:378, 1972 336. Cornblath WT, Eggenberger ER: Progressive visual loss from giant cell arteritis despite high-dose intravenous
methylprednisolone. Ophthalmology 104:854, 1997 337. Gardiner PVG, Griffiths ID: Sudden death after treatment with pulsed methylprednisolone. BMJ 300:125, 1990 338. Matteson EL, Gold KN, Bloch DA et al: Long-term survival of patients with giant cell arteritis in the American
College of Rheumatology giant cell arteritis criteria cohort. Am J Med 100:193, 1996 339. van der Veen MJ, Dinant HJ, Vam Booma-Frankfort C et al: Can methotrexate
be used as a steroid sparing agent in the treatment of polymyalgia
rheumatica and giant cell arteritis? Ann Rheum Dis 55:218. 1996 340. Demaziere A: Dapsone in the long-term treatment of temporal arteritis. Am J Med 87:3, 1989 341. Skillern PG, Lockhart G: Optic neuritis and uncontrolled diabetes mellitus in 14 patients. Ann Intern Med 51:468, 1959 342. Lubow M, Makley TA: Pseudopapilledema of juvenile diabetes mellitus. Arch Ophthalmol 85:417, 1971 343. Barr CC, Glaser JS, Blankenship G: Acute disc swelling in juvenile diabetes: clinical profile and natural
history of 12 cases. Arch Ophthalmol 98:2185, 1980 344. Regillo CD, Brown GC, Savino PJ et al: Diabetic papillopathy: patient characteristics and fundus findings. Arch Ophthalmol 113:889, 1995 345. Carroll FD: Optic nerve complications of cataract extraction. Trans Am Acad Ophthalmol Otolaryngol 77:623, 1973 346. Hayreh SS: Anterior ischemic optic neuropathy. IV. Occurrence after cataract extraction. Arch Ophthalmol 98:1410, 1980 347. Serrano LA, Behrens MM, Carroll FD: Postcataract extraction ischemic optic neuropathy. Arch Ophthalmol 100:1177, 1982 348. Hamed LM, Purvin V, Rosenberg M: Recurrent anterior ischemic optic neuropathy in young adults. J Clin Neuroophthalmol 8:239, 1988 349. O'Hara M, O'Connor PS: Migrainous optic neuropathy. J Clin Neuro Ophthalmol 4:85, 1984 350. Katz B: Bilateral sequential migrainous ischemic optic neuropathy. Am J Ophthalmol 99:489, 1985 351. Toshniwal P: Anterior ischemic optic neuropathy secondary to cluster headache. Acta Neurol Scand 73:213, 1986 352. Hayreh SS: Anterior ischemic optic neuropathy. VIII. Clinical features and pathogenesis
of post-hemorrhagic amaurosis. Ophthalmology 94:1488, 1987 353. Williams EL, Hart WM, Tempelhoff R: Postoperative ischemic optic neuropathy. Anesth Analg 80:1018, 1995 354. Hollenhorst RW, Svein HJ, Benoit CF: Unilateral blindness occurring during anesthesia for neurosurgical operations. Arch Ophthalmol 52:819, 1954 355. Katz DM, Trobe JD, Cornblath WT, Kline LB: Ischemic optic neuropathy after lumbar spine surgery. Arch Ophthalmol 112:925, 1994 356. Jaben SL, Glaser JS, Daily M: Ischemic optic neuropathy following general surgical procedures. J Clin Neuroophthalmol 3:239, 1983 357. Johnson MW, Kincaid MC, Trobe JD: Bilateral retrobulbar optic nerve infarctions after blood loss and hypotension: a
clinicopathologic case study. Ophthalmology 94:1577, 1987 358. Connolly SE, Gordon KB, Horton JC: Salvage of vision after hypotension-induced ischemic optic neuropathy. Am J Ophthalmol 117:235, 1994 359. Shapira OM, Kimmel WA, Lindsey PS et al: Anterior ischemic optic neuropathy after open heart operations. Ann Thorac Surg 61:660, 1996 360. Slavin ML: Ischemic optic neuropathy after cardiac arrest. Am J Ophthalmol 104:435, 1987 361. Knox DL, Hanneken AM, Hollows FC et al: Uremic optic neuropathy. Arch Ophthalmol 106:50, 1988 362. Hamed LM, Winward KE, Glaser JS et al: Optic neuropathy in uremia. Am J Ophthalmol 108:30, 1989 363. Michaelson C, Behrens M, Odel J: Bilateral anterior ischemic optic neuropathy associated with optic disc
drusen and systemic hypotension. Br J Ophthalmol 73:767, 1989 364. Waybright EA, Selhorst JB, Combs J: Anterior ischemic optic neuropathy with internal carotid artery occlusion. Am J Ophthalmol 93:42, 1982 365. Brown GC: Anterior ischemic optic neuropathy occurring in association with carotid
artery obstruction. J Clin Neuroophthalmol 6:39, 1986 366. Bogousslavsky J, Regli F, Zografos L et al: Optico-cerebral syndrome: simultaneous hemodynamic infarction of optic
nerve and brain. Neurology 37:263, 1987 367. Leonard TJK, Sanders MD: Ischaemic optic neuropathy in pulseless disease. Br J Ophthalmol 67:389, 1983 368. Hall S, Barr W, Lie JT et al: Takayasu arteritis: a study of 32 North American patients. Medicine 64:89, 1995 369. Liebermann MF, Shahi A, Grenn WR: Embolic ischemic optic neuropathy. Am J Ophthalmol 86:206, 1978 370. Tomsak RL: Ischemic optic neuropathy associated with retinal embolism. Am J Ophthalmol 99:590, 1985 371. Beck RW, Gamel JW, Willcourt RJ et al: Acute ischemic optic neuropathy in severe preeclampsia. Am J Ophthalmol 90:342, 1980 372. DeFrancisco M, Savino PJ, Schatz NJ: Optic atrophy in acute intermittent porphyria. Am J Ophthalmol 87:221, 1979 373. Manor RS, Axer-Siegal R, Cohenn S et al: Bilateral anterior ischemic optic neuropathy, pseudoxanthoma elasticum, and
platelet hyperaggregability. Neuroophthalmology 6:173, 1986 374. Slavin ML, Barondes MJ: Ischemic optic neuropathy in sickle cell disease. Am J Ophthalmol 105:212, 1988 375. Perlman JI, Forman S, Gonzalez ER. Retrobulbar ischemic optic neuropathy
associated with sickle cell disease. J Neuroophthalmol 14:45, 1994 376. Sklar EML, Schatz NJ, Glaser JS et al: MR of vasculitis-induced optic neuropathy. AJNR Am J Neuroradiol 17: 121, 1996 377. Ahmadieh H, Roodpeyma S, Azarmina M et al: Bilateral simultaneous optic neuritis in chidhood systemic lupus erythematosus. J Neuroophthalmol 14:84, 1994 378. Frohman LP, Lama P: Annual review of systemic diseases: 1991996. I. J Neuroophthalmol 18:67, 1998 379. Tesar JT, McMillan V, Molina R et al: Optic neuropathy and central nervous system disease assoiciated with Sjögren's
syndrome. Am J Med 92:686, 1992 380. Hutchinson CH: Polyarteritis nodosa presenting as posterior ischemic optic neuropathy. J R Soc Med 77:1043, 1984 381. Massry GG, Chung SM, Selhorst JB: Optic neuropathy, headache, and diplopia with MRI suggestive of cerebral
arteritis in relapsing polychondritis. J Neuroophthalmol 15:171, 1995 382. Schmidt MH, Fox AJ, Nicolle DA: Bilateral anterior ischemic optic neuropathy as a presentation of Takayasu's
disease. J Neuroophthalmol 17:156, 1997 383. Isayama Y, Takahashi T, Inoue M et al: Posterior ischemic optic neuropathy. III. Clinical diagnosis. Ophthalmologica 187:141, 1983 384. Sawle GV, Sarkies NJC: Posterior ischaemic optic neuropathy due to internal artery occlusion. Neuroophthalmology 7:349, 1987 385. Sommer A, Tielsch JM, Katz J et al: Relationship between intra-ocular pressure and primary open-angle glaucoma
among white and black Americans. Arch Ophthalmol 109:10980, 1991 386. Caprioli J: Recognizing structural damage to the optic nerve head and nerve fiber layer
in glaucoma. Am J Ophthalmol 124:516, 1997 387. Kalenak JW, Kosmorsky GS, Hassenbusch SJ: Compression of the intracranial optic nerve mimicking unilateral normal-pressure
glaucoma. J Clin Neuroophthalmol 12: 230, 1992 388. Greenfield DS, Siatkowski RM, Glaser JG et al: The cupped disc: who needs neuroimaging? Ophthalmology 105:1866, 1998 389. Siegner SW, Netland PA: Optic disc hemorrhages and progression of glaucoma. Ophthalmology 103:1014, 1996 390. Brown GC, Shields JA: Tumors of the optic nerve head. Surv Ophthalmol 29:239, 1985 391. Dutton JJ: Gliomas of the anterior visual pathway. Surv Ophthalmol 38:427, 1994 392. Listernick R, Louis DN, Packer RJ et al: Optic pathway gliomas in children with neurofibromatosis 1: consensus statement
from NF1 Optic Pathway Glioma Task Force. Ann Neurol 41:143, 1997 393. Anderson DR, Spencer WH: Ultrastructural and histochemical observations of optic nerve gliomas. Arch Ophthalmol 83:324, 1970 394. Grimson BS, Perry DD: Enlargement of the optic disk in childhood optic nerve tumors. Am J Ophthalmol 97: 627, 1984 395. McDonnell P, Miller NR: Chiasmatic and hypothalamic extension of optic nerve glioma. Arch Ophthalmol 101:1412, 1983 396. Imes RK, Hoyt WF: Magnetic resonance imaging signs of optic nerve gliomas in neurofibromatosis 1. Am J Ophthalmol 111:729, 1991 397. Brodsky MC: The “pseudo-CSF” signal of orbital optic glioma on magnetic
resonance imaging: a signature of neurofibromatosis. Surv Ophthalmol 38:213, 1993 398. Gans MS, Frazier Byrne S, Glaser JS: Standardized a-scan echography in optic nerve disease. Arch Ophthalmol 105:1232, 1987 399. Sadun F, Hinton DR, Sadun AA: Rapid growth of an optic nerve ganglioglioma in a patient with neurofibromatosis 1. Ophthalmology 103:794, 1996 400. Jenkin D, Angyalfi S, Becker L et al: Optic glioma in children: surveillance, resection or irradiation. Int J Radiat Oncol Biolm Phys 25:215, 1993 401. Dutton JJ: Optic nerve sheath meningiomas. Surv Ophthalmol 37:167, 1992 402. Karp LA, Zimmerman LE, Borit A et al: Primary intraorbital meningiomas. Arch Ophthalmol 91:24, 1974 403. Ing EB, Garrity JA, Cross SA et al: Sarcoid masquerading as optic nerve sheath meningioma. Mayo Clin Proc 72:38, 1997 404. Zimmerman CF, Schatz NJ, Glaser JS: Magnetic resonance imaging of optic nerve meningiomas. Ophthalmology 97:585, 1990 405. Lindblom B, Truwit CL, Hoyt WF: Optic nerve sheath meningioma: definition of intraorbital, intracanalicular, and
intracranial components with magnetic resonance imaging. Ophthalmology 99:560, 1992 406. Eng TY, Albright NW, Kuwahara G et al: Precision radiation therapy for optic nerve sheath meningiomas. Int J Rad Oncol Biol Phys 22:1093, 1992 407. Lee AG, Woo SY, Miller NR et al: Improvement in visual function in an eye with presumed optic nerve sheath
meningioma after treatment with three-dimensional conformal radiation
therapy. J Neuroophthalmol 16:247, 1996 408. Hsu DW, Efird JT, Hedley-White ET: Progesterone and estrogen receptors in meningiomas: prognostic considerations. J Neurosurg 86:113, 1997 409. Olsen ME, Chernik NL, Posner JB: Infiltration of the leptomeninges by systemic cancer: a clinical and pathologic
study. Arch Neurol 30:122, 1974 410. Little JR, Dale AJD, Okazaki H: Meningeal carcinomatosis. Arch Neurol 30:138, 1984 411. Christmas NJ, Mead MD, Richardson EP, Albert DM: Secondary optic nerve tumors. Surv Ophthalmol 36:196, 1991 412. McFadzean R, Brosnahan D, Doyle D et al: A diagnostic quartet in leptomeningeal infiltration of the optic nerve
sheath. J Neuroophthalmol 14:175, 1994 413. Krol G, Sze G, Malkin M et al: MR of cranial and spinal meningeal carcinomatosis: comparison with CT and
myelography. AJNR Am J Neuroradiol 9:709, 1988 413a. de la Sayette V, Bertran F, Honnorat J et al: Paraneoplastic cerebellar syndrome and optic neuritis with anti-CV2 antibodies: clinical
response to excision of the primary tumor. Arch Neurol 55:405, 1998 414. Siatkowski RM, Lam BL, Schatz NJ et al: Optic neuropathy in Hodgkin's disease. Am J Ophthalmol 114:625, 1992 415. Strominger MB, Schatz NJ, Glaser JS: Lymphomatous optic neuropathy. Am J Ophthalmol 116:774, 1993 416. Kashani AA, Kerman BM: Central nervous system malignant B-cell lymphoma identified with standardized
echography of the optic nerve. J Neuroophthalmol 17:243, 1997 417. Kodsi SR, Younge BR, Leavitt JA et al: Intracranial plasma cell granuloma presenting as an optic neuropathy. Surv Ophthalmol 38:70, 1993 418. Nikaido H, Mishima H, Ono H et al: Leukemic involvement of the optic nerve. Am J Ophthalmol 105:294, 1988 419. Kaikov Y: Optic nerve head infiltration in acute leukemia in children: an indication
for emergency optic nerve radiation therapy. Med Pediatr Oncol 826:101, 1996 420. Cramer SC, Glaspy JA, Efird JT et al: Chronic lymphocytic leukemia and the central nervous system: a clinical
and pathologic study. Neurology 46:19, 1996 421. Purvin V, vanDyk HJL: Primary reticulum cell sarcoma of the brain presenting as steroid-responsive
optic neuropathy. J Clin Neuroophthalmol 4:15, 1984 422. Wagoner MD, Gonder JR, Albert DM: Intraocular reticulum cell sarcoma. Ophthalmology 87:724, 1980 423. Khawly JA, Rubin P, Petros W et al: Retinopathy and optic neuropathy in bone marrow transplantation for breast
cancer. Ophthalmology 103:87, 1996 423a. Purvin, V: Anterior ischemic optic neuropathy secondary to interferon-alpha. Arch Ophthalmol 113:1041, 1995 424. Caraceni A, Martini C, Spatti G et al: Recovering optic neuritis during systemic cisplatin and carboplatin chemotherapy. Acta Neurol Scand 96:260, 1997 425. DeLano MC, Fun FY, Zinreich SJ: Relationship of the optic nerves to the posterior paranasal sinuses: a
CT anatomic study. AJNR Am J Neuroradiol 17:669, 1996 426. Buss DR, Tse Dt, Farris BK: Ophthalmic complications of sinus surgery. Ophthalmology 97:612, 1990 427. Hayman, Carter K, Schiffman JS et al: A sellar misadventure: imaging considerations. Surv Ophthalmol 41:252, 1996 428. Goodwin JA, Glaser JS. Chiasmal syndrome in sphenoid sinus mucocele. Ann
Neurol 4:440, 1978 429. Johnson LN, Hepler RS, Yee RD et al: Sphenoid sinus mucocele (anterior clinoid variant) mimicking diabetic ophthalmoplegia
and retrobulbar neuritis. Am J Ophthalmol 102:111, 1986 430. Dooley DP, Hollsten DA, Grimes SR et al: Indolent orbital apex syndrome caused by occult mucormycosis. J Clin Neuroophthalmol 12:245, 1992 431. Hutnik, Nicolle DA, Munoz DG: Orbital aspergillosis: a fatal masquerader. J Neuroophthalmol 17:257, 1997 432. Frohman LP, Lama P: Annual review of systemic diseases: 1991996. J Neuroophthalmol 18:67, 1998 433. Newman NJ, Slamovits TL, Friedland S et al: Neuro-ophthalmic manifestations of meningocerebral inflammation from the
limited form of Wegener's granulomatosis. Am J Ophthalmol 120:613, 1995 434. Goldberg RA, Weisman JS, McFarland JE et al: Orbital inflammation and optic neuropathies associated with chronic sinusitis
of intranasal cocaine abuse: possible role of contiguous inflammation. Arch Ophthalmol 107:831, 1989 435. Harbison JW, Lessell S, Selhorst JB: Neuro-ophthalmology of sphenoid sinus carcinoma. Brain 107:855, 1984 436. Berman EL, Chu A, Wirtschafter JD et al: Esthesioneuroblastoma presenting as sudden unilateral visual loss. J Clin Neuroophthalmol 12:31, 1992 437. Neigel JM, Rootman J, Belkin RI et al: Dysthyroid optic neuropathy: the crowded orbital apex syndrome. Ophthalmology 95:1515, 1988 438. Glatt HJ: Optic nerve dysfunction in thyroid eye disease: a clinician's perspective. Radiology 200:26, 1996 439. Birchall D, Goodall KL, Noble JL et al: Graves' ophthalmopathy: intracranial prolapse on CT as an indicator of
optic nerve compression. Radiology 200:123, 1996 440. Hufnagel TJ, Hickey WF, Cobbs WH et al: Immunohistochemical and ultrastructural studies on the exenterated orbital
tissues of a patient with Graves' disease. Ophthalmology 91:1411, 1984 441. Bartley GB, Fatourechi V, Kadrmas EF et al: Clinical features of Graves' ophthalmopathy in an incidence cohort. Am J Ophthalmol 121:284, 1996 442. Trobe JD, Glaser JS, LaFlamme P: Dysthyroid optic neuropathy: clinical profile and rationale for management. Arch Ophthalmol 179:285, 1978 443. Kao SCS, Kendler DL, Nugent RA et al: Radiotherapy in the management of thyroid orbitopathy: computed tomography
and clinical outcomes. Arch Ophthalmol 111:819, 1993 444. Jacobson DM, Warner JJ, Broste SK: Optic nerve contact and compression by the carotid artery in asymptomatic
patients. Am J Ophthalmol 123:677, 1997 445. Lindenberg R, Walsh FB, Sacks JG: Neuropathology of Vision: An Atlas. Philadelphia, Lea & Febiger, 1973 446. Golnik KC, Hund PW, Stroman GA et al: Magnetic resonance imaging in patients with unexplained optic neuropathy. Ophthalmology 103:515, 1996 447. Colapinto EV, Cabeen MA, Johnson LN: Optic nerve compression by a dolichoectatic internal carotid artery: case
report. Neurosurgery 39:604, 1996 448. Miller NR, Savino PJ, Schneider T: Rapid growth of an intracranial aneurysm causing apparent retrobulbar optic
neuritis. J Neuroophthalmol 15:212, 1995 449. Chan JW, Hoyt WF, Ellis WG et al: Pathogenesis of acute monocular blindness from leaking anterior communicating
artery aneurysms: report of six cases. Neurology 48:680, 1997 450. Samples JR, Younge BR: Tobacco-alcohol amblyopia. J Clin Neuroophthalmol 1:213, 1981 451. Frisen L: Fundus changes in acute malnutritional optic neuropathy. Arch Ophthalmol 101:577, 1983 452. Kupersmith MJ, Weiss PA, Carr RE: The visual-evoked potential in tobacco-alcohol and nutritional amblyopia. Am J Ophthalmol 95:307, 1983 453. van Noort BAA, Bos PJM, Klopping C et al: Optic neuropathy from thiamine deficiency in a patient with ulcerative
colitis. Doc Ophthalmol 67:45, 1987 454. Stambolian D, Behrens MM: Optic neuropathy associated with B12 deficiency. Am J Ophthalmol 83:465, 1977 455. Troncoso J, Mancall EL, Schatz NJ: Visual evoked responses in pernicious anemia. Arch Neurol 36:168, 1979 456. Rizzo JF, Lessell S: Tobacco amblyopia. Am J Ophthalmol 116:84, 1993 457. Cuba Neuropathy Field Investigation Team: Epidemic optic neuropathy in
Cuba: clinical characterization and risk factors. N Engl J Med 333:1176, 1955 458. Freeman AG: Optic neuropathy and chronic cyanide toxicity. Lancet 1:441, 1986 459. McKellar MJ, Hidajat RR, Elder MJ: Acute ocular methanol toxicity: clinical and electrophysiologic features. Aust N Z J Ophthalmol 3:225, 1997 460. Sharpe JA, Hostovsky M, Bilbao JM et al: Methanol optic neuropathy: a histopathological study. Neurology 32: 1093, 1982 461. Fasler JJ, Rose FC: West Indian amblyopia. Postgrad Med J 56:494, 1980 462. Grant WM: Toxicology of the Eye. Springfield, IL, Charles C Thomas, 1986 463. Kumar A, Sandramouli S, Verma L et al: Ocular ethambutol toxicity: is it reversible? J Clin Neuroophthalmol 13:15, 1993 464. Helm G, Holland G: Ocular tuberculosis. Surv Ophthalmol 38:230, 1993 465. Salmon JF, Carmichael TR, Welsh NH: Use of contrast sensitivity measurement in the detection of subclinical
ethambutol toxic optic neuropathy. Br J Ophthalmol 71: 192, 1987 466. Ricoy JR, Ortega A, Cabello A: Subacute myelo-optic neuropathy (SMON). J Neurol Sci 53:241, 1982 467. Pittman FE, Westphal MC: Optic atrophy following treatment with diiodohydroxyquin. Pediatrics 53:81, 1974 468. Godel V, Nemet P, Lazar M: Chloramphenicol optic neuropathy. Arch Ophthalmol 98:1417, 1980 469. Klingele TG, Burde RM: Optic neuropathy associated with penicillamine therapy in a patient with
rheumatoid arthritis. J Clin Neuroophthalmol 4:75, 1984 470. Ehyai A, Freemon FR: Progressive optic neuropathy and sensorineural hearing loss due to chronic
glue sniffing. J Neurol Neurosurg Psychiatry 46:349, 1983 471. Adams JW, Bofenkamp TM, Kobrin J et al: Recurrent acute toxic optic neuropathy secondary to 5-FU. Cancer Treat Rep 68:565, 1984 472. Pickrell L, Purvin V: Ischemic optic neuropathy secondary to intracarotid infusion of BCNU. J Clin Neuroophthalmol 7:87, 1987 473. Slamovits TL, Burde RM, Klingele TG: Bilateral optic atrophy caused by chronic oral ingestion and topical application
of hexachlorophene. Am J Ophthalmol 89:676, 1980 474. Coskucan NM, Jabs DA, Dunn JP et al: The eye in bone marrow transplantation. VI. Retinal complications. Arch Ophthalmol 112:372, 1994 475. Gittinger JW, Asdourian GK: Papillopathy caused by amiodarone. Arch Ophthalmol 105:349, 1987 476. Garrett SN, Kennedy JJ, Schiffman JS: Amiodarone optic neuropathy. J Clin Neuroophthalmol 8:105, 1988 477. Matthews GP, Sandberg MA, Berson EL: Foveal cone electro-retinograms in patients with central visual loss of
unexplained etiology. Arch Ophthalmol 110:1568, 1992 478. Steinsapir KD, Goldberg RA: Traumatic optic neuropathy. Surv Ophthalmol 38:487, 1994 479. Kline LB, McCluskey MM, Skalka HW: Imaging techniques in optic nerve evulsion. J Clin Neuroophthalmol 8:281, 1988 480. Brodsky MC, Wald KJ, Chen S et al: Protracted posttraumatic optic disc swelling. Ophthalmology 102:1628, 1995 481. Quigley HA, Davis EB, Anderson DR: Descending optic nerve degeneration in primates. Invest Ophthalmol Vis Sci 16:861, 1977 482. Cheney ML, Blair PA: Blindness as a complication of rhinoplasty. Arch Otolaryngol Head Neck Surg 113:768, 1987 483. Callahan MA: Prevention of blindness after blepharoplasty. Ophthalmology 90:1047, 1983 484. Horton JC, Hoyt WF, Foreman DS et al: Confirmation by magnetic resonance imaging of optic nerve injury after
retrobulbar anesthesia. Arch Ophthalmol 114:351, 1996 485. Devoto MH, Kersten RC, Zalta AH et al: Optic nerve injury after retrobulbar anesthesia. Arch Ophthalmol 115:687, 1997 486. Liu C, Youl B, Moseley I: Magnetic resonance imaging of the optic nerve in extremes of gaze: implications
for the positioning of the globe for retrobulbar anaesthesia. Br J Ophthalmol 76:728, 1992 487. Hollenhorst RW, Svien HJ, Benold CF: Unilateral blindness occurring during anesthesia for neurosurgical operations. Arch Ophthalmol 52:819, 1954 488. Jampol LM, Goldbaum M, Rosenberg M, Bahr R: Ischemia of ciliary arterial circulation from ocular compression. Arch Ophthalmol 93:1311, 1975 489. Chou P-I, Sadun AA, Lee H: Vasculature and morphometry of the optic canal and intracanalicular optic
nerve. J Neuroophthalmol 15:186, 1995 490. Lessell S: Indirect optic nerve trauma. Arch Ophthalmol 107:382, 1989 491. Cook MW, Levin LA, Joseph MP et al: Traumatic optic neuropathy: a meta-analysis. Arch Otolaryngol Head Neck Surg 122:389, 1996 492. Ross HS, Rosenberg S, Friedman AH: Delayed radiation necrosis of the optic nerve. Am J Ophthalmol 76:683, 1973 493. Shukovsky LJ, Fletcher GH: Retinal and optic nerve complications in a high dose irradiation technique
of ethmoid sinus and nasal cavity. Radiology 104:629, 1972 494. Salz JJ, Donin JF: Blindness after burns. Can J Ophthalmol 7:243, 1972 | 
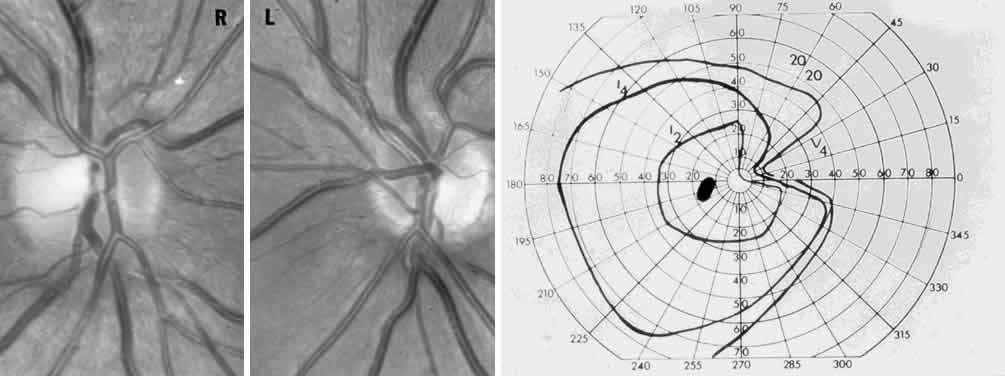
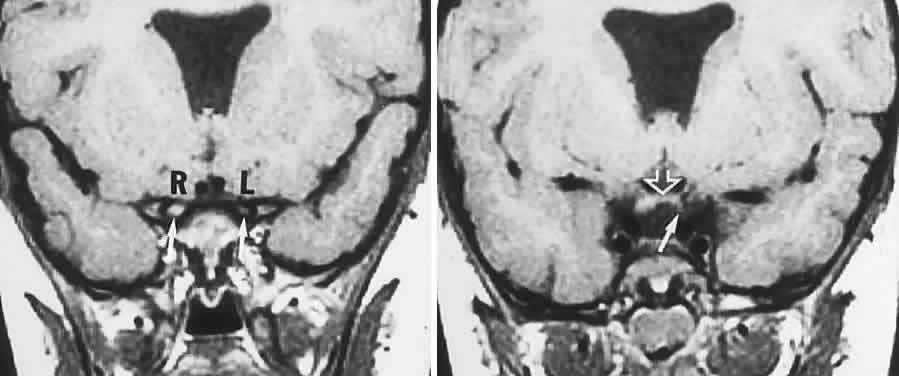
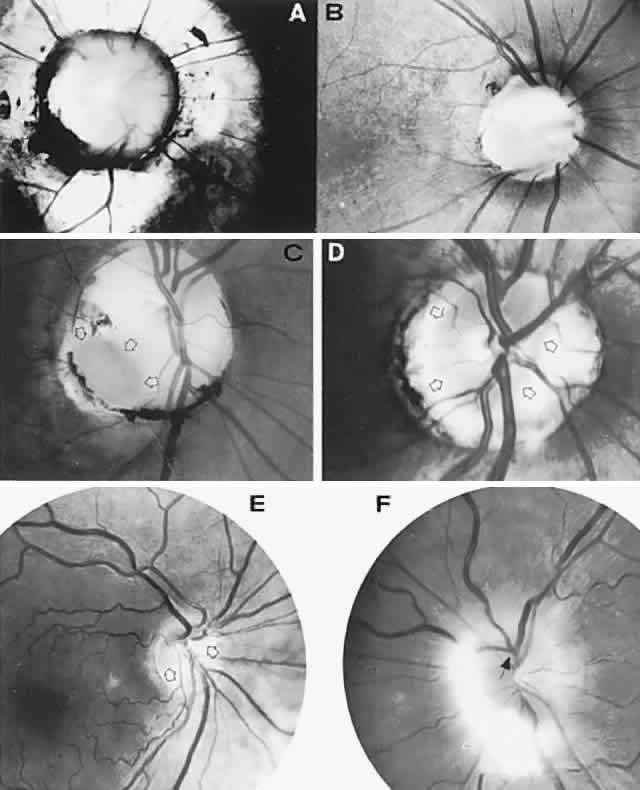
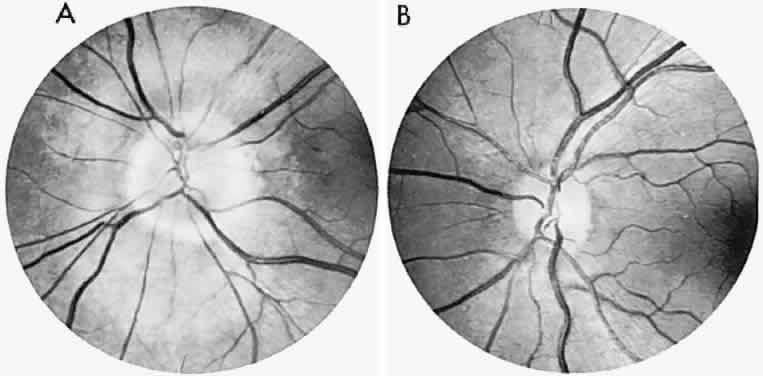
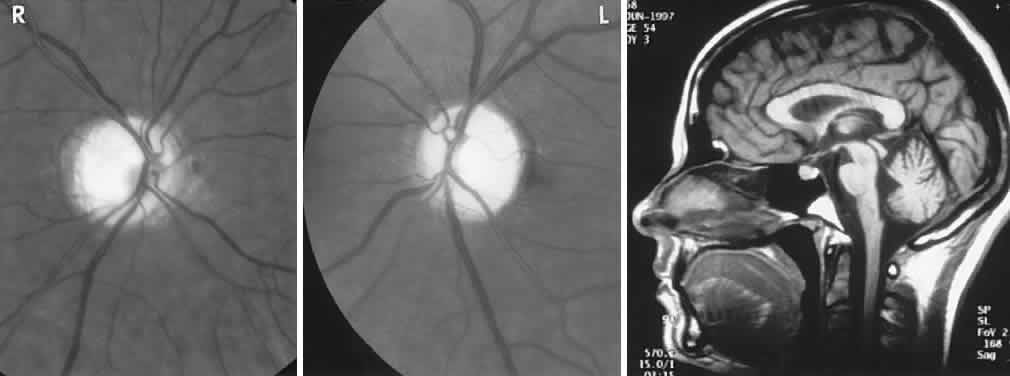
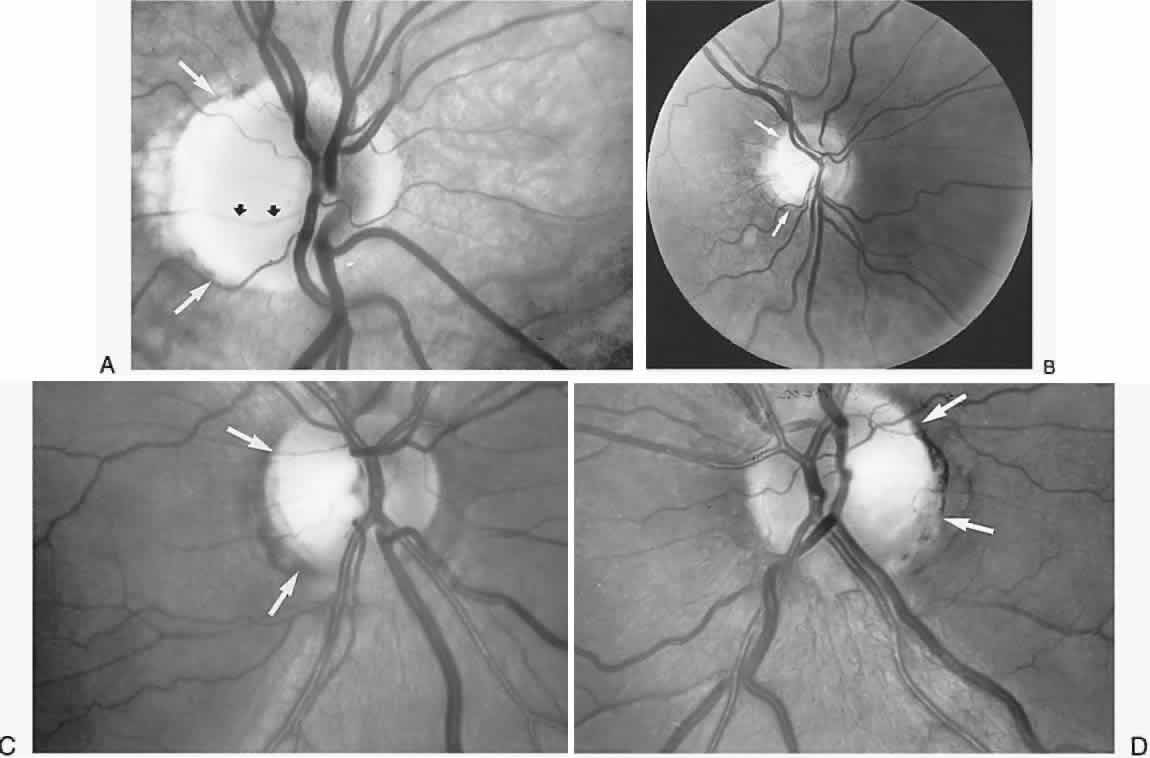

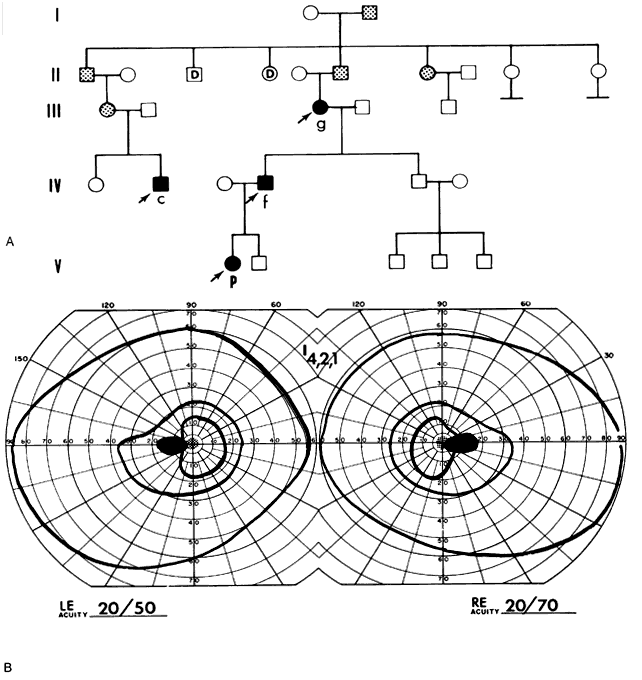
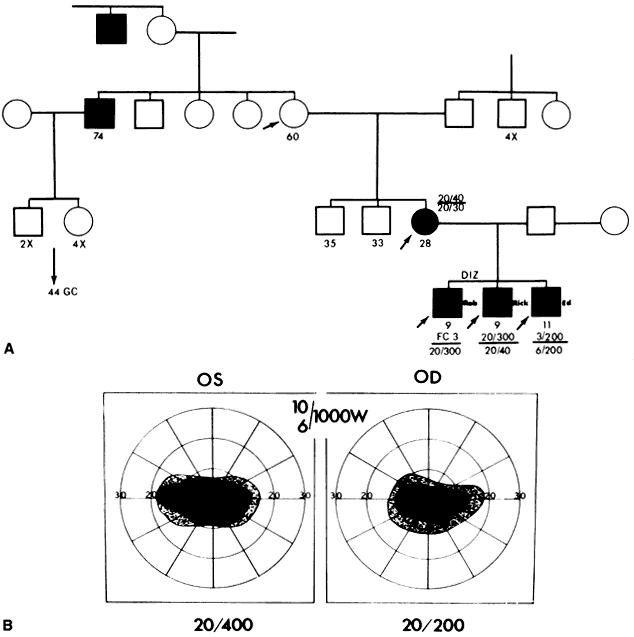
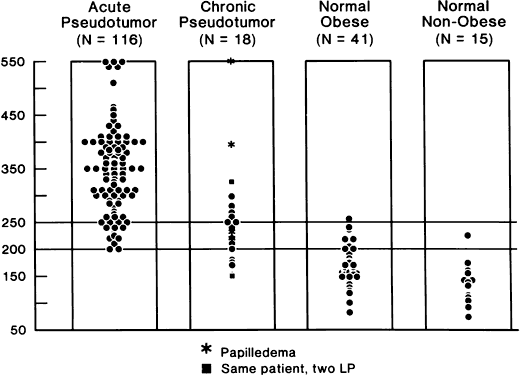
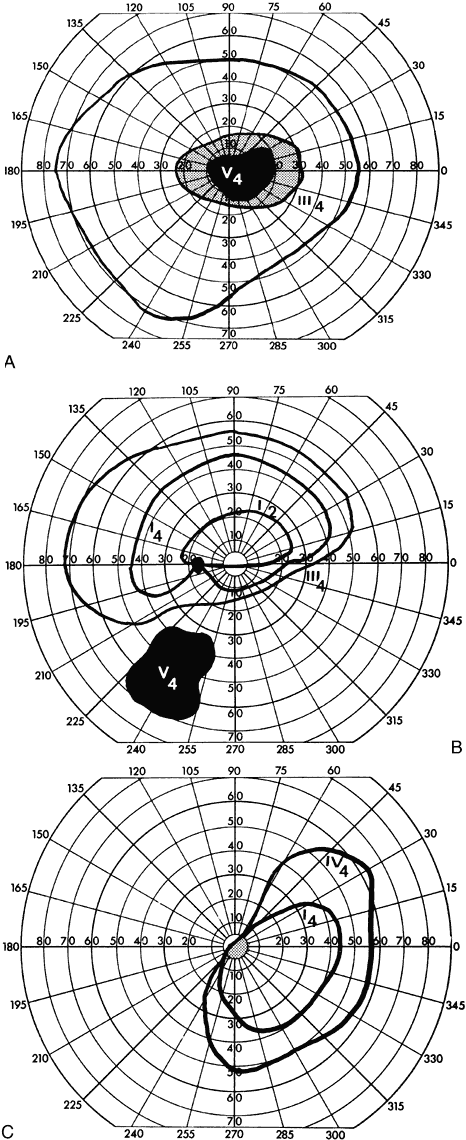
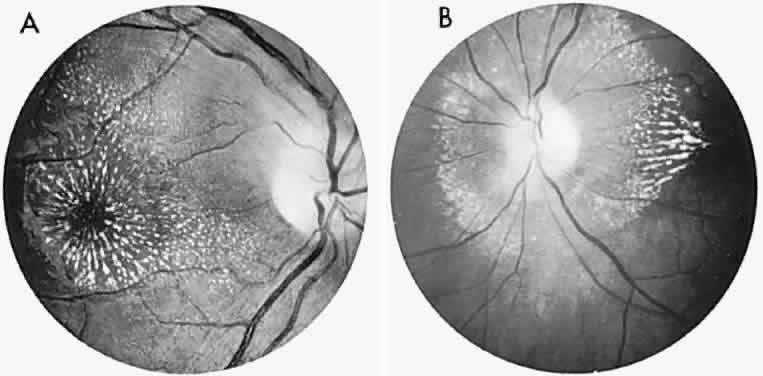
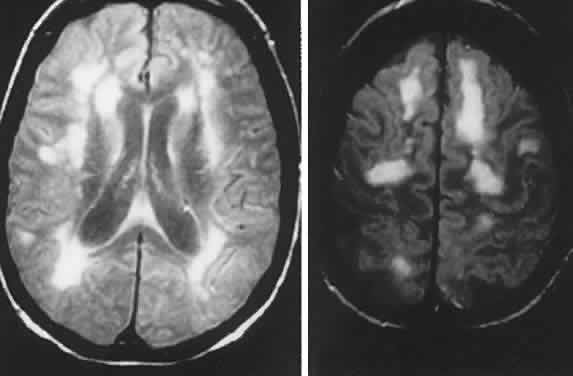

 40%
40% 40
40